Introduction
Lung cancer is the most common cause of
cancer-associated mortality worldwide (1). The majority (80–85%) of all lung
cancers are non-small cell lung cancer (NSCLC) (2), which is characterized by multiple
mutations in the epidermal growth factor receptor (EGFR) gene
(3). Since mutations in EGFR
constitutively act as an active receptor tyrosine kinase in NSCLC
cells, several tyrosine kinase inhibitors, including gefitinib,
have been developed and used as chemotherapeutic drugs to treat
patients with NSCLC (4). Despite the
initial clinical success of tyrosine kinase inhibitors (TKIs),
acquired resistance to TKIs has developed in numerous patients with
NSCLC (5). Much of the acquired
resistance to TKIs has been associated with a secondary T790M
mutation in EGFR (6,7). To overcome resistance to TKIs, several
combined NSCLC treatments, such as erlotinib and cetuximab
(8), afatinib and cetuximab
(9), yuanhuadine and gefitinib
(10), and metformin and gefitinib
(11) have been proposed and
studied). However, these therapeutic approaches often result in
renewed drug resistance by activating alternative survival pathways
(12–14).
Hypoxia is a characteristic of solid tumors,
including NSCLC, that directly stimulates the malignant properties
of cancer (15). In this tumor
microenvironment, hypoxia-inducible factors (HIFs) are activated,
and activated HIFs induce the expression of multiple genes
associated with angiogenesis, metabolic regulation, cell apoptosis
and tumor survival (16). The
essential roles of HIFs in blood vessel formation and the recovery
of the tumor blood supply make tumors difficult to treat, leading
to resistance to radiotherapy, chemotherapy and immunotherapy
(17). HIFs are heterodimeric
transcription factors consisting of a HIF-α (HIF-α) and a
constitutive β (HIF-β) subunits (18). The HIF-1α protein is strictly
regulated by oxygen concentration in the tumor microenvironment
(19). Due to hypoxia-specific
expression and activity of HIF-1α in tumorigenesis and escape from
cancer therapy, HIF-1α may be a promising therapeutic target
(20).
Gefitinib showed substantial efficacy in patients
with NSCLC and active EGFR mutations (21). However, almost all patients who
experience a marked response to gefitinib eventually develop
progressive disease (21). This type
of resistance has been observed in clinical trials, and includes
primary and acquired resistance (22). Primary resistance usually occurs in
patients with wild-type EGFR and other gene mutations downstream of
the EGFR signaling pathway, such as the KRAS mutation (23). The acquired mutation is mainly due to
the mutation T790M in the tyrosine kinase functional domain of EGFR
(21). The T790M mutation is located
in the ATP-/drug-binding cleft and triggers resistance by blocking
the binding of gefitinib and the kinase domain (21).
In the present study, gefitinib was selected as the
representative EGFR-TKI, and the differences between normal NSCLC
and gefitinib-resistant (GR) NSCLC cell lines were investigated,
focusing on HIF-1α protein expression and hypoxia-induced
angiogenesis. The results showed that HIF-1α protein expression
level and hypoxia-induced angiogenesis were increased in GR NSCLC
cell lines. These results suggested that HIF-1α directly or
indirectly regulated gefitinib resistance in NSCLC cell lines and
could be a novel therapeutic target for combination treatment with
EGFR-TKIs.
Materials and methods
Materials
Gefitinib was purchased from Sigma Aldrich; Merck
KGaA. The HIF-1α antibodies were purchased from BD Pharmingen (BD
Biosciences) and NOVUS Biologicals LLC.. The CD31 antibody was
obtained from BD Pharmingen (BD Biosciences). The antibody against
α-tubulin, MTT and cobalt chloride (CoCl2) were all
purchased from Sigma Aldrich; Merck KGaA. Matrigel was purchased
from BD Pharmingen; BD Biosciences.
Cell culture, hypoxia treatment and
transfection
The human NSCLC PC9 and HCC827 cell lines were
purchased from Asan Medical Center and the American Type Culture
Collection, respectively. The PC9/GR and HCC827/GR cell lines were
generated as previously described (24,25). All
the NSCLC cell lines were maintained in RPMI-1640 medium (Welgene,
Inc.) supplemented with 10% FBS (HyClone; Cytiva) and 1%
penicillin/streptomycin (Corning Life Sciences). The human dermal
microvascular endothelial cell line (HMEC-1) was kindly provided by
Dr Fransisco Candal Center for Disease Control and Prevention and
maintained in MCDB131 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (HyClone; Cytiva), 1%
penicillin/streptomycin, 10 mM L-glutamate (Gibco; Thermo Fisher
Scientific, Inc.), 10 ng/ml epidermal growth factor (Sigma Aldrich;
Merck KGaA) and 1 µg/ml hydrocortisone in a 37°C humidified
incubator with 5% CO2 or in a hypoxic chamber (1%
O2, 5% CO2 and 94% N2; InvivO2;
The Baker Company). The cell lines were inspected for mycoplasma
contamination using a light microscope.
The vectors, pGIPZ, pGIPZ-V2LHS-132152 and
pGIPZ-V2LHS-236718 were purchased from Open Biosystems Inc.,
(Thermo Fisher Scientific Inc.) and the latter 2 were used for
short hairpin (sh)RNA vectors against human HIF-1A. pcDNA-HIF-1α
for the overexpression of human HIF-1α and the pcDNA3.1 vector were
a kind gift from Professor Gregg L. Semenza (Department of
Medicine, Johns Hopkins School of Medicine, Baltimore, MD, USA).
The PC9 cell line, in a 60-mm culture dish were transfected with 5
µg pGIPZ, pGIPZ-V2LHS-132152, pGIPZ-V2LHS-236718, pcDNA3.1 or
pcDNA- HIF-1α using 4 µg/ml polyethyleneimine (Sigma Aldrich; Merck
KGaA) at 37°C for 6 h. pGIPZ or pcDNA3.1 was transfected and used
as the control or mock. The media were replaced with fresh complete
media, 6 h after transfection. The cells were used for subsequent
experimentation 24 h after transfection.
MTT assay
The PC9, PC9/GR, HCC827 and HCC827/GR cells
(1×104 cells per well) were plated in a 24-well plated
and incubated with 0.1 µM gefitinib for 48 or 72 h in a 37°C
humidified incubator with 5% CO2. MTT (0.1 mg/ml) was
added to each well and incubated at 37°C for 2 h, then dimethyl
sulfoxide was added. The absorbance was measured at 560 nm using an
iMark microplate absorbance reader (Bio-Rad Laboratories). All the
data are presented as the mean ± SEM from three wells and from 3
independent experiments.
Colony formation assay
A total of 50 PC9 cells were plated on 60-mm dishes
and were treated with increasing concentrations of gefitinib
(10–200 nM) for 2 weeks at 37°C in a humidified incubator with 5%
CO2. Following the incubation, RPMI-1640 medium
(Welgene, Inc.) with 200 nM gefitinib and 10% FBS (HyClone; Cytiva)
was removed, the cells were rinsed with PBS, fixed in acetic
acid:methanol (1:7, vol/vol) for 5 min, and stained with crystal
violet staining solution for 10 min, both at room temperature (RT).
The number of colonies was counted under a light microscope. The
data are presented as the mean ± SEM from 3 independent
experiments.
Western blot analysis
The PC9, PC9/GR, HCC827 and HCC827/GR cell lines
were incubated in a hypoxic chamber (37°C; 1% O2, 5%
CO2 and 94% N2; InvivO2; The Baker Company)
for 2, 8, 24 and 48 h. The cells were harvested and lysed in lysis
buffer (50 mM Tris, 150 mM NaCl and 1% NP-40) supplemented with a
protease inhibitor cocktail and phosphatase inhibitors (1 mM sodium
orthovanadate and 10 mM sodium fluoride). The BCA method was used
to determine the concentrations of the cell extracts. The cell
extracts (30 µg/lane) were separated using 9% SDS-PAGE, then
transferred to PVDF membranes (EMD Millipore). The membrane was
blocked with 5% skimmed milk in TBS containing 0.1% Tween-20 for 1
h at room temperature, then incubated overnight at 4°C with the
appropriate primary antibodies. Next, the membrane was incubated
with a HRP-conjugated secondary antibody (1:10,000; cat. no.
PI-2000; Vector Laboratories; Maravai Lifesciences) for 1 h at room
temperature. The signal was developed using an ECL western
detection reagent (Thermo Fisher Scientific, Inc.). The following
primary antibodies were used: Anti-HIF-1α (1:3,000; cat. no.
610958) and anti-α-tubulin (1:10,000; cat. no. T5168).
Conditioned media preparations
For preparation of conditioned medium (CM), the
medium from the PC9 and PC9/GR cells was changed with 1% FBS
(HyClone; Cytiva)-containing RPMI-1640 (Welgene, Inc.) and further
incubated for 16 h in a hypoxic chamber (37°C; 1% O2, 5%
CO2 and 94% N2; InvivO2; The Baker Company).
The CM was collected and filtered through a 0.22-µm pore membrane
(EMD Millipore).
Wound healing assay
The HMEC-1 cell line (1×105) were plated
on 24-well plates, cultured, then the monolayer was wounded with a
micropipette tip and images of the cells were captured at 0 h time
point. The attached HMEC-1 cells were incubated with growth medium
mixed with 1 mM thymidine and CM collected from hypoxia-stimulated
PC9 or PC9/GR cells for 16 h. Then, the cells were rinsed with PBS,
fixed in absolute methanol for 5 min, stained with Giemsa
(Sigma-Aldrich; Merck KGaA) for 10 min, both at RT, then images of
each wound were captured at the same location. The migrated cells
that moved beyond the reference line were counted and the number of
migrated cells was divided by the number of unmigrated cells. The
data are presented as the mean ± SEM from 3 independent
experiments. The average migrated HMEC-1 cells, treated with CM
from normoxic PC9 cells, was set to 100%.
Tube formation assay
A total of 200 µl Matrigel was polymerized on
24-well plates at 37°C for 30 min. The HMEC-1 cell line
(1×105) was seeded on the polymerized Matrigel and
incubated with CM from either the hypoxia-stimulated PC9 or PC9/GR
cell lines. After 16 h, morphological changes were observed and the
areas of the tube branches were measured using ImageJ software
v1.52 (National Institutes of Health). The data are presented as
the mean ± SEM from 3 independent experiments. The average areas of
the tube branches treated with CM from normoxic PC9 cells was set
to 100%.
Rat aortic ring sprouting assay
The rat experiments were approved by the Committee
for Care and Use of Laboratory Animals at the Kyung Hee University
(KHUASP-20-289). A 6-week-old male Sprague Dawley rat (150–180 g)
was purchased from Daehan Biolink and anesthetized with 30 mg/kg
Zoletil and 10 mg/kg Rompun (26,27) by
intraperitoneal injection. The aorta was extracted and the
peri-aortic fibroadipose tissue was carefully removed. The rings
were sliced at a thickness of 1 mm and randomly divided into 4
groups (PC9+normoxia, PC9/GR+normoxia, PC9+hypoxia and
PC9/GR+hypoxia; n=3). Each ring was placed on polymerized Matrigel
in each well of a 24-well plate, then covered with an additional 50
µl Matrigel. CM from hypoxia-stimulated PC9 or PC9/GR cell lines
were added to each well for 5 days. Average sprouting was measured
with ImageJ software v1.52 (National Institutes of Health) after
images of the plates were captured. The data are presented as the
mean ± SEM from 3 independent experiments. The average sprouting
with CM from normoxic PC9 cells was set to 100%.
Mouse xenograft tumor model
All the mouse experiments were approved by the
Committee for Care and Use of Laboratory Animals at the Kyung Hee
University [KHUASP(SE)-17-144]. A total of 24 6-week-old male
BALB/c nude mice were purchased from Daehan Biolink. The mice were
housed 2–4 per cage and maintained under a controlled temperature
(23±0.5°C), humidity (50±10%) and a 12:12 h light:dark cycle, and
food, drinking water and litter were changed every 2 days. The
7-week-old male mice (20–22 g) were randomly divided into 2 groups
(PC9 and PC9/GR; n=12), anesthetized with 30 mg/kg Zoletil and 10
mg/kg Rompun (28,29), by intraperitoneal injection, then
injected at a dorsal flank site with the PC9 or PC9/GR cell lines
(5×106 cells per mouse), suspended in Matrigel, to
perform subcutaneous tumorigenesis analysis. Their body weights and
tumor sizes were measured every day for 3 weeks with a vernier
caliper (Mitutoyo Corporation) and a digital balance, respectively.
The tumor volume was calculated using the following formula: Tumor
volume (mm3) = 0.5 (width × length × height). After 3
weeks, the mice were euthanized with sodium pentobarbital (100
mg/kg), by intraperitoneal injection and the death of the mice was
verified 10 min later by loss of movement, breath, heartbeat,
corneal reflex and muscular tension. Before tumor extraction,
subcutaneous tissue attached to each tumor was examined and images
were captured under a light microscope. Microvessels could be
detected by the naked eye. Extracted tumors were frozen with
optimal cutting temperature (OCT) compound (Sigma-Aldrich; Merck
KgaA) at −20°C for 2 h and stored at −80°C until use.
Immunohistochemical and
immunofluorescent staining
OCT compound-frozen tumor tissues were sliced at a
thickness of 10-µm and placed on gelatin-coated glass slides. The
sectioned tissues were fixed with 4% paraformaldehyde at RT for 15
min and incubated with methanol containing 3% hydrogen peroxide for
20 min. To increase tumor permeability, the tissues were incubated
with 0.3% Triton X-100 in PBS at RT for 20 min. The tissues were
blocked with blocking solution [0.1% BSA (Sigma-Aldrich; Merck
KgaA), 0.3% Triton X-100 and 1.5% FBS (HyClone; Cytiva)] at RT for
1 h, then incubated with the appropriate antibodies overnight at
4°C. The tissues were subsequently washed with PBS and incubated
with biotinylated anti-rabbit (cat. no. BA-1000) and anti-rat (cat.
no. BA-4000) IgG (H+L) antibodies (1:500; Vector Laboratories;
Maravai LifeSciences) or fluorescent anti-rabbit (cat. no. A11008)
and anti-rat (cat. no. A21471) IgG (H+L) antibodies (1:500;
Invitrogen; Thermo Fisher Scientific, Inc.) labeled secondary
antibodies.
For biotinylation, the tissues were incubated with
an Elite ABC kit (Vector laboratories, Maravai LifeSciences), and
immunodetected by incubation with 3′3 diaminobenzidine
(Sigma-Aldrich; Merck KgaA) solution at RT for 5 min. For
fluorescence detection, the nuclei were stained with Hoechst 33342
(1:10,000; cat. no. 62249; Thermo Fisher Scientific, Inc.) at RT
for 15 min. The following primary antibodies were used: Anti-HIF-1α
(1:300; cat. no. NB100-479) and anti-CD31 (1:200; cat. no. 550274).
Three random fields of view in each section were calculated using
ImageJ software (National Institutes of Health) and the relative
protein expression level of each protein was quantified according
to integrated optical intensity from 3 independent experiments.
Statistical analysis
All the experiments were performed 3 times
independently and the data are presented as the mean and SEM using
SPSS software (v25; IBM Corp). Differences between 2 groups were
evaluated using an unpaired Student's t-test differences between 3
groups were evaluated by one-way ANOVA followed by a Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant significance.
Results
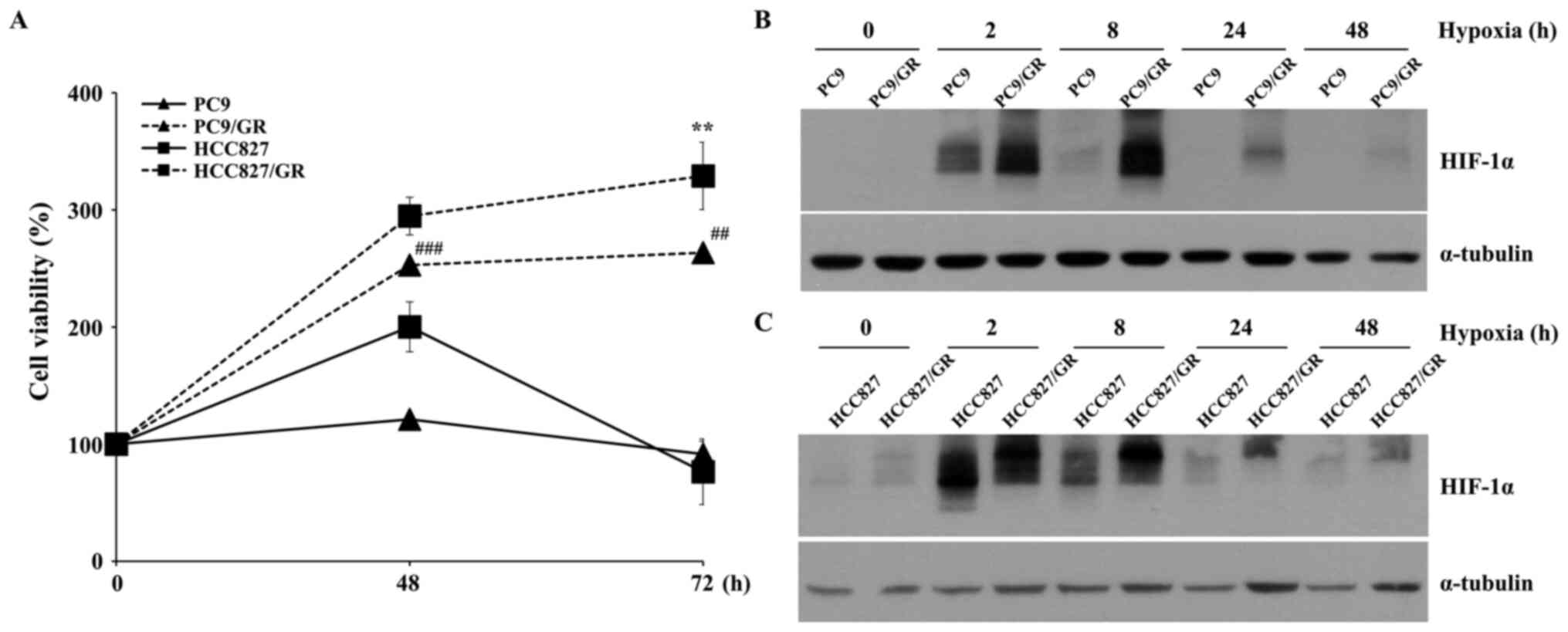
HIF-1α protein expression levels were
increased in GR NSCLC cell lines
To investigate the characteristics of GR NSCLC cell
lines, PC9/GR and HCC827/GR were used in the present study. The GR
cell lines were verified by analyzing the viabilities of these
cells following treatment with gefitinib (Fig. 1A). Since HIF-1α is a key factor in
tumor progression (30), HIF-1α
protein expression levels in the PC9 and HCC827 cell lines were
compared with that in the PC9/GR and HCC827/GR cell lines,
respectively. As shown in Fig. 1B,
the HIF-1α protein expression level in the PC9/GR cell line was
higher compared with that in the PC9 cell line at all hypoxic
exposure times tested. The same experimental results were observed
between the HCC827 and HCC827/GR cell lines (Fig. 1C), indicating that hypoxia-induced
HIF-1α protein expression levels were upregulated in GR NSCLC cell
lines, and the PC9 cell line was selected for further analysis.
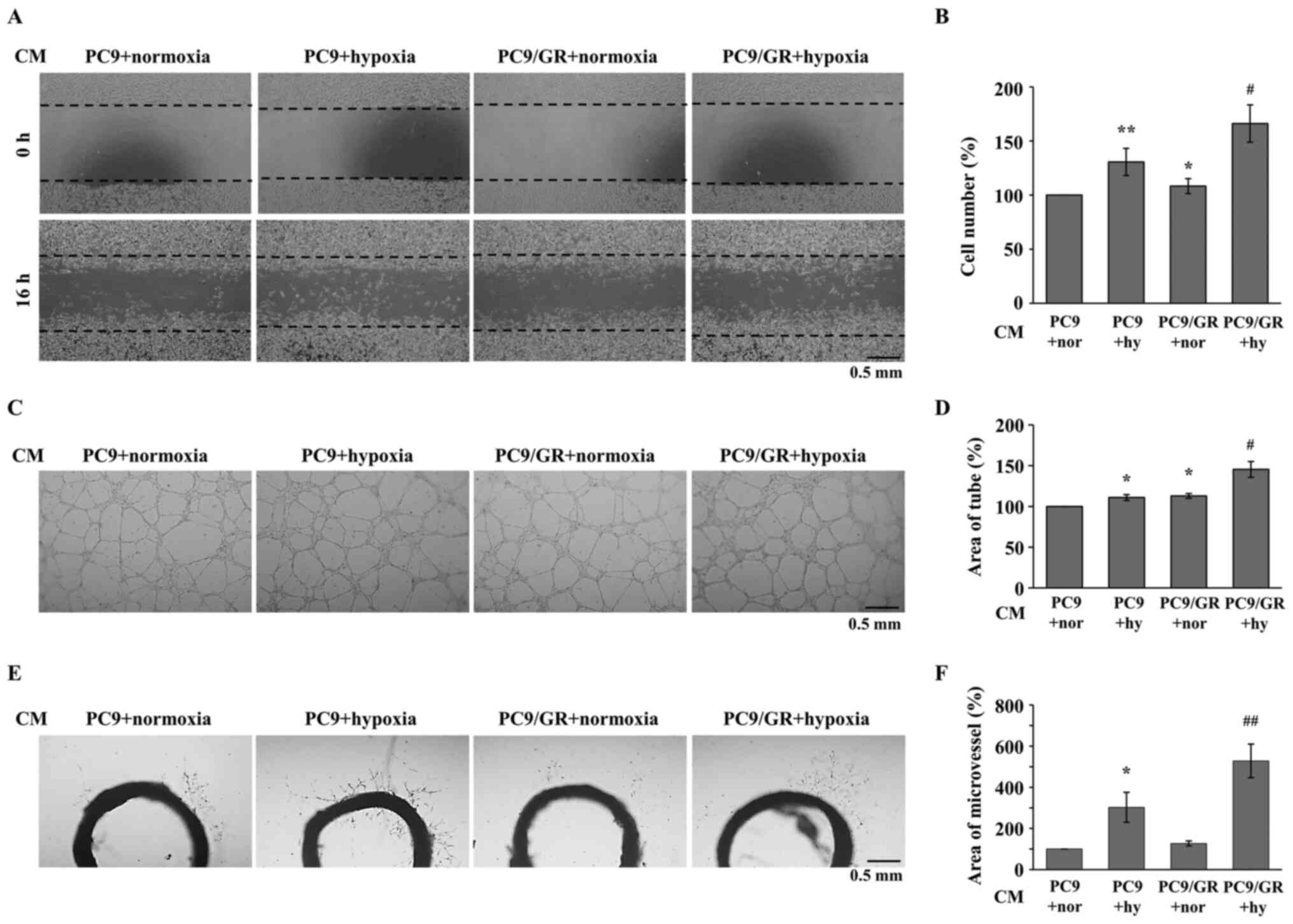
GR cell lines induce angiogenesis
under hypoxic conditions
As HIF-1α mainly regulates hypoxia-induced
angiogenesis during tumorigenesis (30,31), the
angiogenic properties of GR NSCLC cell lines were analyzed using
several angiogenesis assays and endothelial cells. The PC9 and
PC9/GR cells were incubated under hypoxic conditions for 16 h, and
CM was collected and administered to human microvascular
endothelial cells, HMEC-1. As shown in Fig. 2A and B, the CM from
hypoxia-stimulated PC9 cells induced the migration of the HMEC-1
cells. Furthermore, the induced migratory activity of the HMEC-1
cells was increased by CM from hypoxia-stimulated PC9/GR cells. To
examine the tube-forming activities of the endothelial cells, a
tube formation assay was performed using the HMEC-1 cells, treated
with the aforementioned CM. Consistent with the migration assay, CM
from hypoxia-stimulated PC9/GR cells increased hypoxia-induced tube
formation (Fig. 2C and D). To
confirm the increase in hypoxia-induced angiogenic activities in
the HMEC-1 cells treated with PC9/GR CM, an ex vivo rat
aortic ring sprouting assay was performed. As shown in Fig. 2E and F, the number of microvessels
sprouting from the aortic ring was increased in the cells treated
with CM from hypoxia-stimulated PC9/GR cells compared with that in
cells treated with CM from hypoxia-stimulated PC9 cells.
Furthermore, the hypoxic induction of angiogenic activities in the
HMEC-1 treated with PC9/GR CM were higher compared with that in the
HMEC-1 cells treated with PC9 CM (Fig.
2B, D and F). These results suggested that hypoxia-induced
angiogenesis was increased by the PC9/GR cell line compared with
that in the PC9 cell line, and that GR NSCLC cell lines could
stimulate angiogenesis during tumorigenesis.
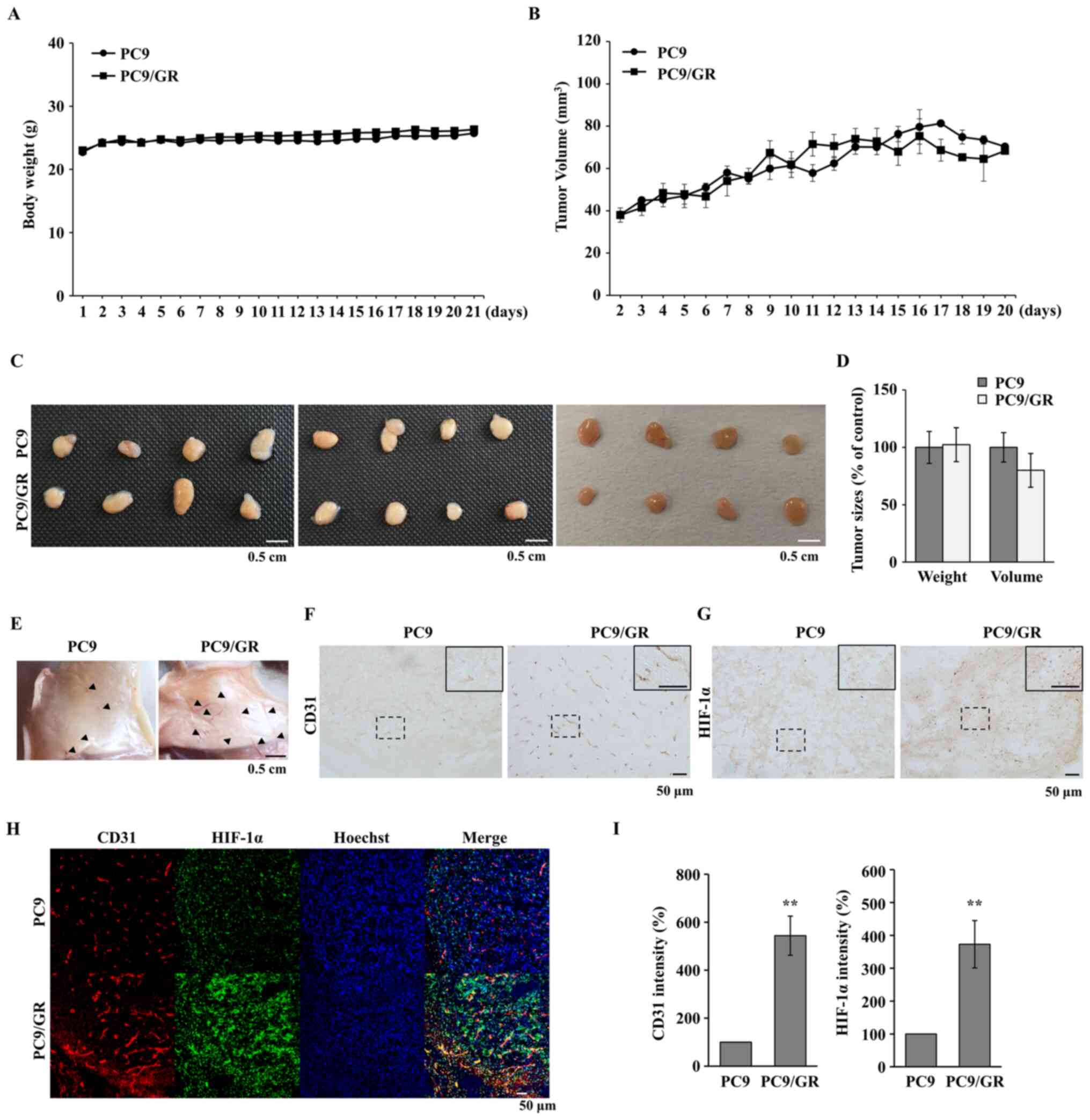
Tumor angiogenesis is increased in the
GR cell lines
Tumor angiogenesis in GR tumors was subsequently
investigated, as angiogenesis is an essential event in tumor
progression (32). Tumor formation
was induced by subcutaneously injecting PC9 and PC9/GR cells into
BALB/c nude mice. As shown in Fig.
3A, the PC9 and PC9/GR tumors had a similar growth pattern and
the body weight of the mice is shown in Fig. 3B. When the isolated tumors, 3 weeks
after injection, were examined, there were no significant
differences in tumor volume and weight between the PC9 and PC9/GR
groups (Fig. 3C and D). The
microvessels in the subcutaneous tissue attached to the tumors were
also analyzed and the number of microvessels near the
PC9/GR-derived tumors was increased compared with that near the
PC9-derived tumors (Fig. 3E). To
evaluate vessel formation within the tumors, it was examined
whether the endothelial cells were present inside the tumors by
staining for CD31, which is a specific marker of endothelial cells.
As shown in Fig. 3F, the CD31 signal
was increased in the PC9/GR tumors. Furthermore, it was found that
the HIF-1α signal was also increased in the PC9/GR tumors (Fig. 3G). To confirm the association between
HIF-1α expression and blood vessel formation, fluorescent
double-staining was performed in the PC9 and PC9/GR tumors. In the
PC9/GR tumor tissues, CD31 expression was increased around the
HIF-1α-expressing region (Fig. 3H and
I), indicating that HIF-1α-induced angiogenesis was stimulated
in GR tumors.
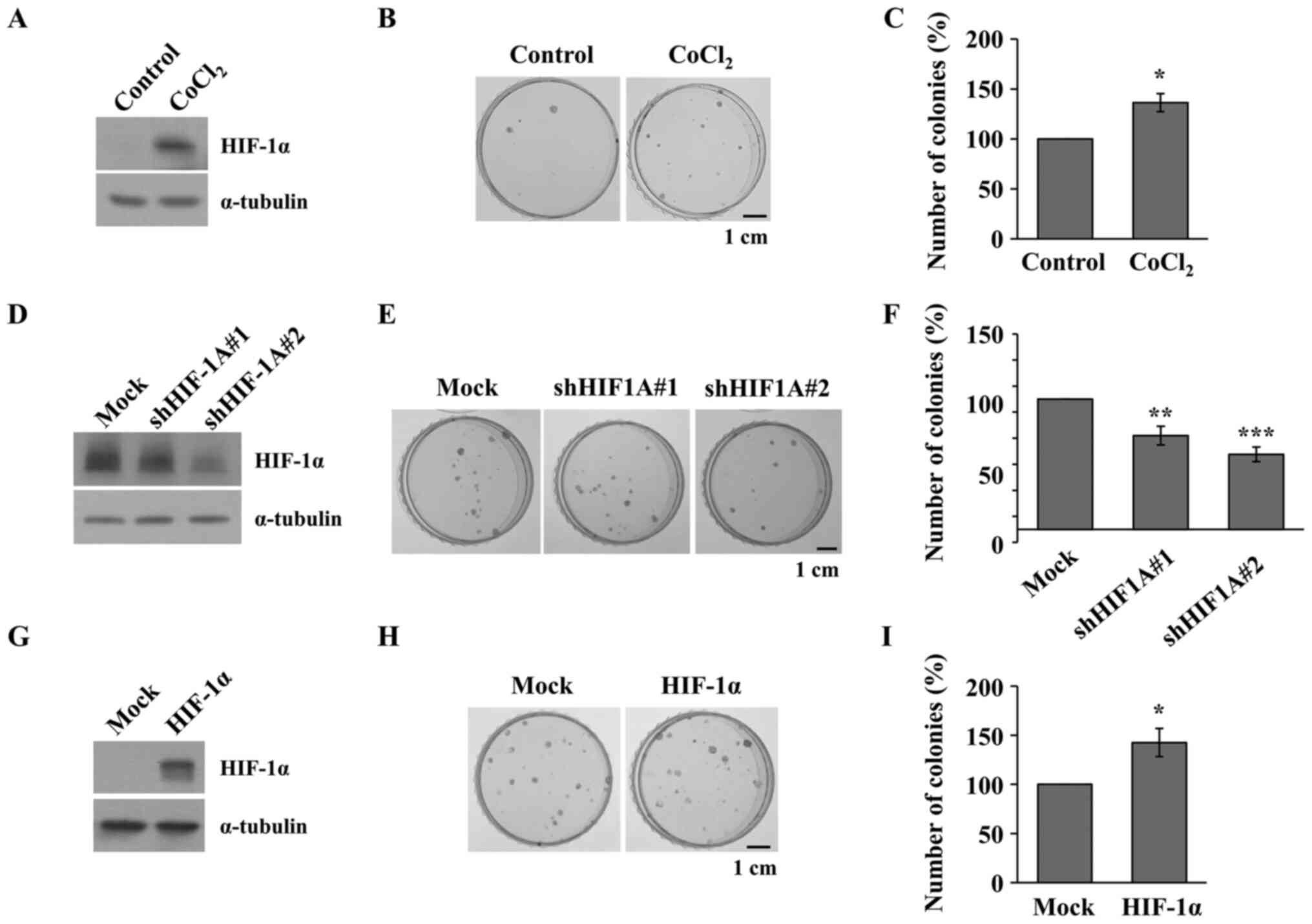
Inhibiting HIF-1α attenuates the
acquisition of GR in the NSCLC cell lines
To demonstrate the association between HIF-1α
expression and gefitinib resistance, HIF-1α-regulated PC9 cells
treated with CoCl2, and transfected with shHIF-1A vector
or HIF-1α overexpression vector were treated with increasing
concentrations of gefitinib (from 10 to 200 nM) for 2 weeks in the
colony formation assay. The PC9 cells were treated with
CoCl2 to induce the upregulation of HIF-1α expression
(Fig. 4A) and a colony formation
assay was performed following CoCl2 and gefitinib
treatment. As shown in Fig. 4B and
C, CoCl2 increased the number of PC9 colonies
following gefitinib treatment. Next, shRNA vectors against HIF-1A
were transfected into the PC9 cells (Fig. 4D) and were subsequently treated with
CoCl2 following which a colony formation assay was
performed under CoCl2 and gefitinib treatment. As shown
in Fig. 4E and F, the colony forming
activity was significantly inhibited by HIF-1α knockdown. On the
contrary, HIF-1α overexpression vector was transfected into the PC9
cells (Fig. 4G) and a colony
formation assay was performed under gefitinib treatment. As shown
in Fig. 4H and I, number of colonies
was significantly increased by HIF-1α overexpression, suggesting
that HIF-1α induced gefitinib resistance in NSCLC cell lines.
Discussion
Chemotherapy using EGFR-TKIs has successfully
improved the survival time in patients NSCLC; however, there are
limitations to the use of EGFR-TKIs due to the acquisition of
resistance (33). Gefitinib is a
first-line treatment that has been used for patients with NSCLC,
and acquired resistance inevitably develops (34). GR NSCLC promoted cell proliferation
and exhibited more aggressive clinical progression (35). HIF-1 is a primary transcription
factor that is activated by hypoxia, and induces the expression of
various genes associated with angiogenesis, proliferation and
survival during tumor progression (36). In addition, the accumulation of
HIF-1α correlated with radiotherapy resistance and drug resistance
to various cytotoxic agents (37–42). In
the present study, it was found that GR tumors had increased HIF-1α
protein expression level and tumor angiogenesis in NSCLC. This
suggested that inhibition of HIF-1α may inhibit angiogenesis in GR
NSCLC. Furthermore, in the PC9 cells treated with gefitinib, the
regulation of HIF-1α controlled the acquisition of resistance.
These findings indicate that HIF-1α could also be a potential
target for overcoming acquired gefitinib resistance in NSCLC.
Due to signaling complexities and an increasing
trend in anticancer drug resistance, a molecular target that plays
a central role in diverse oncogenic signaling pathways has been
investigated (43). A promising
anticancer drug target, is HIF-1, which promotes physiological
changes associated with therapeutic resistance, including the
limitation of drug accumulation within cells and the regulation of
the cellular response to chemotherapeutic agents (44–46).
Previous studies have demonstrated that activation of HIF-1α
inhibited doxorubicin-mediated apoptosis in human osteosarcoma
(47), was associated with cisplatin
resistance through the regulation of the glutamate-cysteine ligase
modifier subunit and multidrug resistance (MDR)-associated proteins
in lung cancer (48), and mediated
resistance to cetuximab by the induction of glycolysis in head and
neck squamous cell carcinoma cells (49,50).
Numerous studies have suggested that HIF-1α was associated with the
induction of the MDR1 gene, which encodes for and results in the
overexpression of phospho-glycoprotein, a predominant membrane
transporter associated with chemotherapy resistance (51). HIF-1α also induced the expression of
pyruvate dehydrogenase kinase (PDK)-1 and −3, and the upregulation
of PDK3 was associated with drug resistance in colon cancer
(52). However, the mechanism by
which HIF-1α mediates drug resistance in cancer is not fully
understood. It was previously reported that HIF-1α expression was
decreased in NSCLC cell lines following gefitinib treatment
(53,54), and activation of HIF-1α in the HCC827
cell line stimulated tumorigenic activities, including
proliferation and migration, even though gefitinib was administered
for ~48 h (54). YC-1 enhanced the
antitumor activity of gefitinib and reversed sensitivity to
gefitinib in GR HCC827 cell lines (55,56). In
the present study, the characteristics of GR NSCLC cell lines were
analyzed and it was found that HIF-1α expression and angiogenic
activities were increased in GR NSCLC cell lines and GR tumors
compared with that in the parent NSCLC cell lines. Furthermore,
overexpression of HIF-1α increased the number of colonies formed by
gradually increasing concentrations of gefitinib-treated PC9 cells
for 2 weeks; in contrast, knockdown of HIF-1α expression by shRNA
vectors decreased the colony number. From the results, we
hypothesized that regulation of HIF-1α may modulate the acquisition
of gefitinib resistance in NSCLC cell lines. In further studies,
the related mechanism by which HIF-1α regulates the acquisition of
gefitinib resistance should be examined.
Several studies support an association between EGFR
activation and HIF-1α expression, which could be important in tumor
progression (57–60). The EGF and EGFR signaling pathways
might induce the translation of HIF-1α (57) and HIF-1α mediated angiogenesis by
upregulating angiogenic factors, such as VEGF in tumor cells
(61). In accordance with previous
studies, the results in the present study indicated the induction
of hypoxia-mediated tumor angiogenesis in GR NSCLC cell lines
(Fig. 2). Previous studies have
demonstrated that gefitinib reduced HIF-1α and VEGF protein
expression levels in EGFR-sensitive NSCLC cell lines (57,62).
HIF-1α expression is reduced by gefitinib; however, HIF-1α enhanced
gefitinib resistance (54). We
propose a possible mechanism to explain how downregulated HIF-1α
controls gefitinib resistance. First, PI3K-AKT-FRAP signaling
increases the rate of HIF-1α synthesis by other receptor and
non-receptor tyrosine kinases, including HER2 and VSRC (63). Second, HIF-1α may increase c-Jun
protein expression level, and a positive feed-forward loop exists
between HIF-1α and c-Jun, in which constitutive activation of the
JNK-c-Jun pathway can upregulate HIF-1α protein levels (64). Third, recent studies have revealed
that HIF-1α was a direct transcriptional regulator of EGFR
(59). EGFR enhances HIF-1α
expression via a positive feed-forward loop (59). The induction of HIF-1α promotes EGFR
transcription, and ultimately (59),
EGFR mutations may be generated during the EGFR transcriptional
process.
In conclusion, the results from the present study
demonstrated that HIF-1α was increased in GR NSCLC cell lines and
lead to induction of angiogenic effects in a mouse xenograft model
and in angiogenesis assays in vitro. In addition, the
regulation of HIF-1α controlled the proliferative ability of the
PC9 cells under gefitinib treatment for two weeks. Therefore,
HIF-1α could be a potential target for overcoming acquired
gefitinib resistance using other EGFR-TKIs.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Research
Foundation of Korea grant funded by the Korea government
(NRF-2021R1A2C1003297) and by a grant from the Basic Research
Program through the National Research Foundation funded by the
Ministry of Science and ICT (2019R1F1A1041812) of Republic of
Korea.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SHL and JWJ designed the experiments. JEC, WYB and
JSC performed the experiments and analyzed the data. WYB and JWJ
wrote the paper. JEC, WYB, JSC, and JWJ confirm the authenticity of
the data in the present manuscript. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Committee for Care and Use of Laboratory Animals at the Kyung Hee
University [KHUASP(SE)-17-144, 11-24-2017].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
GR
|
gefitinib-resistant
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jackson AL, Zhou B and Kim WY: HIF,
hypoxia and the role of angiogenesis in non-small cell lung cancer.
Expert Opin Ther Targets. 14:1047–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nan X, Xie C, Yu X and Liu J: EGFR TKI as
first-line treatment for patients with advanced EGFR
mutation-positive non-small-cell lung cancer. Oncotarget.
8:75712–75726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dong L, Lei D and Zhang H: Clinical
strategies for acquired epidermal growth factor receptor tyrosine
kinase inhibitor resistance in non-small-cell lung cancer patients.
Oncotarget. 8:64600–64606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma C, Wei S and Song Y: T790M and acquired
resistance of EGFR TKI: A literature review of clinical reports. J
Thorac Dis. 3:10–18. 2011.PubMed/NCBI
|
|
8
|
Regales L, Gong Y, Shen R, de Stanchina E,
Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O,
Mellinghoff IK, et al: Dual targeting of EGFR can overcome a major
drug resistance mutation in mouse models of EGFR mutant lung
cancer. J Clin Invest. 119:3000–3010. 2009.PubMed/NCBI
|
|
9
|
Janjigian YY, Smit EF, Groen HJ, Horn L,
Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, et al:
Dual inhibition of EGFR with afatinib and cetuximab in kinase
inhibitor-resistant EGFR-mutant lung cancer with and without T790M
mutations. Cancer Discov. 4:1036–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim D, Bach DH, Fan YH, Luu TT, Hong JY,
Park HJ and Lee SK: AXL degradation in combination with EGFR-TKI
can delay and overcome acquired resistance in human non-small cell
lung cancer cells. Cell Death Dis. 10:3612019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pan YH, Jiao L, Lin CY, Lu CH, Li L, Chen
HY, Wang YB and He Y: Combined treatment with metformin and
gefitinib overcomes primary resistance to EGFR-TKIs with EGFR
mutation via targeting IGF-1R signaling pathway. Biologics.
12:75–86. 2018.PubMed/NCBI
|
|
12
|
Zhuang H, Bai J, Chang JY, Yuan Z and Wang
P: MTOR inhibition reversed drug resistance after combination
radiation with erlotinib in lung adenocarcinoma. Oncotarget.
7:84688–84694. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mok TSK, Kim SW, Wu YL, Nakagawa K, Yang
JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S, et al: Gefitinib Plus
chemotherapy versus chemotherapy in epidermal growth factor
receptor mutation-positive non-small-cell lung cancer resistant to
first-line gefitinib (IMPRESS): Overall survival and biomarker
analyses. J Clin Oncol. 35:4027–4034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhuang H, Shi S, Guo Y and Wang Z:
Increase of secondary mutations may be a drug-resistance mechanism
for lung adenocarcinoma after radiation therapy combined with
tyrosine kinase inhibitor. J Cancer. 10:5371–5376. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Muz B, de la Puente P, Azab F and Azab AK:
The role of hypoxia in cancer progression, angiogenesis,
metastasis, and resistance to therapy. Hypoxia (Auckl). 3:83–92.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Majmundar AJ, Wong WJ and Simon MC:
Hypoxia-inducible factors and the response to hypoxic stress. Mol
Cell. 40:294–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tatum JL, Kelloff GJ, Gillies RJ, Arbeit
JM, Brown JM, Chao KS, Chapman JD, Eckelman WC, Fyles AW, Giaccia
AJ, et al: Hypoxia: Importance in tumor biology, noninvasive
measurement by imaging, and value of its measurement in the
management of cancer therapy. Int J Radiat Biol. 82:699–757. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ziello JE, Jovin IS and Huang Y:
Hypoxia-inducible factor (HIF)-1 regulatory pathway and its
potential for therapeutic intervention in malignancy and ischemia.
Yale J Biol Med. 80:51–60. 2007.PubMed/NCBI
|
|
19
|
Jing X, Yang F, Shao C, Wei K, Xie M, Shen
H and Shu Y: Role of hypoxia in cancer therapy by regulating the
tumor microenvironment. Mol Cancer. 18:1572019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin X, Dai L, Ma Y, Wang J and Liu Z:
Implications of HIF-1α in the tumorigenesis and progression of
pancreatic cancer. Cancer Cell Int. 20:2732020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li H, Zhou S, Li X, Wang D, Wang Y, Zhou C
and Schmid-Bindert G: Gefitinib-resistance is related to BIM
expression in non-small cell lung cancer cell lines. Cancer Biother
Radiopharm. 28:115–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharma P, Hu-Lieskovan S, Wargo JA and
Ribas A: Primary, adaptive, and acquired resistance to cancer
immunotherapy. Cell. 168:707–723. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He J, Jin S, Zhang W, Wu D, Li J, Xu J and
Gao W: Long non-coding RNA LOC554202 promotes acquired gefitinib
resistance in non-small cell lung cancer through upregulating
miR-31 expression. J Cancer. 10:6003–6013. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nigro A, Ricciardi L, Salvato I, Sabbatino
F, Vitale M, Crescenzi MA, Montico B, Triggiani M, Pepe S, Stellato
C, et al: Enhanced expression of CD47 is associated with off-target
resistance to tyrosine kinase inhibitor gefitinib in NSCLC. Front
Immunol. 10:31352020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Del Signore A, Gotti C, Rizzo A, Moretti M
and Paggi P: Nicotinic acetylcholine receptor subtypes in the rat
sympathetic ganglion: Pharmacological characterization, subcellular
distribution and effect of pre- and postganglionic nerve crush. J
Neuropathol Exp Neurol. 63:138–150. 2004. View Article : Google Scholar
|
|
27
|
Kim AR, Kim JH, Kim A, Sohn Y, Cha JH, Bak
EJ and Yoo YJ: Simvastatin attenuates tibial bone loss in rats with
type 1 diabetes and periodontitis. J Transl Med. 16:3062018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pacioni S, Rueger MA, Nisticò G, Bornstein
SR, Park DM, McKay RD and Androutsellis-Theotokis A: Fast, potent
pharmacological expansion of endogenous
hes3+/sox2+ cells in the adult mouse and rat
hippocampus. PLoS One. 7:e516302012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwon TR, Han SW, Kim JH, Lee BC, Kim JM,
Hong JY and Kim BJ: Polydeoxyribonucleotides improve diabetic wound
healing in mouse animal model for experimental validation. Ann
Dermatol. 31:403–413. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maxwell PH, Dachs GU, Gleadle JM, Nicholls
LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW and Ratcliffe PJ:
Hypoxia-inducible factor-1 modulates gene expression in solid
tumors and influences both angiogenesis and tumor growth. Proc Natl
Acad Sci USA. 94:8104–8109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carmeliet P, Dor Y, Herbert JM, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, et al: Role of HIF-1alpha in hypoxia-mediated apoptosis,
cell proliferation and tumour angiogenesis. Nature. 394:485–490.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Folkman J: What is the evidence that
tumors are angiogenesis dependent? J Natl Cancer Inst. 82:4–6.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin JJ and Shaw AT: Resisting resistance:
Targeted therapies in lung cancer. Trends Cancer. 2:350–364. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao J, Li HR, Jin C, Jiang JH and Ding JY:
Strategies to overcome acquired resistance to EGFR TKI in the
treatment of non-small cell lung cancer. Clin Transl Oncol.
21:1287–1301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qi M, Tian Y, Li W, Li D, Zhao T, Yang Y,
Li Q, Chen S, Yang Y, Zhang Z, et al: ERK inhibition represses
gefitinib resistance in non-small cell lung cancer cells.
Oncotarget. 9:12020–12034. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Semenza GL: HIF-1 and human disease: One
highly involved factor. Genes Dev. 14:1983–1991. 2000.PubMed/NCBI
|
|
37
|
Chen L, Feng P, Li S, Long D, Cheng J, Lu
Y and Zhou D: Effect of hypoxia-inducible factor-1alpha silencing
on the sensitivity of human brain glioma cells to doxorubicin and
etoposide. Neurochem Res. 34:984–990. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nardinocchi L, Puca R, Sacchi A and
D'Orazi G: Inhibition of HIF-1alpha activity by
homeodomain-interacting protein kinase-2 correlates with
sensitization of chemoresistant cells to undergo apoptosis. Mol
Cancer. 8:12009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hao J, Song X, Song B, Liu Y, Wei L, Wang
X and Yu J: Effects of lentivirus-mediated HIF-1alpha knockdown on
hypoxia-related cisplatin resistance and their dependence on p53
status in fibrosarcoma cells. Cancer Gene Ther. 15:449–455. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu L, Ning X, Sun L, Zhang H, Shi Y, Guo
C, Han S, Liu J, Sun S, Han Z, et al: Hypoxia-inducible factor-1
alpha contributes to hypoxia-induced chemoresistance in gastric
cancer. Cancer Sci. 99:121–128. 2008.PubMed/NCBI
|
|
41
|
Sasabe E, Zhou X, Li D, Oku N, Yamamoto T
and Osaki T: The involvement of hypoxia-inducible factor-1alpha in
the susceptibility to gamma-rays and chemotherapeutic drugs of oral
squamous cell carcinoma cells. Int J Cancer. 120:268–277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brown LM, Cowen RL, Debray C, Eustace A,
Erler JT, Sheppard FC, Parker CA, Stratford IJ and Williams KJ:
Reversing hypoxic cell chemoresistance in vitro using genetic and
small molecule approaches targeting hypoxia inducible factor-1. Mol
Pharmacol. 69:411–418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
von Manstein V, Yang CM, Richter D, Delis
N, Vafaizadeh V and Groner B: Resistance of cancer cells to
targeted therapies through the activation of compensating signaling
loops. Curr Signal Transduct Ther. 8:193–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Unruh A, Ressel A, Mohamed HG, Johnson RS,
Nadrowitz R, Richter E, Katschinski DM and Wenger RH: The
hypoxia-inducible factor-1 alpha is a negative factor for tumor
therapy. Oncogene. 22:3213–3220. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Verduzco D, Lloyd M, Xu L, Ibrahim-Hashim
A, Balagurunathan Y, Gatenby RA and Gillies RJ: Intermittent
hypoxia selects for genotypes and phenotypes that increase
survival, invasion, and therapy resistance. PLoS One.
10:e01209582015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhao Q, Li Y, Tan BB, Fan LQ, Yang PG and
Tian Y: HIF-1α induces multidrug resistance in gastric cancer cells
by inducing miR-27a. PLoS One. 10:e01327462015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roncuzzi L, Pancotti F and Baldini N:
Involvement of HIF-1α activation in the doxorubicin resistance of
human osteosarcoma cells. Oncol Rep. 32:389–394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li F, Zhu T, Cao B, Wang J and Liang L:
Apatinib enhances antitumour activity of EGFR-TKIs in non-small
cell lung cancer with EGFR-TKI resistance. Eur J Cancer.
84:184–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu H, Lu Y, Xie Y, Qiu S, Li X and Fan Z:
Rational combination with PDK1 inhibition overcomes cetuximab
resistance in head and neck squamous cell carcinoma. JCI Insight.
4:42019. View Article : Google Scholar
|
|
50
|
Lu H, Liang K, Lu Y and Fan Z: The
anti-EGFR antibody cetuximab sensitizes human head and neck
squamous cell carcinoma cells to radiation in part through
inhibiting radiation-induced upregulation of HIF-1α. Cancer Lett.
322:78–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Comerford KM, Wallace TJ, Karhausen J,
Louis NA, Montalto MC and Colgan SP: Hypoxia-inducible
factor-1-dependent regulation of the multidrug resistance (MDR1)
gene. Cancer Res. 62:3387–3394. 2002.PubMed/NCBI
|
|
52
|
Lu CW, Lin SC, Chien CW, Lin SC, Lee CT,
Lin BW, Lee JC and Tsai SJ: Overexpression of pyruvate
dehydrogenase kinase 3 increases drug resistance and early
recurrence in colon cancer. Am J Pathol. 179:1405–1414. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH, Yoo YD and Lee JC: Gefitinib circumvents
hypoxia-induced drug resistance by the modulation of HIF-1alpha.
Oncol Rep. 21:801–807. 2009.PubMed/NCBI
|
|
54
|
Jin Q, Zhou J, Xu X, Huang F and Xu W:
Hypoxia-inducible factor-1 signaling pathway influences the
sensitivity of HCC827 cells to gefitinib. Oncol Lett. 17:4034–4043.
2019.PubMed/NCBI
|
|
55
|
Hu H, Miao XK, Li JY, Zhang XW, Xu JJ,
Zhang JY, Zhou TX, Hu MN, Yang WL and Mou LY: YC-1 potentiates the
antitumor activity of gefitinib by inhibiting HIF-1α and promoting
the endocytic trafficking and degradation of EGFR in
gefitinib-resistant non-small-cell lung cancer cells. Eur J
Pharmacol. 874:1729612020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jin Q, Zheng J, Chen M, Jiang N, Xu X and
Huang F: HIF-1 inhibitor YC-1 reverses the acquired resistance of
EGFR-mutant HCC827 cell line with MET amplification to gefitinib.
Oxid Med Cell Longev. 2021:66338672021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao
GD and Maity A: EGFR tyrosine kinase inhibitors decrease VEGF
expression by both hypoxia-inducible factor (HIF)-1-independent and
HIF-1-dependent mechanisms. Cancer Res. 66:3197–3204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee SH, Koo KH, Park JW, Kim HJ, Ye SK,
Park JB, Park BK and Kim YN: HIF-1 is induced via EGFR activation
and mediates resistance to anoikis-like cell death under lipid
rafts/caveolae-disrupting stress. Carcinogenesis. 30:1997–2004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Peng XH, Karna P, Cao Z, Jiang BH, Zhou M
and Yang L: Cross-talk between epidermal growth factor receptor and
hypoxia-inducible factor-1alpha signal pathways increases
resistance to apoptosis by up-regulating survivin gene expression.
J Biol Chem. 281:25903–25914. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Swinson DE and O'Byrne KJ: Interactions
between hypoxia and epidermal growth factor receptor in
non-small-cell lung cancer. Clin Lung Cancer. 7:250–256. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tang N, Wang L, Esko J, Giordano FJ, Huang
Y, Gerber HP, Ferrara N and Johnson RS: Loss of HIF-1alpha in
endothelial cells disrupts a hypoxia-driven VEGF autocrine loop
necessary for tumorigenesis. Cancer Cell. 6:485–495. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee JG and Wu R: Erlotinib-cisplatin
combination inhibits growth and angiogenesis through c-MYC and
HIF-1α in EGFR-mutated lung cancer in vitro and in vivo. Neoplasia.
17:190–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Laughner E, Taghavi P, Chiles K, Mahon PC
and Semenza GL: HER2 (neu) signaling increases the rate of
hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel
mechanism for HIF-1-mediated vascular endothelial growth factor
expression. Mol Cell Biol. 21:3995–4004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Meng S, Wang G, Lu Y and Fan Z: Functional
cooperation between HIF-1α and c-Jun in mediating primary and
acquired resistance to gefitinib in NSCLC cells with activating
mutation of EGFR. Lung Cancer. 121:82–90. 2018. View Article : Google Scholar : PubMed/NCBI
|