Introduction
Autophagy is an important cellular degradation
process that is used as a cell compensatory mechanism to various
stress conditions, such as nutritional starvation, oxidation, DNA
replication and endoplasmic reticulum (ER) and bacterial invasion
stress (1–7). Autophagy is generally considered as an
important mechanism of cell survival. However, excessive autophagic
activity causes severe damage of subcellular structures and results
in cell death. Therefore, autophagy serves roles in both cell
survival and cell death. Currently, the mechanisms underlying the
autophagic regulation in cell death and survival are not fully
understood.
The role of autophagy in antitumor drug resistance
has been extensively studied (8–10). It is
well documented that autophagy is an important process for cancer
cell drug resistance, as determined by studies using both targeted
therapy and chemotherapy-based approaches (11,12).
Therefore, autophagy is considered as a cellular process that
promotes tumor cell survival and resistance to antitumor drugs.
Currently, suppression of autophagy has emerged as a novel strategy
in cancer therapy, which has been applied in multiple types of
cancer, such as renal, liver and prostate cancer (13–16).
In the present study, the prostate cancer DU145 cell
line was used, which lacks the expression of autophagy related 5
(ATG5) and is therefore defective in ATG5-dependent autophagy
(17,18). The experiments aimed to determine the
effects of ATG5-dependent autophagy on cell proliferation and
migration, and the cytotoxicity of chemotherapeutic drugs by the
overexpression of ATG5 in DU145 cells. The results indicated that
restoration of autophagy by overexpression of ATG5 enhanced the
cytotoxic effects of the chemotherapeutic drugs, docetaxel and
valproic acid (VPA), and of the ER stress inducers, brefeldin A,
tunicamycin and thapsigargin. The data demonstrated the role of the
ATG5-dependent autophagy in the chemotherapeutic drug resistance of
cancer cells.
Materials and methods
Materials
Anti-ATG5 (C-1; cat. no. sc-133158; 1:500),
anti-MAP-LC3-β (G-9; cat. no. sc-376404; 1:500) and
anti-sequesteome 1 (SQSTM1; D-3; cat. no. SC-28359; 1:500) primary
antibodies were purchased from Santa Cruz Biotechnology, Inc.
Anti-β-actin (ACTB; cat. no. 100166-MM10; 1:500) primary antibody
was obtained from Sino Biological, Inc., and the anti-clathrin
primary antibody was obtained from Covance, Inc. The PCR primers
were synthesized by Sangon Biotech Co., Ltd. Atorvastatin calcium
was purchased from Sigma-Aldrich (Merck KGaA). Rapamycin and
bortezomib were purchased from LC Laboratories. Chloroquine was
obtained from Cell Signaling Technology, Inc. EGF was obtained from
PeproTech, Inc., and docetaxel from Absin Bioscience Inc. Valproic
acid (VPA), brefeldin A, tunicamycin and thapsigargin were obtained
from APeXBIO Technology LLC. The Annexin V-FITC apoptosis detection
kit (cat. no. AD10) was purchased from Dojindo Molecular
Technologies, Inc. All cancer cell lines were purchased from the
American Type Culture Collection.
Cell culture and treatment
The prostate cancer cell lines, DU145, PC3 and
22RV1, and the lung cancer cell lines, A549, NCI-H1650 and
NCI-H1975, were cultured in DMEM (Wisent, Inc.; cat. no.
319-005-CL) supplemented with 10% heat-inactivated FBS (Shanghai
ExCell Biology, Inc.) and 100 U/ml penicillin and streptomycin. The
cells were incubated at 37°C with 5% CO2. Treatment with
the chemicals, inhibitors or EGF was performed at the
concentrations and time points prior to the cell harvest as
indicated in the figure legends.
Cell lysate preparation and
immunoblotting
The preparation of the cell lysates was performed on
ice. The cells were rinsed once with cold PBS following removal of
the culture medium, lysed using the ice-cold mammalian cell lysis
buffer (100 mM NaCl, 40 mM Hepes, pH 7.4, 25 mM glycerol phosphate,
1% Triton X-100, 1 mM EDTA, 1 mM sodium orthovanadate, 10 µg/ml
leupeptin and 10 µg/ml aprotinin) and incubated on rocking plates
at 4°C for 30 min. The cell lysates were centrifuged at 15,000 × g
in a microcentrifuge for 15 min at 4°C before use. The SDS-PAGE
lysate samples were prepared by addition of 5X SDS sample buffer
directly to the lysates, followed by vortexing and denaturation at
100°C for 5–10 min. Following electrophoresis on SDS gels (10–14%),
the separated proteins were transferred to a PVDF membrane (EMD
Millipore; cat. no. IPFL00010). The membrane was blocked with 1%
BSA (Sigma-Aldrich; Merck KGaA) for 1 h at 22°C and incubated with
the aforementioned primary antibodies (anti-ATG5, anti-SQSTM1,
anti-LC3 and anti-ACTB) overnight at 4°C. Following washing of the
membrane 4 times with 1X TBS-Tween-20 (containing 0.1% Tween-20)
for 7 min, the membrane was incubated with HRP-conjugated secondary
antibodies (1:10,000; Thermo Fisher Scientific, Inc.; anti-mouse
cat. no. 31430; anti-rabbit. cat. no. 31460) for 2 h at 22°C. The
protein bands were visualized using the Western lightning ECL
Detection kit (Beyotime Institute of Biotechnology).
Immunofluorescence staining
The cells were cultured in a 12-well plate on
coverslips (MatTek) to 50–70% confluence. The cells were rinsed
with cold PBS twice, fixed with 4% paraformaldehyde at 22°C for 30
min and permeabilized with 0.5% Triton X-100 in PBS at 22°C for 20
min. Following washing with PBS three times, the cells were
incubated with the aforementioned primary antibodies (anti-LC3 or
anti-ATG5; 1:100) at 4°C overnight. The cells were washed with PBS
three times and incubated with a Texas red-conjugated (for
anti-ATG5) or an Oregon green-conjugated (for anti-LC3) secondary
antibody (1:500; Thermo Fisher Scientific, Inc.; cat. nos. T6390
and O6381, respectively) and DAPI (1:10,000) at 37°C for 1–2 h.
Following washing with PBS three times, the coverslips containing
cells with fluorescent labels were mounted on microscopic slides
and their images were captured using a Zeiss LSM710 confocal
microscope (Zeiss AG; cat. no. MA 01960; magnification, ×600).
Construction of the ATG5 plasmid in
the lentiviral expression vector
The human ATG5 cDNA was subcloned into the FUW-HA
lentiviral expression vector (Addgene, Inc.) to establish stable
ATG5-overexpressing DU145 cell lines. The ATG cDNA was amplified
from a human cDNA library with primers for
hATG5-forward-BamH1
(5′-AATCGACTGGATCCATGACAGATGACAAAGATGT-3′) and for
hATG5-reverse-EcoR1 (5′-ATCGACGAATTCTCAATCTGTTGGCTGTGG−3′)
by PCR and inserted into the BamHI/EcoR1 sites of the
FUW-HA lentiviral expression vector. PCR was performed using
MegaFi™ Fidelity 2X PCR MasterMix (Applied Biological Materials,
Inc.; cat. no. G897) using the following conditions: 5 min at 94°C
for initial denaturation followed by 22 cycles of 30 sec at 94°C
for denaturation, 30 sec at 56°C for annealing and 30 sec at 72°C
for elongation, and 10 min at 72°C for final extension.
Lentiviral particle package and
infection
The lentiviral plasmid (FUW-HA-ATG5; 1.5 µg) was
co-transfected with the psPAX2 (Addgene, Inc.; cat. no. 12260; 1.0
µg) and the pMD2.G (Addgene, Inc.; cat. no. 12259; 0.5 µg)
packaging plasmids into 293T cells using Lipofectamine®
2000 Transfection reagent (Thermo Fisher Scientific, Inc.; cat no.
11668030) for 8–12 h. The culture medium containing viral particles
was collected every 24 h thrice. The medium was centrifuged at 200
× g for 5 min at 22°C and used for infecting DU145 cells in the
presence of 6 µg/ml polybrene (Sigma-Aldrich; Merck KGaA). The
infected DU145 cells were selected by 2.5 µg/ml puromycin for three
days to obtain the ATG5-overexpressing cell line.
Cell proliferation and migration
assays
A total of 2.5×104 DU145 cells
[transfected with the empty vector control FUW-Mr2tTA (Addgene,
Inc.; cat. no. 20342) or FUW-HA-ATG5] were seeded in each well of a
12-well culture plate. Following culture for 24, 48, 72 or 96 h,
the cells were counted using a light phase microscope
(magnification, ×200) with a hemocytometer. The cell proliferation
was evaluated by the increase in cell number. The proliferation
assay was repeated at least three times. Cell migration was
determined using a wound healing assay as described previously
(19). Briefly, DU145 cells
(1×106) were seeded into 6-well plates and cultured in
the standard culture medium (DMEM plus 10% FBS). When the cells
reached 90–95% confluence, a straight scratch line was made in the
cell monolayer using a pipette tip. The cells were subsequently
cultured for 36 h at 37°C in the standard culture medium with 10%
FBS for sustaining normal cell migration during wound healing
(19). For stimulation of cell
migration using EGF, EGF (100 ng/ml) was directly added into the
culture medium during the migration assay. Images of the migrated
cells in the scratched zone were captured using a light phase
microscope (magnification, ×100). The recovered area of the
migrated cells in the scratched zone was quantified using ImageJ
software (Version 1.53; National Institutes of Health) and was used
for evaluation of the migration rate.
Apoptosis assay
DU145 cells (5×104) were seeded in each
well of a 12-well culture plate. Following incubation of the cells
for 16 h, they were treated with docetaxel (30 nM) and thapsigargin
(250 nM) for 48 h at 37°C. Following removal of the culture medium,
the cells were rinsed with PBS and incubated with 100 µl 1X Annexin
V binding solution and 5 µl Annexin V-FITC per well at 22°C for 15
min. The stained apoptotic cells were observed and quantified using
a Nikon Eclipse TE2000 inverted fluorescence microscope (Nikon
Corporation; cat. no. NY 1174; magnification, ×100).
Statistical analysis
The data from three independent experiments were
analyzed with the statistical software SPSS (version 19.0; IBM
Corp.) using the Student's t-test (unpaired). The data were
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Prostate cancer DU146 cell line is
defective in the induction of autophagy due to loss of ATG5
expression
To identify specific autophagy-defective cancer cell
lines, several prostate and lung cancer cell lines were screened by
assessing the expression levels of ATG5. The data indicated that
ATG5 was not detectable in DU145 and NCI-H1650 cells (Fig. 1A). DU145 and NCI-H1650 cells have
been previously reported as autophagy-defective cell lines, lacking
the expression of ATG5 and ATG7 (17,20). To
verify whether DU145 and NCI-H1650 were autophagy-defective cell
lines, the PC3 and DU145 prostate cancer cells, along with
NCI-H1650 lung cancer cells, were incubated with the lysosomal
inhibitor chloroquine, which is known to block the autophagic flux
and cause accumulation of LC3-II (21). The specific proteasomal inhibitor
bortezomib was used as a control. A marked accumulation of LC3-II
was observed in PC3 cells following treatment with chloroquine,
while no LC3-II expression was detected in either DU145 or
NCI-H1650 cells following treatment with chloroquine (Fig. 1B), indicating the lack of autophagic
flux for both DU145 and NCI-H1650 cells. In addition, the protein
levels of the autophagy receptor SQSTM1, which is degraded by
autophagy (1), were markedly higher
in DU145 and NCI-H1650 cells than in PC3 cells (Fig. 1B), suggesting a defect in autophagy
in both DU145 and NCI-H1650 cells. As the defect in autophagy of
the lung cancer NCI-H1650 cells has been well characterized
(20), the present study focused on
the prostate cancer DU145 cells. The defect in autophagy was
further confirmed following treatment of DU145 cells with
atorvastatin, a hydroxylmethylglutaryl co-enzyme A reductase
inhibitor that has been shown to induce autophagy (22). Treatment of the two ATG5-expressing
prostate cancer PC3 and 22RV1 cell lines with atorvastatin induced
an increase of LC3-II expression compared with DMSO, but this
effect was not noted in DU145 cells (Fig. 1C). Treatment of the cells with
chloroquine caused a marked accumulation of LC3-II in PC3 and 22RV1
cells, whereas this effect was not observed in DU145 cells
(Fig. 1C). These data confirmed that
DU145 cells were defective in the induction of autophagy.
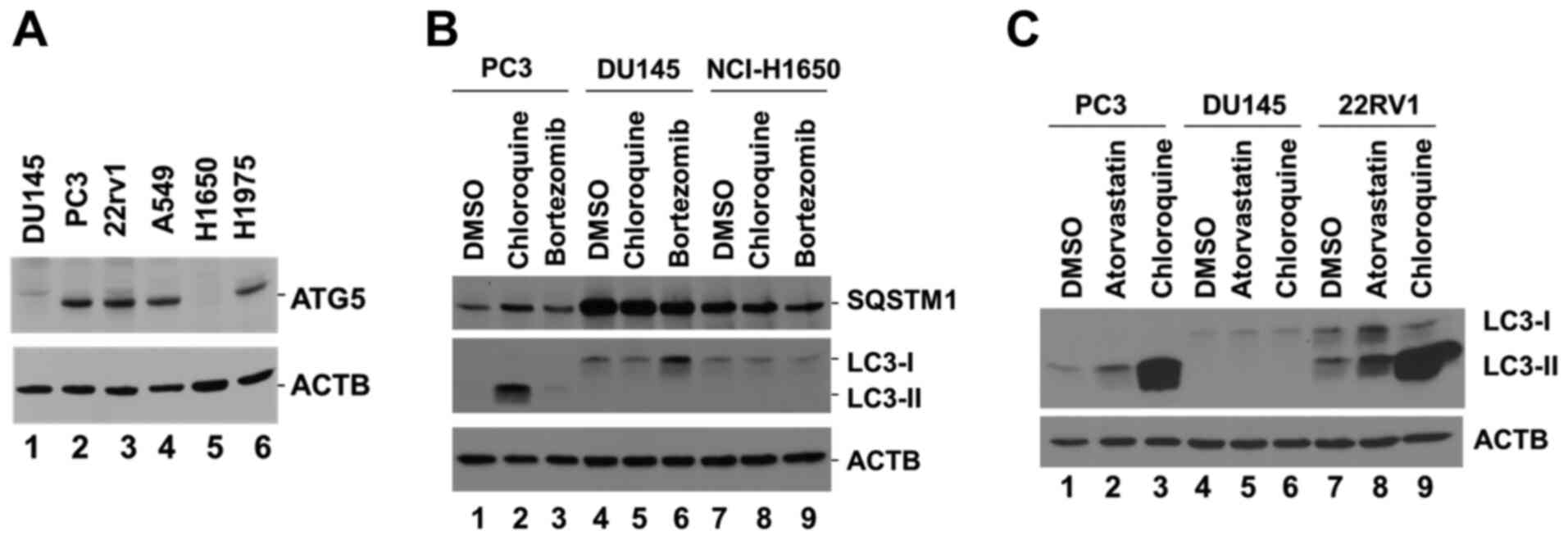 | Figure 1.DU145 cells lack ATG5 expression and
are defective in the induction of autophagy. (A) Detection of ATG5
protein expression in cell lysates derived from three prostate
cancer cell lines, DU145, PC3 and 22RV1, and three lung cancer cell
lines, A549, NCI-H1650 and NCI-H1975, using western blotting. (B)
Detection of LC3-II and SQSTM1 protein expression in PC3, DU145 and
NCI-H1650 cell lysates following treatment of the corresponding
cells with the lysosomal inhibitor chloroquine (50 µM) or the
proteasomal inhibitor bortezomib (10 µM) for 24 h. (C) Detection of
LC3-II protein expression in PC3, DU145 and 22RV1 cell lysates
following treatment of the cells with the lysosomal inhibitor
chloroquine (50 µM) or the hydroxylmethylglutaryl Co-enzyme A
reductase inhibitor atorvastatin (10 µM) for 48 h. β-actin was used
as a loading control. ATG5, autophagy related 5; SQSTM1,
sequesteome 1. |
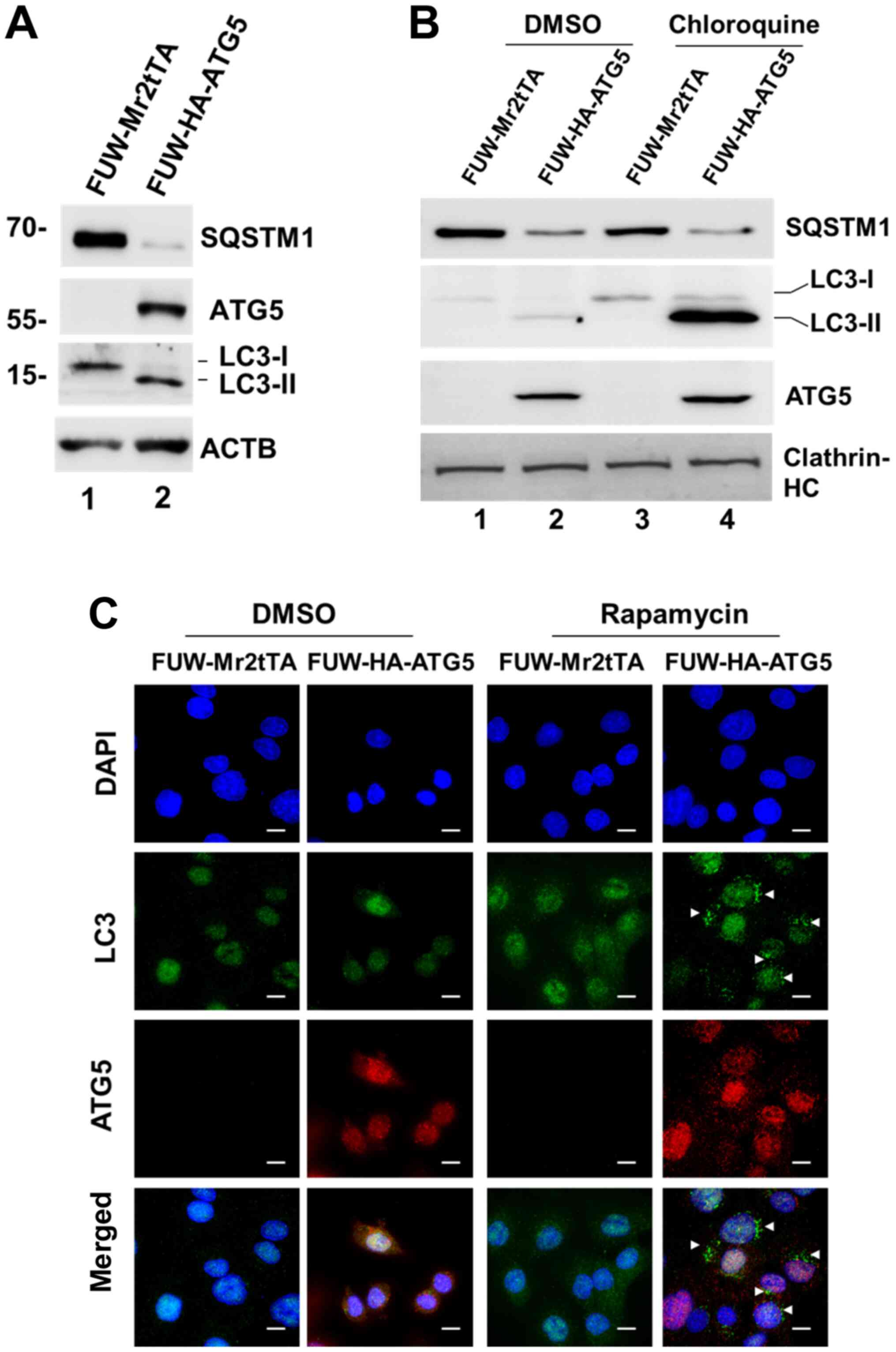
Overexpression of ATG5 restores
autophagy in DU145 cells
To confirm that loss of ATG5 expression was the
cause for the defect in autophagy of DU145 cells, a lentiviral
mammalian expression system was employed and ATG5 expression was
restored in the cells (Fig. 2A).
Restoration of ATG5 expression induced the expression of the
autophagosomal protein LC3-II, indicating that autophagy was
resumed (Fig. 2A). In addition, the
expression levels of the selective autophagic adaptor protein,
SQSTM1, were markedly decreased following ATG5 overexpression,
which indicated that the autophagic degradation was active
(Fig. 2A). As expected, when the
ATG5-expressing cells were treated with chloroquine, LC3-II
expression was markedly increased (Fig.
2B), indicating that the autophagic flux was active in the
cells. Furthermore, autophagosomes were observed in the
ATG5-expressing cells following their treatment with the autophagic
inducer rapamycin, whereas these changes were not noted in the
vector control cells (Fig. 2C).
Overall, these data demonstrated that loss of ATG5 expression was
the cause for the defect in autophagy in DU145 cells.
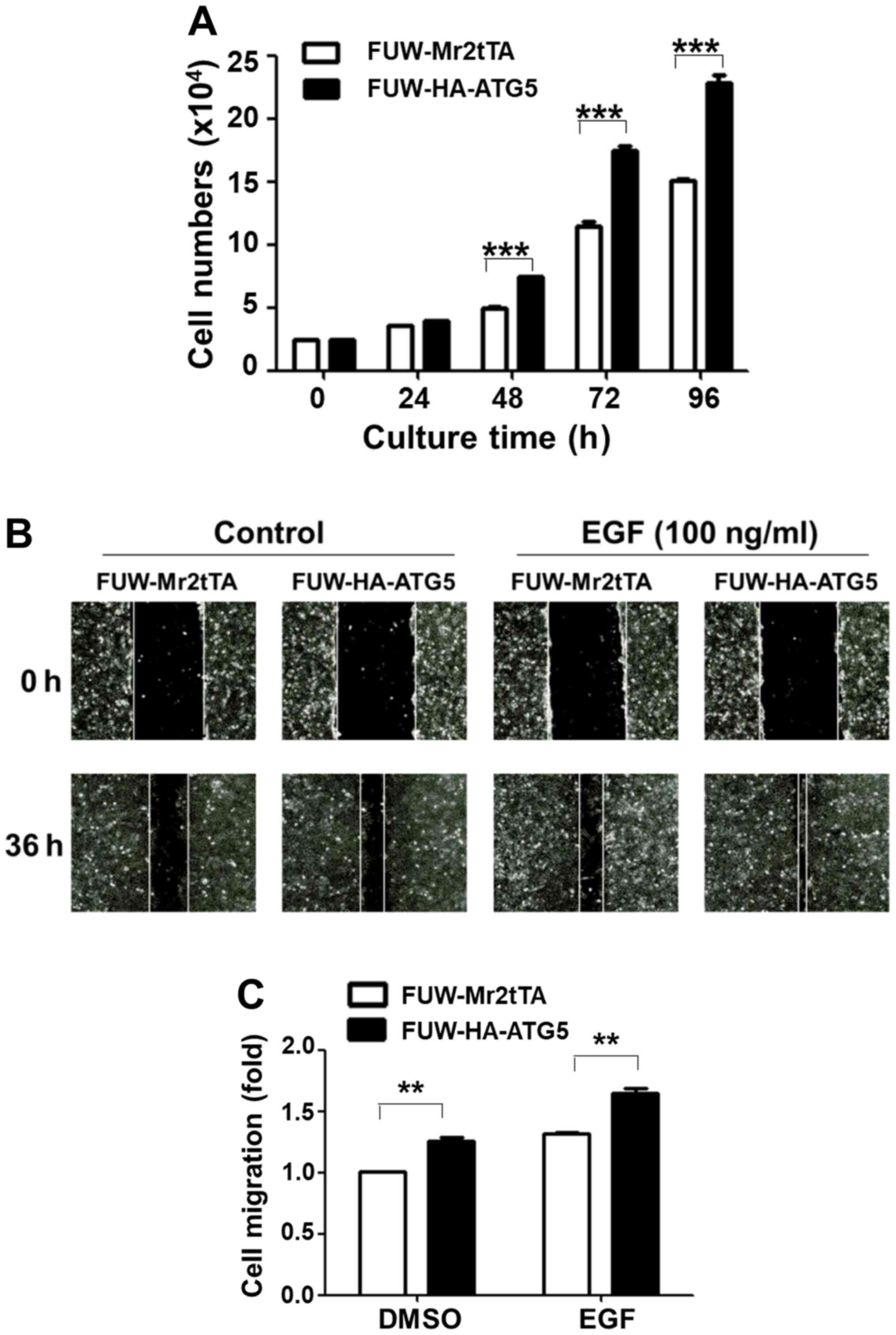
Restoration of autophagy in DU145
cells enhances cell proliferation and migration
The effects of the restoration of autophagy were
subsequently examined with regard to DU145 cell proliferation and
migration. Ectopic expression of ATG5 significantly increased the
cell proliferation rate after 2 days of culture compared with the
vector control cells (Fig. 3A). The
wound healing assay was used to detect cell migration rate in both
the control and the ATG5-expressing cell lines. The data indicated
that ATG5 overexpression in DU145 cells significantly promoted both
the basal and the EGF-stimulated cell migration rate (Fig. 3B and C). These data indicated that
restoration of autophagy in DU145 cells facilitated cell
proliferation and migration.
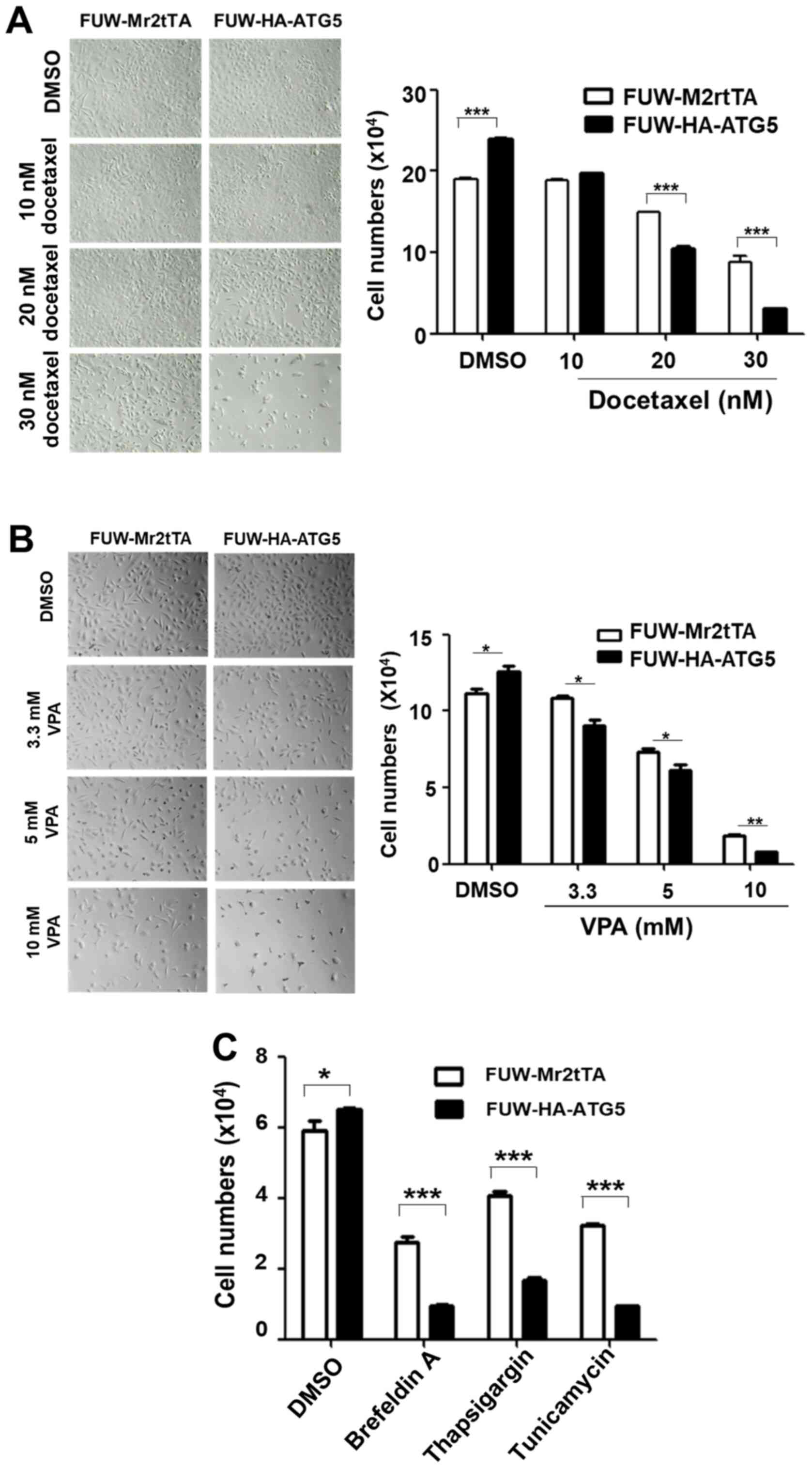
Restoration of autophagy enhances the
sensitization of DU145 cells to the cytotoxicity caused by the
chemotherapeutic drug docetaxel and ER stress-associated drugs
To examine the effects of autophagy on the efficacy
of chemotherapeutic drugs and cellular stress resistance, the
proliferation rate of the ATG5-expressing cells was compared with
that of the vector control cells following treatment with the
chemotherapeutic drugs, docetaxel and VPA, or the ER stressors,
thapsigargin, tunicamycin and brefeldin A. Notably, ≥20 nM
docetaxel, ≥3.3 mM VPA, 250 nM thapsigargin, 2 µM tunicamycin and
500 nM brefeldin A significantly inhibited the proliferation of the
ATG5-expressing cells compared with that of the vector control
cells (Fig. 4A-C), indicating that
restoring autophagy in DU145 increased the sensitivity of the cells
to the cellular stress caused by chemotherapeutic drugs or ER
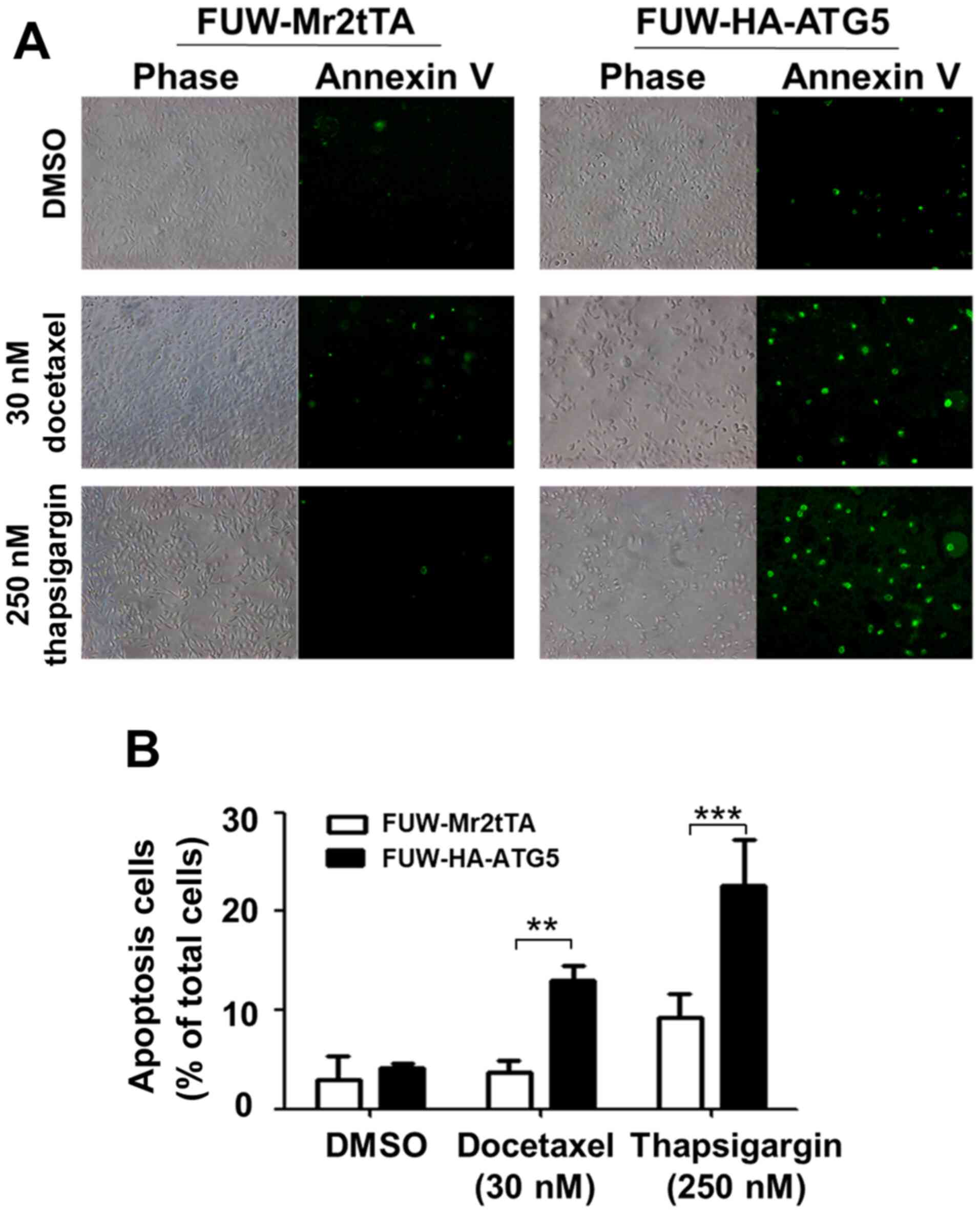
stressors. The effects of the restored autophagy on cell survival
were further investigated following treatment with the
chemotherapeutic drug docetaxel and the ER stressor thapsigargin.
The treatment of the ATG5-expressing cells with these compounds
caused a significant increase in apoptosis compared with that in
the vector control cells (Fig. 5A and
B), indicating that restoring autophagy in DU145 cells resulted
in sensitization to the stress signal-induced apoptosis.
Discussion
Previous studies have demonstrated that DU145 cells
lack expression of ATG5 and are defective in the induction of
autophagy (17,18). Restoration of ATG5 reverses the
autophagic response to VPA (17).
The results of the present study confirmed that DU145 cells lacked
ATG5 expression and were defective in ATG5-dependent autophagy.
Restoration of ATG5 expression reversed autophagy and enhanced cell
proliferation and migration, suggesting that the ATG5-dependent
autophagy may benefit the cancer cells and promote cancer
progression. Notably, restoration of the ATG5-dependent autophagy
in DU145 cells increased the sensitivity of the cells to the
cytotoxicity induced by the chemotherapeutic drugs, docetaxel and
VPA, and the ER stressors, brefeldin A, tunicamycin and
thapsigargin, compared with the effects in the vector control
cells, suggesting that ATG5-dependent autophagy sensitized cells to
the antitumor effects of the chemotherapeutic drugs and ER
stressors. It is well documented that autophagy is an important
cellular process required to counteract various stress conditions
and maintain cell survival in cancer cells (1–7). It has
been proposed to use autophagy inhibitors combined with
chemotherapeutic drugs to overcome the drug resistance in cancer
(23). However, the present study
revealed a novel role of autophagy in sensitization of cancer cells
to the cytotoxicity caused by chemotherapeutic drugs or ER
stressors, raising a concern when applying the therapeutic strategy
of inhibiting autophagy to overcome the chemoresistance in patients
with cancer. The current observation of the sensitization to
cytotoxicity of chemotherapeutic drugs or ER stressors by autophagy
was limited to DU145 cells. To conclude the role of autophagy in
sensitizing cytotoxicity of chemotherapeutic drugs or ER stressors
in prostate cancer, more prostate cancer cell lines should be
investigated.
Notably, the current finding on the sensitization
effect of the restored autophagy on the cytotoxicity of VPA has not
been observed in a previous study (17). This discrepancy may result from the
difference in re-expression level of ATG5 in DU145 cells between
the studies. It seems that the restored level of ATG5 in DU145
cells in the present study was higher than that in a study by
Ouyang et al (17), which may
yield differential sensitivity of the restored autophagy response
to cytotoxicity of VPA. Further studies may need to establish the
connection between intensity of autophagic activity and sensitivity
of cancer cells to chemotherapeutic drugs.
The current study did not explain the mechanism by
which DU145 and NCI-H1650 cells, which are defective in the
ATG5-dependent autophagy, were able to maintain cellular
homeostasis and respond to various stress signals. These cells may
have developed an ATG5-independent autophagic pathway in order to
sustain their autophagic function. Previous studies have identified
an ATG5-independent autophagic pathway (known as alternative
autophagy) that is dependent on RAB9A member RAS oncogene family
and utilizes the endosome- or Golgi-derived vesicles for substrate
degradation (24,25). A previous study has revealed that
this alternative autophagic pathway serves an important role in
genotoxic stress (26).
Additionally, it has been proposed that alternative autophagy is a
complementary autophagic process to the ATG5-dependent autophagy
(conventional autophagy) (25).
However, whether alternative autophagy is able to replace the full
function of ATG5-dependent autophagic activities remains
unknown.
The molecular mechanism underlying the effects of
the restored ATG5-dependent autophagy on the cytotoxicity caused by
the chemotherapeutic drugs and the ER stressors is not known. It
may be possible that the restored ATG5-dependent autophagy may
interfere with the established ATG5-independent autophagy and may
cause the cells to be more sensitive to the stresses induced by the
chemotherapeutic drugs and the ER stressors compared with the
effects noted in the parent cells. Stresses were more efficient in
activating apoptosis in the ATG5-expressing DU145 cells than in the
vector control cells. Further clarification of the signaling events
mediated by the restored ATG5-dependent autophagy would enhance the
understanding of the role of this process in sensitizing cells to
the cytotoxicity of the chemotherapeutic drugs and stressors. In
conclusion, the current approach may aid the development of precise
autophagic-associated therapeutic strategies for prostate cancer
therapy.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81372208, 81871888 and
81472558).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KP performed the majority of the experiments. AS
performed the immunofluorescence staining analysis. JZ performed
the experiments on screening cancer cell lines for ATG5 expression.
JG performed the cell culture experiments. YL performed the
experiments on ATG5 cDNA cloning. GS participated in the
experimental data analysis. WY participated in the experimental
design, data analysis and manuscript preparation. QL conceived the
study and participated in the experimental design analysis of the
data and preparation of the manuscript. KP and QL confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vanzo R, Bartkova J, Merchut-Maya JM, Hall
A, Bouchal J, Dyrskjøt L, Frankel LB, Gorgoulis V, Maya-Mendoza A,
Jäättelä M and Bartek J: Autophagy role(s) in response to oncogenes
and DNA replication stress. Cell Death Differ. 27:1134–1153. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mizushima N: The pleiotropic role of
autophagy: From protein metabolism to bactericide. Cell Death
Differ. 12 (Suppl 2):S1535–S1541. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
9
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer treatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
10
|
Li X, Zhou Y, Li Y, Yang L, Ma Y, Peng X,
Yang S, Liu J and Li H: Autophagy: A novel mechanism of
chemoresistance in cancers. Biomed Pharmacother. 119:1094152019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schoenlein PV, Periyasamy-Thandavan S,
Samaddar JS, Jackson WH and Barrett JT: Autophagy facilitates the
progression of ERalpha-positive breast cancer cells to
anti-estrogen resistance. Autophagy. 5:400–403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: Autophagy facilitates the development of breast cancer
resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS
One. 4:e62512009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones TM, Carew JS and Nawrocki ST:
Therapeutic targeting of autophagy for renal cell carcinoma
therapy. Cancers (Basel). 12:11852020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tran E, Chow A, Goda T, Wong A, Blakely K,
Rocha M, Taeb S, Hoang VC, Liu SK and Emmenegger U:
Context-dependent role of ATG4B as target for autophagy inhibition
in prostate cancer therapy. Biochem Biophys Res Commun.
441:726–731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YG and Jeon TI: Modulation of the
autophagy-lysosomal pathway in hepatocellular carcinoma using small
molecules. Molecules. 25:15802020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ouyang DY, Xu LH, He XH, Zhang YT, Zeng
LH, Cai JY and Ren S: Autophagy is differentially induced in
prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing
profiles of ATG5. Autophagy. 9:20–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cristofani R, Montagnani Marelli M,
Cicardi ME, Fontana F, Marzagalli M, Limonta P, Poletti A and
Moretti RM: Dual role of autophagy on docetaxel-sensitivity in
prostate cancer cells. Cell Death Dis. 9:8892018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun A, Yu G, Dou X, Yan X, Yang W and Lin
Q: Nedd4-1 is an exceptional prognostic biomarker for gastric
cardia adenocarcinoma and functionally associated with metastasis.
Mol Cancer. 13:2482014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mandelbaum J, Rollins N, Shah P, Bowman D,
Lee JY, Tayber O, Bernard H, LeRoy P, Li P, Koenig E, et al:
Identification of a lung cancer cell line deficient in
atg7-dependent autophagy. Autophagy. Jun 19–2015.(Epub ahead of
Print). View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parikh A, Childress C, Deitrick K, Lin Q,
Rukstalis D and Yang W: Statin-induced autophagy by inhibition of
geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate.
70:971–981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiao M, Benoit A, Hasmim M, Duhem C, Vogin
G, Berchem G, Noman MZ and Janji B: Targeting cytoprotective
autophagy to enhance anticancer therapies. Front Oncol.
11:6263092021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishida Y, Arakawa S, Fujitani K,
Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y
and Shimizu S: Discovery of Atg5/Atg7-independent alternative
macroautophagy. Nature. 461:654–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimizu S: Biological roles of alternative
autophagy. Mol Cells. 41:50–54. 2018.PubMed/NCBI
|
|
26
|
Torii S, Yamaguchi H, Nakanishi A, Arakawa
S, Honda S, Moriwaki K, Nakano H and Shimizu S: Identification of a
phosphorylation site on Ulk1 required for genotoxic stress-induced
alternative autophagy. Nat Commun. 11:17542020. View Article : Google Scholar : PubMed/NCBI
|