Introduction
Liver cancer, which is one of the most common tumors
worldwide, has become difficult to treat due to its high malignancy
and mortality rates (1). The
mortality rate of liver cancer has increased from 7.5 to 11.2 in
men and from 2.8 to 3.8 in women (per 100,000 persons) between 2000
and 2015, worldwide (2). A high
amount of research has been performed to identify effective
reagents for the treatment liver cancer (3). So far, chemotherapy remains the most
widely used treatment for liver cancer; however, most conventional
chemotherapeutic drugs, including cisplatin, 5-fluorouracil, and
doxorubicin (Dox) exhibit poor efficiency in the treatment of
hepatocellular carcinoma (HCC), with a <10% inhibition growth
rate (4). A previous study has also
confirmed the ineffectiveness of conventional chemotherapy whether
they were administered intravenously or intra-arterially (5). Sorafenib® is the only
tyrosine kinase inhibitor for HCC treatment approved by the Food
and Drug Administration (FDA) of the United States of America;
however, its limited efficacy and adverse side effects, including
hand/food/skin reactions, asthenia, diarrhea, and arterial
hypertension, highlights whether it is suitable for use in a
clinical setting (5,6).
Multidrug resistance (MDR) also contributes to the
intractability of HCC. MDR is defined as a process in which cancer
cells gain resistance to multiple chemotherapeutic drugs with
different structures and mechanisms of action (7). MDR has been reported to be responsible
for >90% of cases in which chemotherapy had failed and the tumor
had recurred (8). Therefore,
identification of efficient drugs to combat MDR has become an
important issue in the medical field. Natural products have great
potential for drug discovery and constitute a large number of
chemotherapy agents in cancer treatment. For example, the discovery
of vinblastine and vincristine have been developed for use as
anticancer agents from natural sources (9). An increasing number of compounds
derived from natural resources have been approved as anticancer
drugs by the FDA, such as camptothecin, paclitaxel, anthracyclines
and taxanes (10–12). Among these, drugs which induce cancer
cell cycle arrest or apoptosis are a big part. It is widely
accepted that cell cycle arrest may result from DNA damage
(13). ATM, a serine/threonine
protein kinase, activates checkpoint signaling upon DNA
double-stranded breaks (DSBs), thereby acting as a DNA damage
sensor and playing a significant role in cell cycle arrest
(14).
Digitoxin is a natural cardiac glycoside derived
from Digitalis (15). As a
potent inhibitor of Na+/K+-ATPase, digitoxin
has been clinically used for congestive heart failure for more than
40 years (16). Previously, a number
of studies have focused on the anticancer potential of digitoxin
and verified notable antitumor activities of digitoxin in lung
cancer (17), pancreatic cancer
(18), glioma (19), liver cancer (20), prostate cancer (21) and melanoma (22). Mechanistic studies have revealed that
the growth inhibitory effect of digitoxin was associated with the
induction of apoptosis (23),
inhibition of epithelial-mesenchymal transition (21) and suppression of cancer cell stemness
(24); however, the underlying
mechanism of action of digitoxin against multidrug-resistant HCC
cells has not been fully elucidated.
In the present study, a library of 78 natural
compounds, including digitoxin was screened in the Dox-resistant
cancer cell line, HepG2/ADM. Further investigations demonstrated
that digitoxin displayed an inhibitory effect on
multidrug-resistant HepG2/ADM cells through G2/M cell
cycle arrest via the serine/threonine-protein kinase ATR
(ATR)-serine/threonine-protein kinase Chk2 (CHK2)-M-phase inducer
phosphatase 3 (CDC25C) signaling pathway and mitochondrial
apoptosis. The findings of the present study suggested that
digitoxin may be developed into a chemotherapeutic agent for
patients with HCC.
Materials and methods
Reagents and antibodies
A library of 78 natural compounds was obtained from
Target Molecule Corp. Digitoxin (≥98% pure) was purchased from
Baoji Herbest Bio-Tech Co., Ltd. MTT was supplied by Sigma-Aldrich
(Merck KGaA). An Annexin-V-FITC/propidium iodide (PI) staining
assay kit was obtained from Beyotime Institute of Biotechnology.
The bicinchoninic protein assay kit (BCA) was purchased from Thermo
Fisher Scientific Inc., while PI and 4′,6-dimidyl-2-phenylindole
(DAPI) were purchased from Roche Diagnostics (Shanghai) Co. Ltd.
Primary antibodies against cyclin-dependent kinase 1 (CDK1, #9116),
cyclin B1 (#4138), phosphorylated (p)-CDK1 (Thr14) (#2543),
p-histone H2AX (γH2AX, #9718), ATR (#2790), p-ATR (Ser428) (#2853),
CHK2 (#6334), p-Chk2 (Thr68) (#2197), CDC25C (#4688), p-CDC25C
(Thr48) (#12028), Bax (#5023), Bcl-2 (#15071), cytochrome c
(#11940), caspase-9 (#9508) and-3 (#9662), cleaved-caspase-3
(#9579) and −9 (#20750), cleaved poly (ADP-ribose) polymerase
(PARP) (#5625), β-actin (#4970) and the horse-radish peroxidase
(HRP)-conjugated secondary antibodies (Anti-mouse IgG, #7076;
Anti-rabbit IgG, #7074), Alexa Fluor 647-conjugated anti-rabbit IgG
(H+L) (#4414) were obtained from Cell Signaling Technology Inc.,
(dilution of primary antibodies, 1:1,000; dilution of secondary
antibodies, 1:2,000).
Cell line and cell culture
The Dox-resistant human HCC cell line, HepG2/ADM was
provided by Professor Kwok-Pui Fung (The Chinese University of Hong
Kong, Hong Kong, China). HepG2/ADM cells were cultured in RPMI 1640
medium supplemented with Dox (1.2 µM, Sigma-Aldrich), 1%
penicillin-streptomycin (PS), and 10% fetal bovine serum (FBS) to
maintain the multidrug-resistant characteristics of the HepG2/ADM
cell line. RPMI 1640 medium, PS, and FBS were supplied by Thermo
Fisher Scientific Inc.. Cells were incubated at 37°C in a
humidified incubator with 5% CO2.
Compound library screening
The cytotoxicity screening of the 78 natural
compounds in the library against HepG2/ADM cells was performed via
the MTT assay. Cells (5,000/well) were seeded into 96-well plates
and cultured overnight at 37°C. After treatment with 78 natural
compounds (0.1 µM) for 72 h at 37°C, respectively, cells were
incubated with 20 µl MTT (5 mg/ml) at 37°C for 3 h. The formazan
crystals were dissolved in 100 µl dimethlysulfoxide (DMSO) and the
absorbance of each well was recorded at 595 nm wavelengths using a
microplate reader (Beckman Coulter Inc.).
Cell viability assay
Viability of HepG2/ADM cells was determined using a
MTT assay. Cells (5,000/well) were seeded in 96-well plates and
cultured overnight. Following treatment with digitoxin at
concentrations ranging from 3.906–1,000.000 nM for 24, 48 and 72 h,
respectively, cells were exposed to 20 µl MTT (5 mg/ml) and
incubated at 37°C for 3 h. The formazan crystals were dissolved
with 100 µl DMSO and the absorbance was measured at 595 nm using a
microplate reader (Beckman Coulter Inc.). As previously described
(25), cells treated with medium
containing 0.2% DMSO for 24, 48 or 72 h were considered as 100%
viable, respectively.
Cell cycle analysis
HepG2/ADM cells (3×105/well) were seeded
in 6-well plates and cultured overnight, then treated with
digitoxin at 4, 20 and 100 nM for 24 h or 20 nM digitoxin for 12,
24 and 36 h, respectively. Following fixation and permeabilization
with pre-cooled 75% ethanol at 4°C overnight, cells were stained
with 0.2 mg/ml PI and 0.1 mg/ml RNase in the dark at room
temperature for 15 min. The PI fluorescence of the cells was
analyzed using an EPICS-X flow cytometry (Beckman Coulter, Inc.)
Then, the phase distribution of cell cycle was analyzed using
ModFit LT v3.1 software (Verity Software House, Inc.).
Western blot analysis
Following treatment with digitoxin for 24 h,
HepG2/ADM cells were collected using trypsinization. Total cellular
protein was extracted using the radioimmunoprecipitation assay
(RIPA) lysis buffer (containing 1 mM PMSF, 1X phosphatase inhibitor
and 1X protease inhibitor, Beyotime Institute of Biotechnology).
Then, a BCA assay kit was used to quantify the protein
concentration. Proteins (30 µg/lane) were separated using 12%
SDS-PAGE gels and then transferred onto PVDF membranes. The
membranes were blocked with 5% skimmed milk at room temperature for
1 h and probed with the primary antibodies at a dilution of 1:1,000
overnight at 4°C. After incubation with secondary antibody at a
dilution of 1:2,000 for 1 h at room temperature, the protein bands
were visualized using an ECL detection kit (Millipore, Merck KGaA)
and quantified using the ImageJ software v1.8.0 (National
Institutes of Health). β-actin was used as the loading control.
Immunofluorescence
HepG2/ADM cells were treated with digitoxin for 24
h. Then, cells were fixed with 4% paraformaldehyde (PFA) for 15 min
at room temperature. After blocking with 5% bovine serum albumin
(BSA; Sigma-Aldrich; Merck KGaA) containing 0.4% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 1 h at room temperature, the cells
were incubated with the γH2AX primary antibody (1:1,000) overnight
at 4°C and Alexa Fluor 647-conjugated secondary antibody (1:2,000)
for 1 h at room temperature. Fluorescence was observed using a
confocal microscope with a 40× magnification (Axio Vert.A1; Zeiss
GmbH).
Annexin-V-FITC/PI staining assay
Following treatment with digitoxin for 24 and 48 h,
respectively, HepG2/ADM cells were collected and stained with
Annexin-V-FITC/PI for 15 min in darkness at room temperature. The
cell apoptotic rates were analyzed using an Epics XL flow cytometer
(Beckman Coulter Inc.). The following wavelengths were used 488
(excitation) and 525 (emission) nm for Annexin V-FITC; and 488
(excitation) and 620 nm (emission) for PI. The data was quantified
using the FlowJo v7.6 software (FlowJo LLC).
Statistical analysis
All experiments were performed at least three times.
Results are presented as the mean ± SEM. GraphPad Prism v7.0
(GraphPad Software Inc.) was used for statistical analysis. One-way
analysis of variance (ANOVA) followed by a Tukey's post hoc test
was used for multiple comparison. P<0.05 was considered to
indicate a statistically significant difference.
Results
Digitoxin shows cytotoxicity towards
HepG2/ADM cells
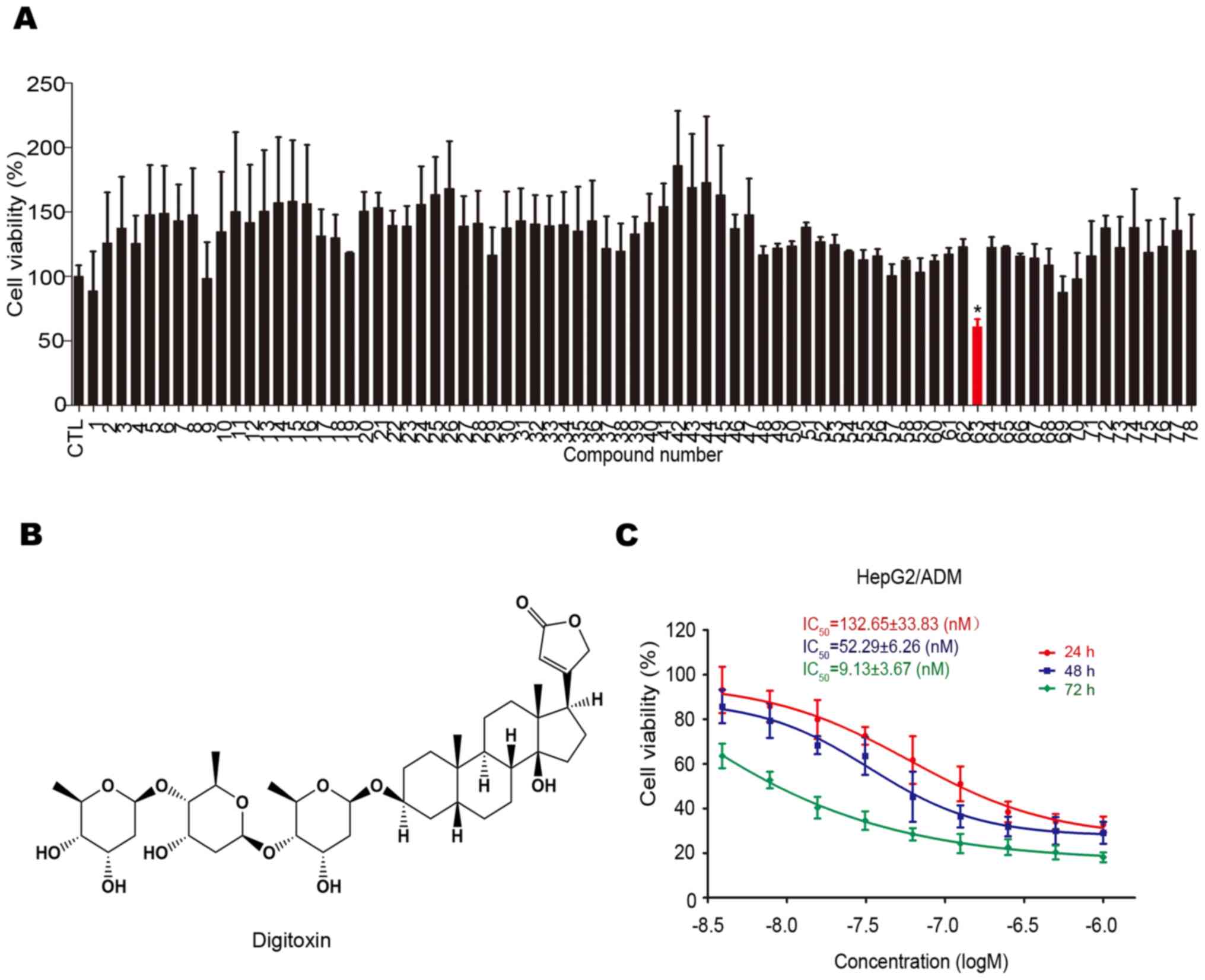
The cytotoxicity of 78 natural compounds on
HepG2/ADM cells were determined using the MTT assay. As shown in
Fig. 1A, digitoxin (No. 63; chemical
structure shown in Fig. 1B) was
found to have a greater cytotoxic effect on HepG2/ADM cells.
Subsequently, the anti-HCC effect of digitoxin on HepG2/ADM cells
was assessed using the MTT assay. As shown in Fig. 1C, digitoxin decreased the viability
of HepG2/ADM cells in a dose-dependent manner, with IC50
values of 132.65±33.83, 52.29±6.26 and 9.13±3.67 nM following
treatment for 24, 48 and 72 h, respectively. The concentrations
used in the cell viability assay were based on the results of the
pre-experiment.
Digitoxin blocks the HepG2/ADM cell
cycle at G2/M phase
To investigate whether the inhibitory effect of
digitoxin on HepG2/ADM cells was associated with cell cycle arrest,
DNA content analysis was performed using flow cytometry. The cell
population at G2/M phase increased from 23.61 to 46.87%
with 0, 12, 24, and 36 h (0 h as the CTL group) following 20 nM
digitoxin treatment, while 0, 4, 20, and 100 nM digitoxin treatment
(0 nM as the CTL group) for 24 h also resulted in an increase in
the number of cells at G2/M phase from 23.64 to 41.40%,
indicating that digitoxin induced G2/M cell cycle arrest
in HepG2/ADM cells (Fig. 2A and B).
CDK1 and cyclin B1, two key regulators of G2/M
transition, have been found to be involved in modulating the cell
cycle by forming the CDK1/Cyclin B1 complex (26). Digitoxin treatment caused significant
downregulation of p-CDK1 (Thr14) and accumulation of cyclin B1
compared with the CTL group, which further confirms that digitoxin
blocked the HepG2/ADM cell cycle at G2/M phase (Fig. 2C and D).
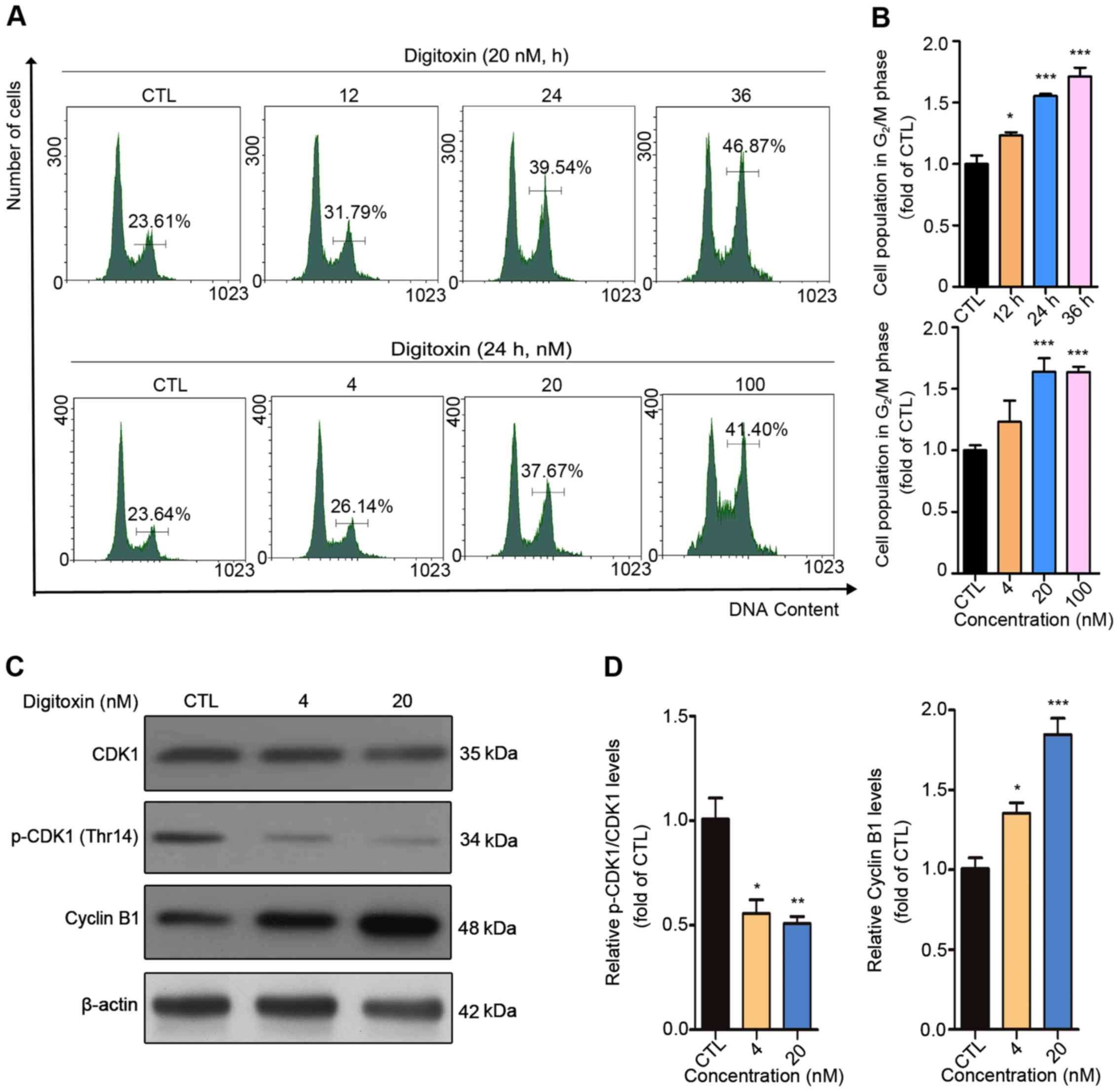 | Figure 2.Digitoxin blocks HepG2/ADM cells in
the G2/M phase of the cell cycle. (A) HepG2/ADM cells
were treated with different concentrations of digitoxin (0, 4, 20,
100 nM) for 24 h or 20 nM of digitoxin for 0, 12, 24 and 36 h, then
the cell cycle distributions were detected using flow cytometry.
The cell population in the G2/M phase was increased
following digitoxin treatment. (B) The cell population in the
G2/M phase was quantified using Prism. Each column
represents the mean ± SEM (n=3). *P<0.05, ***P<0.001 vs. the
control group. (C) HepG2/ADM cells were treated with or without
digitoxin (4 and 20 nM) for 24 h, and the protein expression levels
of CDK1, p-CDK1 (Thr14) and cyclin B1 were measured using western
blot analysis. β-actin served as the loading control.
Digitoxin-induced G2/M phase arrest was associated with
CDK1 and cyclin B1. (D) Quantitative analysis of the relative
protein expression. Data are presented as the mean ± SEM.
*P<0.05, **P<0.01, ***P<0.001 vs. the control group. CTL,
control; CDK1, cyclin-dependent kinase 1; p, phosphorylated. |
It is well-known that cell cycle arrest may result
from DNA damage, in which the serine-protein kinase
(ATM)/ATR-CHK1/CHK2-CDC25C signaling pathway plays significant role
(27). To determine whether DNA
lesions were responsible for the G2/M cell cycle arrest
induced by digitoxin, an immunofluorescent staining assay was
performed to detect the expression level of γH2AX, a marker of DNA
double-stranded break (DSB) (28).
The numbers of punctuate γH2AX foci significantly increased in a
dose-dependent manner following digitoxin treatment compared with
the CTL group. The highest accumulation of γH2AX foci was detected
following treatment with 100 nM digitoxin (Fig. 3A and B). ATM and ATR are pivotal
sensors of the DNA damage response pathway (29) and regulate the cell cycle partly
through activating cell cycle checkpoint kinases CHK1 and CHK2
(30). Active CHK1 and CHK2 decrease
the activity of CDC25C, thus inhibiting the dephosphorylation of
CDK1 to maintain the inactive status of the CDK1-cyclin B1 complex
(31,32). Digitoxin significantly increased the
protein expression levels of p-CHK2 (Thr68) in a dose-dependent
manner as well as p-ATR (Ser428) compared with the CTL group. In
addition, digitoxin inhibited the phosphorylation of CDC25C
(Fig. 3C and D). Taken together,
these results indicated that digitoxin induced G2/M
phase arrest via the ATR-Chk2-Cdc25C signaling pathway following
DNA damage.
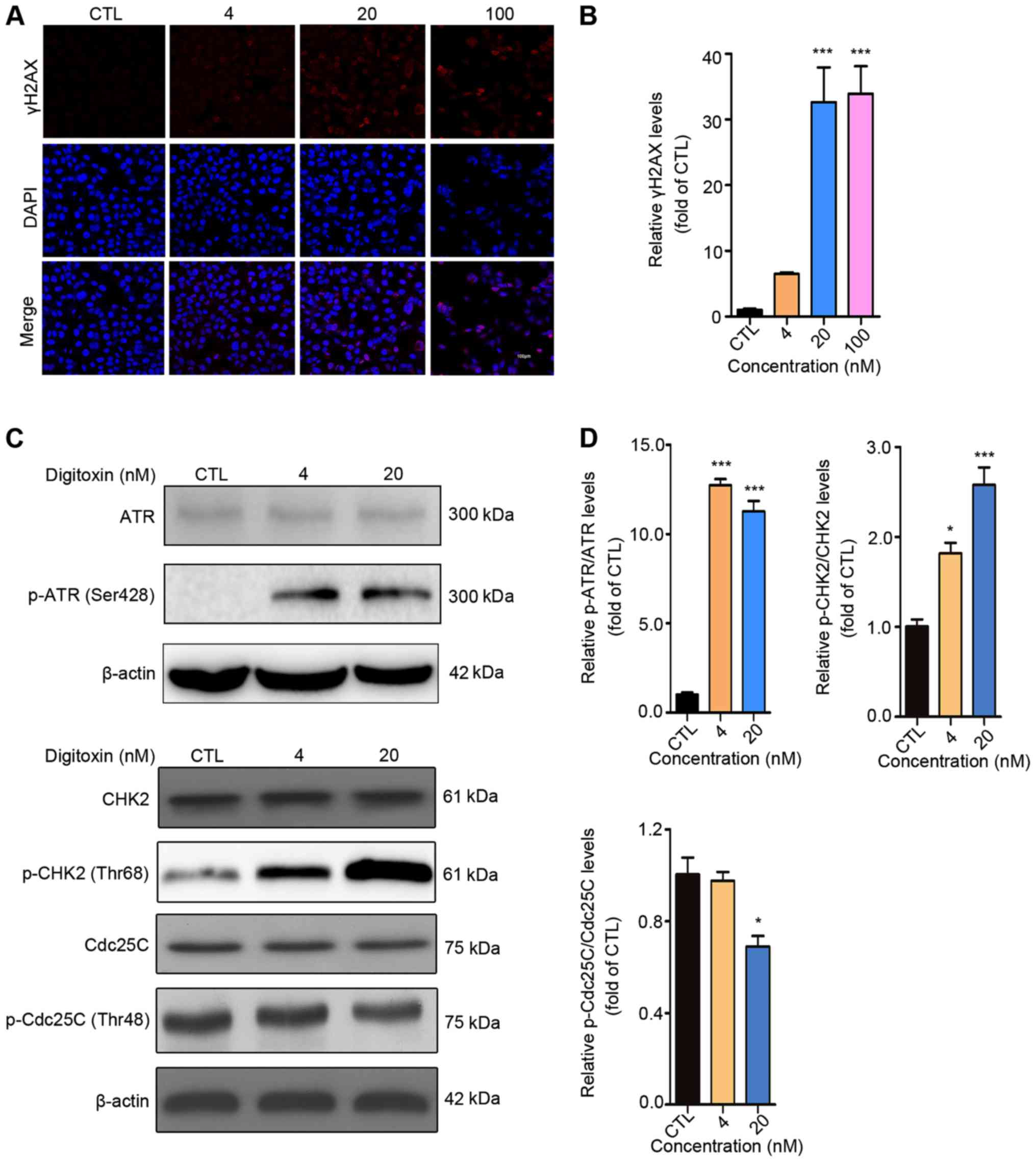 | Figure 3.Digitoxin induces G2/M
phase arrest via the ATR-CHK2-CDC25C signaling pathway. (A)
Digitoxin increased the expression level of γH2AX. HepG2/ADM cells
were treated with or without digitoxin (4, 20 and 100 nM) for 24 h,
then, the expression level of γH2AX was measured using an
immunofluorescence assay. Images were obtained at a magnification
of ×200. (B) γH2AX fluorescence intensity was subsequently
quantified. Data are shown as the mean ± SEM (n=3). ***P<0.001
vs. the control group. (C) The effect of digitoxin on the
ATR-CHK2-CDC25C signaling pathway. HepG2/ADM cells were treated
with or without digitoxin (4 and 20 nM) for 24 h, then western blot
analysis was used to detect the protein expression levels of ATR,
p-ATR (Ser428), CHK2, p-CHK2 (Thr68), CDC25C, p-CDC25C (Thr48).
β-actin was used as the loading control. (D) The relative protein
expression was quantified. Data are presented as the mean ± SEM
(n=3). *P<0.05, ***P<0.001 vs. the control group. γH2AX,
p-histone H2AX; ATR, serine/threonine-protein kinase ATR; CHK2,
serine/threonine-protein kinase chk2; CDC25C, M-phase inducer
phosphatase 3; p, phosphorylated; CTL, control. |
Digitoxin induces HepG2/ADM cell
apoptosis through the mitochondrial apoptotic pathway
To investigate the underlying mechanism of
digitoxin-induced HepG2/ADM cell death, the Annexin V-FITC/PI
staining flow cytometry assay was performed to determine whether
digitoxin induced apoptosis in HepG2/ADM cells. Different
concentrations (CTL, 4, 20, 100 and 500 nM) of digitoxin treatment
resulted in a significant augmentation of apoptotic cells (Fig. 4A and B). The apoptotic ratio of
HepG2/ADM cells increased by approximately 7-fold from 5.68 to
43.15% for 24 h and 13-fold from 5.71 to 73.74% for 48 h. In
addition, the ratio of cleaved caspase-3/caspase-3 and cleaved
caspase-9/caspase-9 increased following different concentrations of
digitoxin in a dose-dependent manner, and the level of cleaved PARP
also increased, confirming that the apoptosis-related genes in the
caspase family were activated (Fig. 4E
and F). Mitochondrial apoptosis is regarded as the most common
mode of cell apoptosis, which is characterized by the release of
cytochrome c and changes in the interaction between Bcl-2
(apoptosis inhibitor) and Bax (apoptosis promotor) (33,34). As
expected, digitoxin slightly increased the protein expression level
of Bax and notably decreased the expression level of Bcl-2. As a
consequence, the value of Bax/Bcl-2 increased approximately 58-fold
by 20 nM digitoxin treatment compared with the CTL group. Since the
level of Bcl-2 after 20 nM digitoxin treatment is very low, higher
concentrations of digitoxin were not used in this assay. In
addition, digitoxin treatment increased the protein expression
level of cytochrome c (Fig. 4C
and D). Taken together, these results indicated that digitoxin
induced HepG2/ADM cell apoptosis through the mitochondrial
apoptotic pathway.
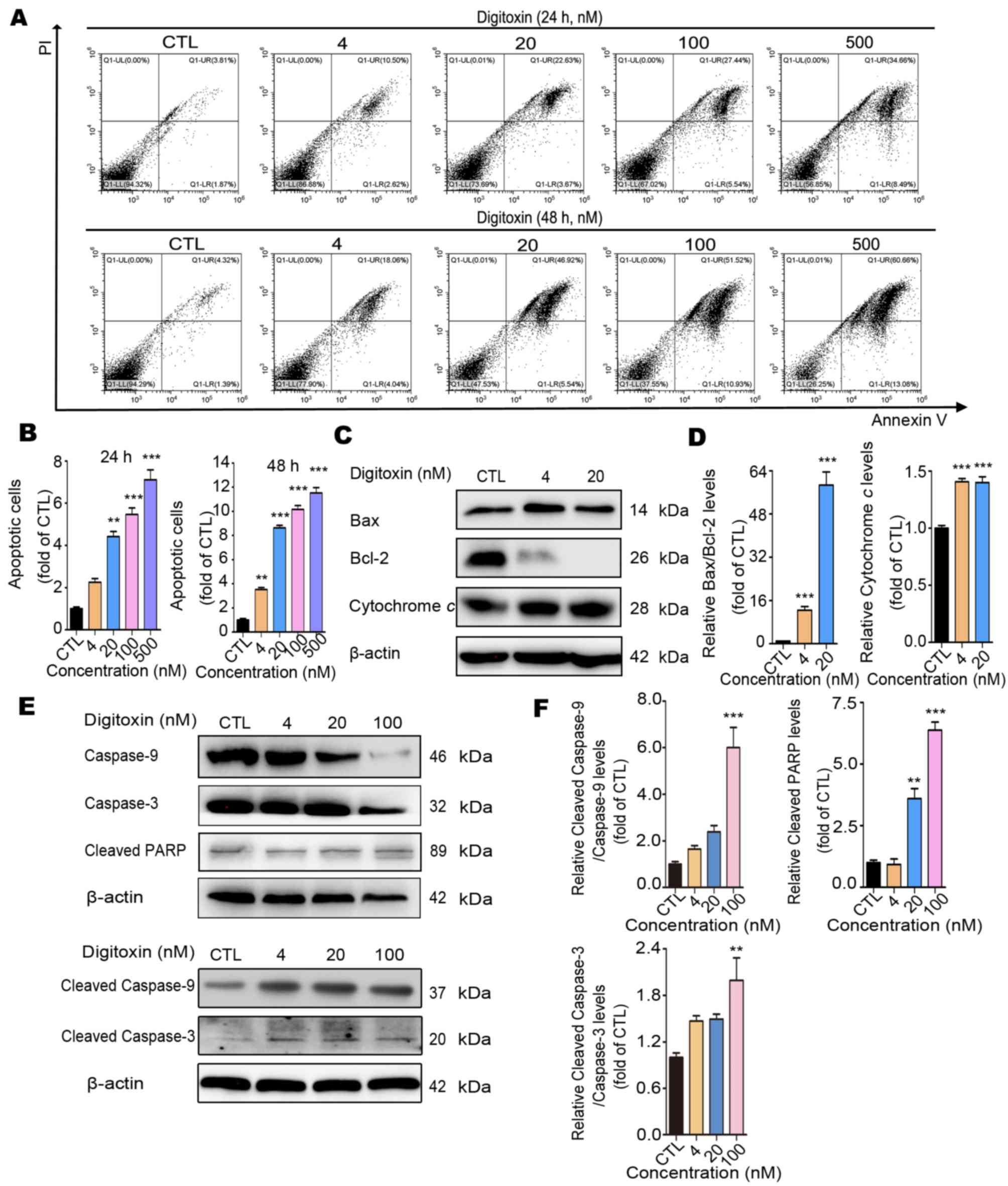 | Figure 4.Digitoxin induces HepG2/ADM cell
apoptosis via the mitochondrial apoptotic pathway. (A) Following
treatment with different concentrations (0, 4, 20, 100 and 500 nM)
of digitoxin for 24 or 48 h, the apoptotic ratio of HepG2/ADM cells
was determined using the Annexin V-FITC/PI staining assay.
Digitoxin induced HepG2/ADM cell apoptosis. (B) The data of
apoptotic cells was quantified and illustrated as the mean ± SEM
(n=3). **P<0.01, ***P<0.001 vs. the control group. (C) The
effect of digitoxin on the protein expression levels of
mitochondrial apoptosis-related proteins. Following treatment with
or without digitoxin (4 and 20 nM) for 24 h, the expression levels
of Bax, Bcl-2 and cytochrome c were determined using western
blot analysis. β-Actin was used as the loading control. (D) The
relative protein expression levels were quantified, and the data
are presented as the mean ± SEM (n=3). ***P<0.001 vs. the
control group. (E) The effect of digitoxin on the expression levels
of the apoptosis-related proteins. Following treatment with or
without digitoxin (4, 20 and 100 nM) for 24 h, the expression
levels of caspase-3 and −9, cleaved-caspase-3, and −9, and cleaved
PARP were detected using western blot analysis. β-actin was used as
the loading control. (F) The relative protein expression levels
were quantified, and the data are presented as the mean ± SEM
(n=3). **P<0.01, ***P<0.001 vs. the control group. PARP,
cleaved poly (ADP-ribose) polymerase; PI, propidium iodide; CTL,
control group. |
Discussion
Resistance to chemotherapy is the primary problem
for effective treatment in patients with liver cancer (35). A high amount of research has been
performed to overcome the problem of multidrug resistance (36–38);
however, no important breakthroughs have been achieved (39). Thus, there remains an urgent
requirement to identify novel anticancer agents that are effective
against chemotherapy-resistant tumors. In the present study, a
Dox-resistant HCC cell line, HepG2/ADM was used to screen a library
of 78 natural compounds, in which digitoxin was selected due to its
effective anti-HCC action on HepG2/ADM cells. In addition, the
molecular and/or cellular mechanisms of the apoptotic effect of
digitoxin on HepG2/ADM cells were also investigated. The findings
of the present study demonstrated that digitoxin induced
G2/M cell cycle arrest via the ATR-CHK2-CDC25C signaling
pathway, which may result from DNA DSB. Furthermore, digitoxin was
found to induce mitochondrial apoptosis in HepG2/ADM cells. The
notable findings of the present study indicate that digitoxin could
be a potential novel anti-HCC drug, particularly in
chemotherapy-resistant HCC.
Digitoxin, a cardenolide, is an inhibitor of
Na+/K+- ATPase (16). A number of studies have revealed the
anticancer activities of digitoxin against various human cancer
cell lines, including hematological, solid, drug-sensitive and/or
drug-resistant cancer cells in vitro and in vivo
(21,40–42). In
liver cancer, a previous study found that the combination of
sorafenib and digitoxin significantly inhibited primary HepG2 cell
growth, which was potentially through suppression of ERK and
hypoxia signaling (43). To the best
of our knowledge, no study has addressed the effect of digitoxin on
Dox-resistant HepG2/ADM cells. The present study performed purely
in vitro work. In vivo studies are required to
investigate digitoxin efficacy in detail, and whether ERK and
hypoxia signaling pathways play a role in digitoxin-induced
HepG2/ADM cell death also requires further investigation.
Numerous studies performed by different laboratories
have demonstrated that digitoxin induces G2/M cell cycle
arrest in several human cancer cell lines, including the KG1a acute
myelogenous leukemia cell line and the K562 chronic myelogenous
leukemia cell line (41), 786-O and
A498 renal cell carcinoma cell lines (44), and the non-small cell lung cancer
cell lines, NCI-H460 (45) and H1975
(17). Previous mechanistic studies
have revealed that the downregulation of the cyclin B1/CDK1
complex, and the protein expression levels of CHK1/2 and p53, with
the decrease in the protein levels of E3 ubiquitin-protein ligase
CCNB1IP1, cyclin-A1, p21, p27, c-Myc and p-5′AMP-activated protein
kinase catalytic subunit α-2 have been associated with
digitoxin-induced G2/M cell cycle arrest (17,45). The
findings of the present study demonstrated cell cycle arrest at the
G2/M phase in HepG2/ADM cells following digitoxin
treatment; however, reduced level of p-CDK1 (Thr14) and
accumulation of cyclin B1 caused by digitoxin were detected in the
present study (Fig. 2C and D), which
may promote cell cycle progression according to the majority of
studies (27,46). In eukaryotic cells, the expression
level of cyclin B1 is very low in the G1 phase, and
significantly increases during the S phase and peaks at the late
G2 phase and early mitosis. When cells enter late
mitosis, the expression level of cyclin B1 was found to be
significantly decreased (47–51).
Therefore, in the present study the increase of cyclin B1 further
confirms that a higher proportion of HepG2/ADM cells were in the
G2/M phase following digitoxin treatment. With respect
to decreasing level of p-CDK1 (Thr14), some signaling pathways,
which are activated to regulate cell cycle events such as P21 and
P53 (52,53) may be responsible for this phenomenon.
Similar results have been reported in other studies. Lee et
al (54) demonstrated that
p-CDK1 dephosphorylation at Tyr15 and the upregulation of cyclin B1
expression level were detected in 2-methoxyestradiol-induced
G2/M arrest in Jurkat T cells. Mak et al
(55) reported that small-molecule
inhibitors of CHK1 (AZD7762) or WEE1 (MK-1775) induced mitotic
arrest, as characterized by the dephosphorylation of p-CDK1 (Tyr15)
in HeLa cells. Rong et al (56) also found that p-CDK1 was
dephosphorylated at Thr161 following gambogic acid-induced DNA
damage and G2/M arrest in HepG2 and A549 cells. However,
the reasons for these different effects of digitoxin are complex
and require further investigation.
A number of small molecules can arrest the cell
cycle at G1/S or S phase to prevent incorrect DNA
replication or at G2/M phase to prevent entry into
mitosis with damaged DNA (57). The
present study found that digitoxin impeded cell cycle progression
at the G2/M phase, suggesting that digitoxin may not
block DNA replication but induce DNA damage instead. In addition,
the activation of the DNA damage response ATR-CHK2-CDC25C pathway
and the increase of γH2AX (Ser139) were also found, which confirmed
that the molecular mechanism of modulating the cell cycle by
digitoxin was induction of DNA damage.
Previous studies have demonstrated that
Na+/K+-ATPase was considered as a potential
target for cardenolides to combat some cancers (58,59). The
protein expression levels of Na+/K+-ATPase in
tumor tissues, such as HCC, renal carcinoma cells, non-small cell
lung carcinoma, colon carcinoma, prostate carcinoma, and glioma was
higher compared with that in normal tissues (60,61). In
addition, the colocalization of Na+/K+-ATPase
and caveolin on the plasma membrane induced by the knockdown of
apolipoprotein E increased the sensitivity of Hep3B cells towards
cardenolides, confirming the role of
Na+/K+-ATPase in the cytotoxicity of
cardenolides (20). These findings
demonstrate that the anticancer effects of cardenolides were
associated with Na+/K+-ATPase. However, to
the best of our knowledge there is a shortage of literature
demonstrating that digitoxin-induced cell cycle arrest was
attributed to the inhibition of
Na+/K+-ATPase. Therefore, the exact molecular
mechanism of how digitoxin induces DNA DSB, as well as whether
Na+/K+-ATPase inhibition was responsible for
apoptotic effect of digitoxin in HepG2/ADM cells requires further
investigation. Furthermore, in vivo effects and
cardiotoxicity of digitoxin also require further investigation.
In conclusion, our study demonstrated that digitoxin
displays an anti-HCC effect on HepG2/ADM cells through
ATR-CHK2-CDC25C-mediated G2/M cell cycle arrest and
Bax/Bcl-2-mediated mitochondrial apoptosis, making digitoxin a
promising chemotherapeutic agent for the treatment of patients with
HCC.
Acknowledgements
The authors would like to thank Professor Dong-Mei
Zhang (College of Pharmacy, Jinan University) and Dr Jun-Shan Liu
(Traditional Chinese Medicine, Southern Medical University) for
their guidance in the design of the present study.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81803790 and 81703975),
National Natural Science Foundation of Guangdong (grant no.
2020A1515011090), Project of Administration of Traditional Chinese
Medicine of Guangdong Province of China (grant no. 20181069) and
the Fundamental Research Funds for the Central Universities (grant
no. 21618336).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
LJD and FFX designed the study and revised the
manuscript for important intellectual content. YHL and HG performed
the experiments and drafted the manuscript. YQH, JYZ, HZ, MSW, XJL
and QYM made contributions to analysis and interpretation of data.
LC and AYS performed the flow cytometry experiments and analyze the
data. JW, YXL, EXZ and YYC assisted with the revision of the
manuscript and performed experiments to update the data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Altekruse SF, Henley SJ, Cucinelli JE and
McGlynn KA: Changing hepatocellular carcinoma incidence and liver
cancer mortality rates in the United States. Am J Gastroenterol.
109:542–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma J, Siegel RL, Islami F and Jemal A:
Temporal trends in liver cancer mortality by educational attainment
in the United States, 2000–2015. Cancer. 125:2089–2098. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anwanwan D, Singh SK, Singh S, Saikam V
and Singh R: Challenges in liver cancer and possible treatment
approaches. Biochim Biophys Acta Rev Cancer. 1873:1883142020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeo W, Mok TS, Zee B, Leung TW, Lai PB,
Lau WY, Koh J, Mo FK, Yu SC, Chan AT, et al: A randomized phase III
study of doxorubicin versus cisplatin/interferon
alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy
for unresectable hepatocellular carcinoma. J Natl Cancer Inst.
97:1532–1538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bruix J, Reig M and Sherman M:
Evidence-based diagnosis, staging, and treatment of patients with
hepatocellular carcinoma. Gastroenterology. 150:835–853. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu AX, Kudo M, Assenat E, Cattan S, Kang
YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL, et al: Effect of
everolimus on survival in advanced hepatocellular carcinoma after
failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA.
312:57–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mansoori B, Mohammadi A, Davudian S,
Shirjang S and Baradaran B: The different mechanisms of cancer drug
resistance: A brief review. Adv Pharm Bull. 7:339–348. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rajabalian S: Methanolic extract of
Teucrium polium L. potentiates the cytotoxic and apoptotic effects
of anticancer drugs of vincristine, vinblastine and doxorubicin
against a panel of cancerous cell lines. Exp Oncol. 30:133–138.
2008.PubMed/NCBI
|
|
10
|
Khanna C, Rosenberg M and Vail DM: A
review of paclitaxel and novel formulations including those
suitable for use in dogs. J Vet Intern Med. 29:1006–1012. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mukherjee AK, Basu S, Sarkar N and Ghosh
AC: Advances in cancer therapy with plant based natural products.
Curr Med Chem. 8:1467–1486. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gokduman K: Strategies targeting DNA
Topoisomerase I in cancer chemotherapy: Camptothecins, nanocarriers
for camptothecins, organic non-camptothecin compounds and metal
complexes. Curr Drug Targets. 17:1928–1939. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu XY, Zhang S, Wang K, Yang MF, Fan CD
and Sun BL: Caudatin inhibits human glioma cells growth through
triggering DNA damage-mediated cell cycle arrest. Cell Mol
Neurobiol. 35:953–959. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Xiong H and Yang DQ: Functional
switching of ATM: Sensor of DNA damage in proliferating cells and
mediator of Akt survival signal in post-mitotic human neuron-like
cells. Chin J Cancer. 31:364–372. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trenti A, Zulato E, Pasqualini L,
Indraccolo S, Bolego C and Trevisi L: Therapeutic concentrations of
digitoxin inhibit endothelial focal adhesion kinase and
angiogenesis induced by different growth factors. Br J Pharmacol.
174:3094–3106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patel S: Plant-derived cardiac glycosides:
Role in heart ailments and cancer management. Biomed Pharmacother.
84:1036–1041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang YZ, Chen X, Fan XX, He JX, Huang J,
Xiao DK, Zhou YL, Zheng SY, Xu JH, Yao XJ, et al: Compound library
screening identified cardiac glycoside digitoxin as an effective
growth inhibitor of gefitinib-resistant non-small cell lung cancer
via downregulation of α-tubulin and inhibition of microtubule
formation. Molecules. 21:3742016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopez-Lazaro M: Digitoxin as an anticancer
agent with selectivity for cancer cells: Possible mechanisms
involved. Expert Opin Ther Targets. 11:1043–1053. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee DH, Lee CS, Kim DW, Ae JE and Lee TH:
Digitoxin sensitizes glioma cells to TRAIL-mediated apoptosis by
upregulation of death receptor 5 and downregulation of survivin.
Anticancer Drugs. 25:44–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu M, Feng LX, Sun P, Liu W, Mi T, Lei M,
Wu W, Jiang B, Yang M, Hu L, et al: Knockdown of apolipoprotein E
enhanced sensitivity of Hep3B cells to cardiac steroids via
regulating Na+/K+-ATPase signalosome. Mol Cancer Ther.
15:2955–2965. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pollard BS, Suckow MA, Wolter WR, Starr
JM, Eidelman O, Dalgard CL, Kumar P, Battacharyya S, Srivastava M,
Biswas R, et al: Digitoxin inhibits
epithelial-to-mesenchymal-transition in hereditary castration
resistant prostate cancer. Front Oncol. 9:6302019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lopez-Lazaro M, Pastor N, Azrak SS, Ayuso
MJ, Austin CA and Cortes F: Digitoxin inhibits the growth of cancer
cell lines at concentrations commonly found in cardiac patients. J
Nat Prod. 68:1642–1645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang QF, Dalgard CL, Eidelman O, Jozwik C,
Pollard BS, Srivastava M and Pollard HB: Digitoxin induces
apoptosis in cancer cells by inhibiting nuclear factor of activated
T-cells-driven c-MYC expression. J Carcinog. 12:82013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee DH, Cheul Oh S, Giles AJ, Jung J,
Gilbert MR and Park DM: Cardiac glycosides suppress the maintenance
of stemness and malignancy via inhibiting HIF-1α in human glioma
stem cells. Oncotarget. 8:40233–40245. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao N, Wang C, Hu N, Li Y, Liu M, Lei Y,
Chen M, Chen L, Chen C, Lan P, et al: Inhibition of
PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant
cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis.
10:2322019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paruthiyil S, Cvoro A, Tagliaferri M,
Cohen I, Shtivelman E and Leitman DC: Estrogen receptor β causes a
G2 cell cycle arrest by inhibiting CDK1 activity through the
regulation of cyclin B1, GADD45A, and BTG2. Breast Cancer Res
Treat. 129:777–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deng LJ, Peng QL, Wang LH, Xu J, Liu JS,
Li YJ, Zhuo ZJ, Bai LL, Hu LP, Chen WM, et al: Arenobufagin
intercalates with DNA leading to G2 cell cycle arrest via ATM/ATR
pathway. Oncotarget. 6:34258–34275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Löbrich M, Shibata A, Beucher A, Fisher A,
Ensminger M, Goodarzi AA, Barton O and Jeggo PA: gammaH2AX foci
analysis for monitoring DNA double-strand break repair: Strengths,
limitations and optimization. Cell Cycle. 9:662–669. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Durocher D and Jackson SP: DNA-PK, ATM and
ATR as sensors of DNA damage: Variations on a theme? Curr Opin Cell
Biol. 13:225–231. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao H and Piwnica-Worms H: ATR-mediated
checkpoint pathways regulate phosphorylation and activation of
human Chk1. Mol Cell Biol. 21:4129–4139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deng LJ, Hu LP, Peng QL, Yang XL, Bai LL,
Yiu A, Li Y, Tian HY, Ye WC and Zhang DM: Hellebrigenin induces
cell cycle arrest and apoptosis in human hepatocellular carcinoma
HepG2 cells through inhibition of Akt. Chem Biol Interact.
219:184–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu YJ, Zheng B, Wang HY and Chen L: New
knowledge of the mechanisms of sorafenib resistance in liver
cancer. Acta Pharmacol Sin. 38:614–622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ceballos MP, Rigalli JP, Cere LI, Semeniuk
M, Catania VA and Ruiz ML: ABC Transporters: Regulation and
association with multidrug resistance in hepatocellular carcinoma
and colorectal carcinoma. Curr Med Chem. 26:1224–1250. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun W, Chen X, Xie C, Wang Y, Lin L, Zhu K
and Shuai X: Co-Delivery of doxorubicin and Anti-BCL-2 siRNA by
pH-responsive polymeric vector to overcome drug resistance in in
vitro and in vivo HepG2 hepatoma model. Biomacromolecules.
19:2248–2256. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gao DY, Lin Ts T, Sung YC, Liu YC, Chiang
WH, Chang CC, Liu JY and Chen Y: CXCR4-targeted lipid-coated PLGA
nanoparticles deliver sorafenib and overcome acquired drug
resistance in liver cancer. Biomaterials. 67:194–203. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marin JJG, Briz O, Herraez E, Lozano E,
Asensio M, Di Giacomo S, Romero MR, Osorio-Padilla LM,
Santos-Llamas AI, et al: Molecular bases of the poor response of
liver cancer to chemotherapy. Clin Res Hepatol Gastroenterol.
42:182–192. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prassas I, Karagiannis GS, Batruch I,
Dimitromanolakis A, Datti A and Diamandis EP: Digitoxin-induced
cytotoxicity in cancer cells is mediated through distinct kinase
and interferon signaling networks. Mol Cancer Ther. 10:2083–2093.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feng Q, Leong WS, Liu L and Chan WI:
Peruvoside, a cardiac glycoside, induces primitive myeloid leukemia
Cell Death. Molecules. 21:5342016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Felth J, Rickardson L, Rosén J, Wickström
M, Fryknäs M, Lindskog M, Bohlin L and Gullbo J: Cytotoxic effects
of cardiac glycosides in colon cancer cells, alone and in
combination with standard chemotherapeutic drugs. J Nat Prod.
72:1969–1974. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiao Y, Yan W, Guo L, Meng C, Li B, Neves
H, Chen PC, Li L, Huang Y, Kwok HF and Lin Y: Digitoxin synergizes
with sorafenib to inhibit hepatocelluar carcinoma cell growth
without inhibiting cell migration. Mol Med Rep. 15:941–947. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nolte E, Wach S, Silva IT, Lukat S, Ekici
AB, Munkert J, Müller-Uri F, Kreis W, Oliveira Simões CM, Vera J,
et al: A new semisynthetic cardenolide analog 3β-[2-(1-amantadine)-
1-on-ethylamine]-digitoxigenin (AMANTADIG) affects G2/M cell cycle
arrest and miRNA expression profiles and enhances proapoptotic
survivin-2B expression in renal cell carcinoma cell lines.
Oncotarget. 8:11676–11691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Elbaz HA, Stueckle TA, Wang HY, O'Doherty
GA, Lowry DT, Sargent LM, Wang L, Dinu CZ and Rojanasakul Y:
Digitoxin and a synthetic monosaccharide analog inhibit cell
viability in lung cancer cells. Toxicol Appl Pharmacol. 258:51–60.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu JS, Huo CY, Cao HH, Fan CL, Hu JY,
Deng LJ, Lu ZB, Yang HY, Yu LZ, Mo ZX and Yu ZL: Aloperine induces
apoptosis and G2/M cell cycle arrest in hepatocellular carcinoma
cells through the PI3K/Akt signaling pathway. Phytomedicine.
61:1528432019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bloom J and Cross FR: Multiple levels of
cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol.
8:149–160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nurse P: A long twentieth century of the
cell cycle and beyond. Cell. 100:71–78. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fisher DL and Nurse P: A single fission
yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and
mitosis in the absence of G1 cyclins. EMBO J. 15:850–860. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hayles J, Fisher D, Woollard A and Nurse
P: Temporal order of S phase and mitosis in fission yeast is
determined by the state of the p34cdc2-mitotic B cyclin complex.
Cell. 78:813–822. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gould KL and Nurse P: Tyrosine
phosphorylation of the fission yeast cdc2+ protein kinase regulates
entry into mitosis. Nature. 342:39–45. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
He YC, He L, Khoshaba R, Lu FG, Cai C,
Zhou FL, Liao DF and Cao D: Curcumin nicotinate selectively induces
cancer cell apoptosis and cycle arrest through a p53-mediated
mechanism. Molecules. 24:41792019. View Article : Google Scholar
|
|
53
|
Żuryń A, Litwiniec A, Safiejko-Mroczka B,
Klimaszewska- Wiśniewska A, Gagat M, Krajewski A, Gackowska L and
Grzanka D: The effect of sulforaphane on the cell cycle, apoptosis
and expression of cyclin D1 and p21 in the A549 non-small cell lung
cancer cell line. Int J Oncol. 48:2521–2533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee ST, Lee JY, Han CR and Kim YH, Jun do
Y, Taub D and Kim YH: Dependency of 2-methoxyestradiol-induced
mitochondrial apoptosis on mitotic spindle network impairment and
prometaphase arrest in human Jurkat T cells. Biochem Pharmacol.
94:257–269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mak JP, Man WY, Ma HT and Poon RY:
Pharmacological targeting the ATR-CHK1-WEE1 axis involves balancing
cell growth stimulation and apoptosis. Oncotarget. 5:10546–10557.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rong JJ, Hu R, Song XM, Ha J, Lu N, Qi Q,
Tao L, You QD and Guo QL: Gambogic acid triggers DNA damage
signaling that induces p53/p21(Waf1/CIP1) activation through the
ATR-Chk1 pathway. Cancer Lett. 296:55–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Laiho M and Latonen L: Cell cycle control,
DNA damage checkpoints and cancer. Ann Med. 35:391–397. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Piacente S, Masullo M, De Néve N, Dewelle
J, Hamed A, Kiss R and Mijatovic T: Cardenolides from Pergularia
tomentosa display cytotoxic activity resulting from their potent
inhibition of Na+/K+-ATPase. J Nat Prod. 72:1087–1091. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mijatovic T, Dufrasne F and Kiss R:
Cardiotonic steroids-mediated targeting of the Na(+)/K(+)-ATPase to
combat chemoresistant cancers. Curr Med Chem. 19:627–646. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu ZW, Wang FM, Gao MJ, Chen XY, Hu WL and
Xu RC: Targeting the Na(+)/K(+)-ATPase alpha1 subunit of hepatoma
HepG2 cell line to induce apoptosis and cell cycle arresting. Biol
Pharm Bull. 33:743–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mijatovic T, Dufrasne F and Kiss R:
Na+/K+-ATPase and cancer. Pharm Pat Anal. 1:91–106. 2012.
View Article : Google Scholar : PubMed/NCBI
|