Introduction
According to the Cancer Statistics report published
by the American Cancer Society in 2021, lung cancer is the leading
cause of death in both men and women, accounting for 22% of all
cancer deaths (1). In China, lung
cancer ranks first among malignant tumors in incidence and
mortality and has become an important disease that endangers public
health and affects people's quality of life (2). Lung cancer lacks obvious clinical
symptoms in the early stage, and at the same time lacks effective
early screening methods, the 5-year survival rate of lung cancer
patients is less than 15% (3,4). In
addition, metastasis and chemoresistance are important causes of
the high mortality in lung cancer (5,6). Hence,
the identification of specific molecular markers and the
exploration of new effective drug targets for the early diagnosis
and treatment of lung cancer are urgently needed and have extremely
important scientific research significance and clinical application
prospects (7).
Epigenetic events are important in all aspects of
biology, and numerous studies have shown that they serve key roles
in carcinogenesis and tumor progression (8–10).
Various mechanisms contribute to the occurrence of lung cancer,
including DNA methylation. However, the specific regulatory
mechanisms have not been fully elucidated (11). DNA methylation is an important
epigenetic regulation and abnormal methylation can affect gene
expression (12,13). Molecular markers of DNA methylation
for the early diagnosis and prognosis prediction of tumors and
tumor-targeting drugs based on epigenetics have been widely
studied, for example, the application of O-6-methylguanine-DNA
methyltransferase (MGMT) DNA methylation in molecular diagnosis of
glioma (14–16). The identification of DNA methylation
profiles in tumors has laid the foundation for the discovery of new
tumor therapeutic targets.
Tripartite motif containing 58 (TRIM58) is a member
of the tripartite motif (TRIM) family (17). The TRIM protein family is a conserved
protein family that plays important roles in signal transduction,
innate immunity, autophagy, tumors and other functions (18,19). For
instance, TRIM67 inhibits the occurrence and progression of
colorectal cancer by activating the p53 signaling pathway (20). The TRIM family is characterized by 3
domains (from the N-terminus to the C-end): the RING (Really
Interesting New Gene) -finger domain, one or two B-boxes, and one
coiled-coil domain (21). Currently,
more than 80 TRIM proteins have been found in humans, most of which
have the function of E3 ubiquitin ligase and regulate cell
transcription, proliferation and apoptosis through the
ubiquitination of target molecules, thus participating in various
physiological and pathological processes in the body, such as
developmental disorders, viral infections and cancer (22,23).
According to the features of the domains, the TRIM family is
divided into 11 subfamilies and TRIM58 is a member of the C-IV-1
subfamily (C-I to C-XI) (24).
The main subtypes of lung cancer are lung
adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC)
(25). In our previous study, we
collected clinical samples of LUSC for genome-wide DNA methylation
analysis and identified many new epigenetic signatures (26). In the present study 3 methylation
microarray datasets (GSE63384, GSE62948 and GSE32861) of LUAD from
the Gene Expression Omnibus (GEO) database were collected for the
integrated analysis of large samples. Integrating the results of
high-throughput screening, focusing on TRIM58, which was
hypermethylated and downregulated in lung cancer. Notably,
functional studies performed demonstrated that overexpression of
TRIM58 inhibited cell proliferation and migration and promoted cell
apoptosis. These findings suggest that TRIM58 serves a critical
role in the malignant phenotype of lung cancer.
Materials and methods
DNA methylation datasets of lung
cancer
The Gene Expression Omnibus (GEO) datasets
(GSE63384, GSE62948 and GSE32861) were all based on the GPL8490
platform (http://www.ncbi.nlm.nih.gov/geo) (27–29). The
3 datasets selected all comprised of paired samples consisting of
tumor and corresponding NTL tissues. The GSE63384 dataset included
35 stage I LUAD tissues and 35 NTL tissues; GSE62948 included 28
LUAD tissues and 28 NTL tissues and GSE32861 contained 59 LUAD
tissues and 59 NTL tissues. Subsequently, these 3 datasets were
used for ROC analysis.
Genome-wide DNA methylation
analysis
Genome-wide DNA methylation analysis using the R
package version 4.2 (http://www.r-project.org/) (30). The linear models for microarray data
(LIMMA) package (v.3.48.0) in Bioconductor was used for data
processing (31). The
Benjamini-Hochberg procedure in R package was used to calculate the
adjusted P-values (32). Probes with
a adjusted P<0.05 and an absolute β difference ≥ 0.2 were
considered differentially methylated genes (DMGs).
The Cancer Genome Atlas (TCGA) data
and validation
Validation datasets were extracted from the data
portal of TCGA (http://tcga-data.nci.nih.gov) (33). TCGA DNA methylation dataset: A total
of 372 LUSC samples with 43 corresponding NTL samples and 460 LUAD
samples with 32 corresponding NTL samples were used for performing
independent DNA methylation verification. TCGA mRNA expression
dataset: including 502 LUSC samples corresponding to 51 NTL
samples, and 571 LUAD samples corresponding to 58 NTL samples for
mRNA expression detection. In addition, the MethHC browser
(http://methhc.mbc.nctu.edu.tw/php/index.php) was used
to analyze the correlation between DNA methylation and mRNA
expression (34).
Cell culture and transfection
A549, a human lung adenocarcinoma cell line was
purchased from the Chinese Academy of Sciences and cultured at 37°C
with 5% CO2 in RPMI-1640 medium (Invitrogen; Thermo
Fisher Scientific Inc.) containing 10% fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific Inc.).
To investigate the molecular functions of TRIM58,
specific small interfering (si) RNA and overexpression vectors of
TRIM58 were constructed. Transfection was performed 24 h after the
cells were plated. TRIM58-siRNA was synthesized by Guangzhou
RiboBio Co., Ltd. and the target sequence was
5′-GGACTATGAAGCCGGTGAA-3′. For the scrambled siRNA used as the
negative control (NC) (Guangzhou RiboBio Co., Ltd.). The final
siRNA (TRIM58-siRNA or scrambled siRNA) concentration was adjusted
to 50nM and transfected with Lipofectamine® RNAiMAX
(Invitrogen; Thermo Fisher Scientific Inc.). pCDNA3.1-TRIM58 vector
was synthesized by Shanghai GeneChem Co., Ltd. and empty pcDNA3.1
vector was used as the negative control. The final pcDNA3.1 vector
concentration was adjusted to 1μg and transfected with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific Inc.).
After transfection for 48 h at 37°C, cell migration was detected
and cells were collected for RNA and protein extraction.
RNA extraction and reverse
transcription-quantitative (RT-q) PCR
A549 cells were collected. TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific Inc.) was used to
extract total RNA. The PrimeScript™ RT reagent kit (Takara Bio,
Inc.) was used for reverse transcription and the standard SYBR
Green PCR kit (cat. no. RR091A; Takara Bio, Inc.) was used for
RT-qPCR according to the manufacturer's protocol. The PCR
thermocycling conditions were as follows: initial denaturation at
95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C
for 60 sec. GAPDH was used as the internal reference gene and the
data were calculated using the 2−∆∆CT method (35). The primer sequences used were as
follows: GAPDH, forward 5′-GGAAGCTTGTCATCAATGGAAATC-3′ and reverse,
5′-TGATGACCCTTTTGGCTCCC-3′; TRIM58 forward,
5′-ATGAGGAAAGAGTTGGAGGACG-3′ and reverse,
5′-AGCCACGATGCTTCTCAAACTC-3′.
Western blotting
A549 cells were collected and protein was extracted
using RIPA lysis buffer (Abcam). The bbicinchoninic acid (BCA) kits
were used to detect protein concentrations. The total protein (40
µg/lane) was separated by 10% SDS-PAGE and transferred to
nitrocellulose membrane. Subsequently, the membrane was incubated
in a blocking solution (5% skimmed milk) for 2 h at room
temperature. The primary antibody was incubated with the samples at
4°C overnight and then the secondary antibody was incubated with
the samples at room temperature for 2 h. The primary antibodies
used were as follows: GAPDH [cat. no. abs132004; 1: 3000; Absin
(Shanghai) Biotechnology Co. Ltd.] and TRIM58 [cat. no. abs103739;
1: 1000; Absin (Shanghai) Biotechnology Co. Ltd.]. GAPDH was used
as the loading control. Anti-rabbit IgG, HRP-linked Antibody [cat.
no. 7074P2; 1: 1000; Cell Signaling Technology Inc.]. ECL
luminescence reagent [cat. no. abs920; Absin (Shanghai)
Biotechnology Co. Ltd.] was used for protein visualization.
Cell proliferation assay
A549 cells were seeded in 96-well plates and cell
proliferation was detected with CellTiter 96® AQueous
One Solution Cell Proliferation Assay (MTS) (Promega Inc.). The
absorbance value was measured at 490 nm.Proliferation was tested
every 24 h for 5 consecutive days.
Wound healing assay
A549 cells were seeded in 6-well plates
(4×105 cells/well). When confluence exceeded 90%, the
cell monolayer was damaged with sterile pipette tips. Subsequently,
the cells were washed gently and quickly with sterile PBS 3 times
and then replaced with RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific Inc.) containing 2% FBS (Gibco; Themo Fisher Scientific
Inc.) and incubated at 37°C for 48 h. The cells in the scratch area
were observed under a light microscope (magnification, ×100) at 0
and 48 h respectively and the migration distance of cells at each
time point was measured manually.
Transwell migration assay
Migration experiments were conducted using 24-well
transwell chambers with 8-µm aperture (Corning, Inc.). The A549
cells were collected, resuspended with serum-free RPMI-1640 medium
and counted. The cell density was adjusted to 4×105
cells/ml. A total of 100 µl cell suspension was inoculated into the
upper chamber and 600 µl RPMI-1640 medium containing 20% FBS was
added to the lower chamber. Cells were incubated at 37°C for 24 h
and stained with 0.1% crystal violet for 20 min at room
temperature. Under the light microscope (magnification, ×100), a
total of 3 fields were randomly selected for photographing and the
number of migrated cells was counted manually.
Flow cytometry analysis
A549 cells were centrifuged at 300 × g for 5 min at
room temperature and the cell precipitate was collected. Annexin
V-FITC/propidium iodide (PI) Apoptosis detection kit (cat. no.
V13241; Invitrogen; Thermo Fisher Scientific Inc.) was used to
detect cell apoptosis (early apoptosis and late apoptosis)
according to the manufacturer's protocol. Apoptotic analysis was
implemented using flow cytometry (Cytomics FC500, Beckman Coulter,
Inc.). CXP (Beckman Coulter, Inc.) software was used.
Gene set enrichment analysis, protein
interaction and co-expression analysis
Gene set enrichment analysis (GSEA) (https://www.gsea-msigdb.org/gsea/index.jsp) was
performed using mRNA expression data from the TCGA database.
Patients were divided into high expression group and low expression
group according to the median expression value
(LUADmedian=−1.966, LUSCmedian=−2.457).
Protein-Protein Interaction Network analysis was constructed using
the STRING database (https://string-db.org/). Co-expression analysis was
performed on lung cancer samples from the Oncomine database
(https://www.oncomine.org/).
Statistical analysis
SPSS version 18.0 (SPSS Inc.) and GraphPad Prism 5.0
software (GraphPad, Inc.) were used for statistical analysis. All
data was from 3 experimental replicates and presented as the mean ±
SEM. Independent Student's t-test was applied to the comparisons
between groups. Spearman correlation method was used to identify
the correlation between DNA methylation and mRNA expression. ROC
analysis was used to explore the diagnostic value of TRIM58.
P<0.05 was considered to indicate statistical significance.
Results
Identification of novel epigenetic
signatures in lung cancer
In the present study, 3 LUAD methylation profiles
(GSE63384, GSE62948, and GSE32861) were collected from the GEO
database to identify DMGs between tumors and NTL tissues. The
present study focused on probes that were specifically
hypermethylated in tumors. The results demonstrated that 67 probes
were hypermethylated in GSE63384, 489 probes were hypermethylated
in GSE62948 and 767 probes were hypermethylated in GSE32861
(Fig. 1A). Subsequently, an
overlapping analysis of these DMGs was conducted and 55 probes that
were significantly hypermethylated in the 3 lung cancer datasets
were found (Fig. 1A and Table I). Additionally, the 55 probes were
analyzed by two-dimensional hierarchical cluster analysis, which
could clearly distinguish tumor tissues from NTL tissues (Fig. 1B). These results revealed a series of
probes that are hypermethylated in LUAD.
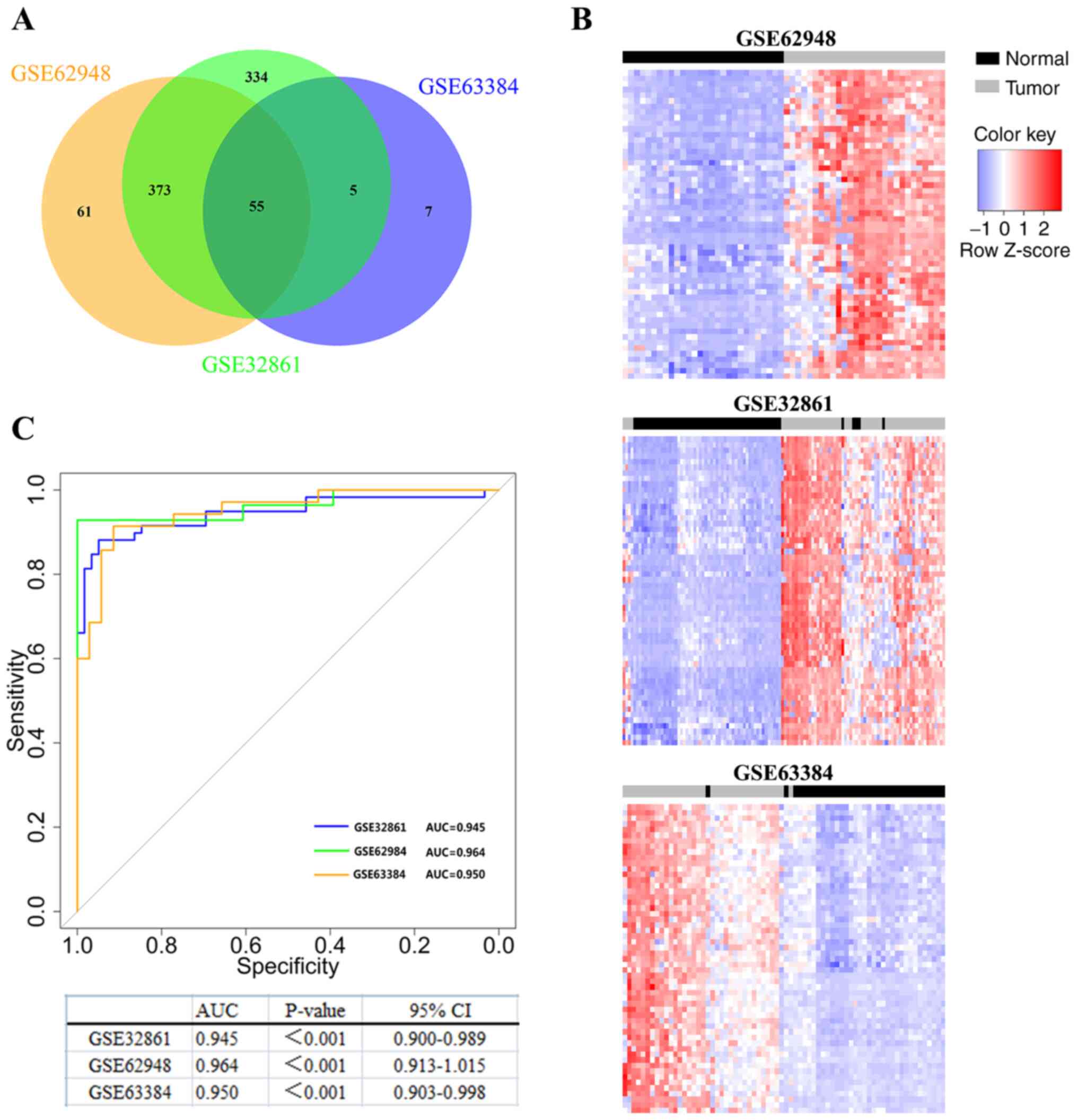 | Figure 1.Identification of novel epigenetic
signatures in lung cancer. (A) Identify differentially methylated
probes through genome-wide DNA methylation analysis. Venn diagram
showing that there are 55 common hypermethylated probes between the
3 LUAD datasets (GSE63384, GSE62948 and GSE32861). (B)
Two-dimensional cluster analysis was performed on the
differentially methylated probes in the 3 datasets. Each row is a
probe; each column is a sample. The blue box, the expression level
is low; the red box, the expression level is high. (C) Diagnostic
value of TRIM58 methylation in lung cancer by ROC curves analysis.
The blue line is GSE32861, the green line is GSE62948, and the
orange line is GSE63384. ROC, receiver operating characteristic;
TRIM58, tripartite motif containing 58; LUAD, lung adenocarcinoma;
AUC, area under curve, CI, confidence interval. |
 | Table I.Basic information of 55 differential
methylation probes. |
Table I.
Basic information of 55 differential
methylation probes.
| Ilmn ID
(Probes) | Gene Symbol | Genbank
Accession | Annotation |
|---|
| cg08572611 | ACTL6B | NM_016188.3 | Actin-like 6B |
| cg10235817 | ADRA2C | NM_000683.3 | Adrenoceptor α
2C |
| cg17619823 | ADRB3 | NM_000025.1 | Adrenoceptor β
3 |
| cg17525406 | AJAP1 | NM_018836.2 | Adherens Junctions
Associated Protein 1 |
| cg20959866 | AJAP1 | NM_018836.2 | Adherens Junctions
Associated Protein 1 |
| cg12111714 | ATP8A2 | NM_016529.3 | ATPase Phospholipid
Transporting 8A2 |
| cg05890484 | BHMT | NM_001713.1 |
Betaine-Homocysteine
S-Methyltransferase |
| cg14419187 | C2orf21 | NM_182587.1 | Unc-80 Homolog,
NALCN Channel Complex Subunit |
| cg03544320 | CRMP1 | NM_001313.3 | Collapsin Response
Mediator Protein 1 |
| cg09229912 | CUTL2 | NM_015267.1 | Cut Like Homeobox
2 |
| cg10303487 | DPYS | NM_001385.1 |
Dihydropyrimidinase |
| cg04048259 | EDN3 | NM_000114.2 | Endothelin 3 |
| cg00027083 | EPB41L3 | NM_012307.2 | Erythrocyte
Membrane Protein Band 4.1 Like 3 |
| cg08575537 | EPO | NM_000799.2 | Erythropoietin |
| cg20723355 | FBXO39 | NM_153230.1 | F-Box Protein
39 |
| cg19831575 | FGF4 | NM_002007.1 | Fibroblast Growth
Factor 4 |
| cg02757432 | GPR26 | NM_153442.1 | G Protein-Coupled
Receptor 26 |
| cg06722633 | GRIK3 | NM_000831.2 | Glutamate
Ionotropic Receptor Kainate Type Subunit 3 |
| cg14859460 | GRM6 | NM_000843.2 | Glutamate
Metabotropic Receptor 6 |
| cg26609631 | GSH1 | NM_145657.1 | GS Homeobox 1 |
| cg10883303 | HOXA13 | NM_000522.2 | Homeobox A13 |
| cg26069745 | HOXA2 | NM_006735.3 | Homeobox A2 |
| cg01354473 | HOXA9 | NM_152739.2 | Homeobox A9 |
| cg01381846 | HOXA9 | NM_152739.2 | Homeobox A9 |
| cg26521404 | HOXA9 | NM_152739.2 | Homeobox A9 |
| cg06760035 | HOXB4 | NM_024015.3 | Homeobox B4 |
| cg08089301 | HOXB4 | NM_024015.3 | Homeobox B4 |
| cg23130254 | HOXD12 | NM_021193.2 | Homeobox D12 |
| cg25574024 | IGF2AS | NM_016412.1 | Insulin-Like Growth
Factor II, Antisense |
| cg23349790 | IGSF21 | NM_032880.2 | Immunoglobin
Superfamily Member 21 |
| cg27409364 | KCNC1 | NM_004976.2 | Potassium
Voltage-Gated Channel Subfamily C Member 1 |
| cg22660578 | LHX1 | NM_005568.2 | LIM homeobox
protein 1 |
| cg04330449 | NEUROG1 | NT_034772.5 | Neurogenin 1 |
| cg22881914 | NID2 | NM_007361.2 | Nidogen 2 |
| cg08441806 | NKX6-2 | NM_177400.1 | NK6 Transcription
Factor Related, Locus 2 |
| cg24194775 | NPR2 | NM_000907.2 | Natriuretic Peptide
Receptor 2 |
| cg00548268 | NPTX2 | NM_002523.1 | Neuronal Pentraxin
2 |
| cg12799895 | NPTX2 | NM_002523.1 | Neuronal Pentraxin
2 |
| cg20291049 | POU3F3 | NM_006236.1 | POU Domain Class 3,
Transcription Factor 3 |
| cg12374721 | PRAC | NM_032391.2 | PRAC1 Small Nuclear
Protein |
| cg09516965 | PTGDR | NM_000953.2 | Prostaglandin D2
Receptor |
| cg08118311 | SALL3 | NM_171999.1 | Spalt Like
Transcription Factor 3 |
| cg15191648 | SALL3 | NM_171999.1 | Spalt Like
Transcription Factor 3 |
| cg02919422 | SOX17 | NM_022454.2 | SRY-Box
Transcription Factor 17 |
| cg02164046 | SST | NM_001048.3 | Somatostatin |
| cg17586860 | SSTR4 | NM_001052.1 | Somatostatin
Receptor 4 |
| cg25720804 | TLX3 | NM_021025.2 | T Cell Leukemia
Homeobox 3 |
| cg14696396 | TM6SF1 | NM_023003.1 | Transmembrane 6
Superfamily Member 1 |
| cg01009664 | TRH | NM_007117.1 | Thyrotropin
Releasing Hormone |
| cg07533148 | TRIM58 | NM_015431.2 | Tripartite Motif
Containing 58 |
| cg07307078 | TUBB6 | NM_032525.1 | Tubulin β 6 |
| cg20616414 | WNK2 | NM_006648.3 | WNK Lysine
Deficient Protein Kinase 2 |
| cg16638540 | ZNF135 | NM_003436.2 | Zinc Finger Protein
135 |
| cg03975694 | ZNF540 | NM_152606.2 | Zinc Finger Protein
540 |
| cg16731240 | ZNF577 | NM_032679.1 | Zinc Finger Protein
577 |
TRIM58 serves as a potential
diagnostic biomarker for lung cancer
ROC analysis was used to evaluate the diagnostic
value of TRIM58 in lung cancer. The area under the curve (AUC)
values of the tumor and NTL groups in the TRIM58 analyses were
significant for all 3 lung cancer datasets and were as follows:
AUCGSE63384=0.950 [P <0.001; 95% confidence interval
(CI), 0.903–0.998]; AUCGSE62948=0.964 (P <0.001; 95%
CI, 0.913–1.015) and AUCGSE32861=0.945 (P<0.001; 95%
CI, 0.900–0.989) (Fig. 1C). The
aforementioned results demonstrated that the methylation level of
TRIM58 can distinguish tumor tissues from normal tissues and that
TRIM58 methylation is a potential marker for the early diagnosis of
lung cancer.
TRIM58 is coordinately hypermethylated
and downregulated in lung cancer
The present study reviewed a large amount of
literature on these 55 hypermethylated probes. Among them, TRIM58
is a member of the TRIM family, which is located on chromosome 1
and on CpG islands (36). Previous
studies have shown that TRIM protein may serve an important role in
tumorigenesis; however, the mechanism by which TRIM58 participates
in the regulation of lung cancer remains unclear (17,37).
Firstly, the results of high-throughput screening
were validated using the TCGA datasets. A total of 372 LUSC samples
with 43 NTL samples and 460 LUAD samples with 32 NTL samples were
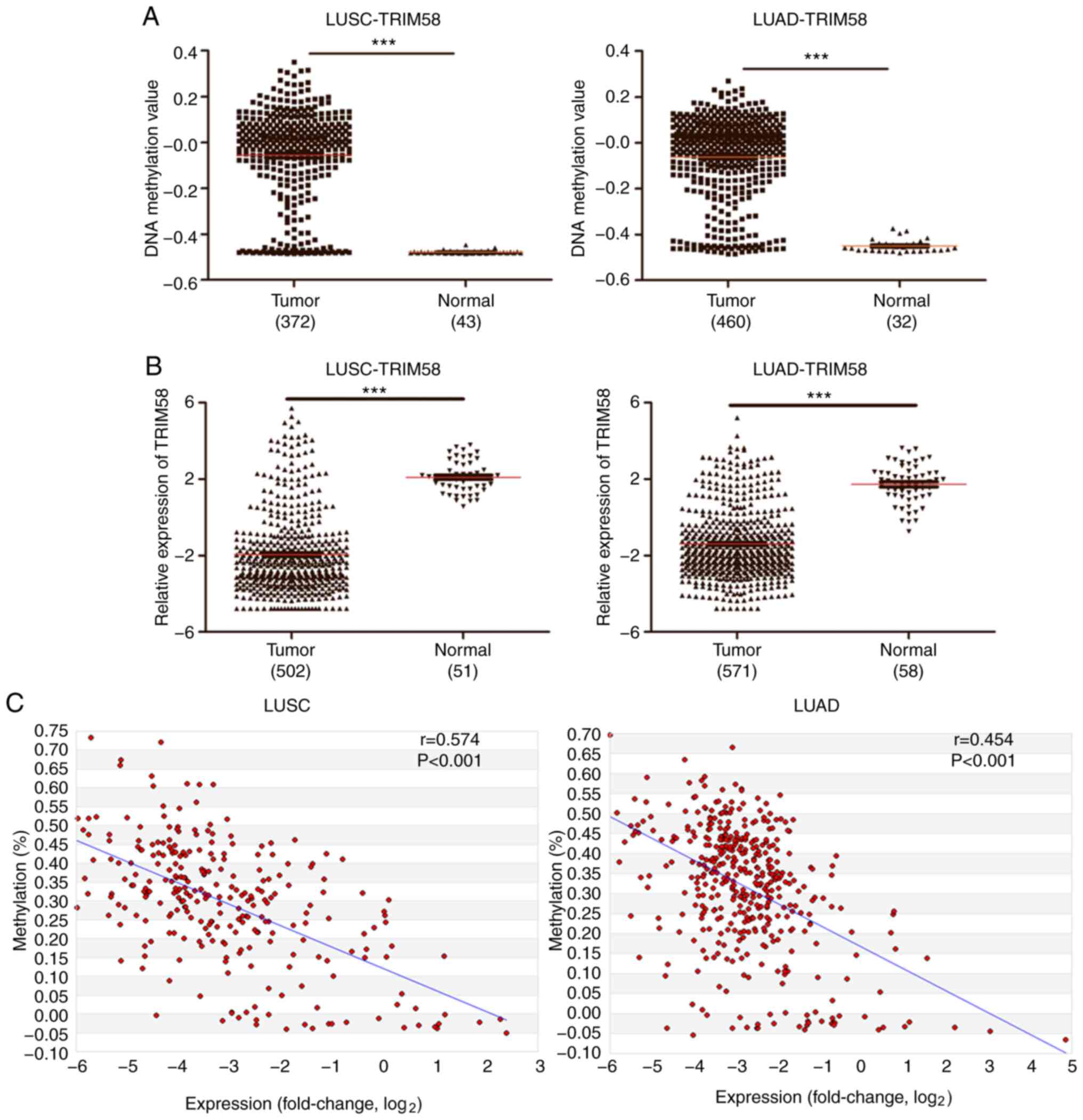
used for independent verification. In both, LUSC and LUAD, TRIM58
was hypermethylated in tumor tissues compared to normal tissues
(Fig. 2A). In contrast, TRIM58 was
downregulated in both LUSC and LUAD compared to normal tissue
(Fig. 2B). TRIM58 was coordinately
hypermethylated and downregulated in lung tumors compared to normal
tissue, indicating that it may be a potential tumor suppressor
gene. Subsequently, the correlation between DNA methylation and
mRNA expression was analyzed in the MethHC database. Scatter plot
analysis revealed that the DNA methylation and mRNA expression
levels of TRIM58 were negatively correlated and the Spearman
correlation coefficient values were rLUSC =0.574
and rLUAD =0.454 (both P<0.001; Fig. 2C). These results suggested that
TRIM58 expression may be regulated by epigenetics, DNA methylation
in particular.
TRIM58 inhibits the malignant
phenotypes of lung cancer cells
To evaluate the molecular functions of TRIM58 in
lung cancer, loss-of-function and gain-of-function assays were
conducted with the A549 cell line. The effects of silencing and
overexpressing TRIM58 on the malignant phenotype were detected.
Firstly, the expression of TRIM58 in A549 cells was silenced using
siRNA. siRNA-TRIM58 was constructed using scramble siRNA as a
negative control (Fig. 3A). The
results demonstrated that compared with the control group, the
siRNA-TRIM58 group exhibited significantly downregulated expression
of TRIM58 (Fig. 3A). The MTS assay
indicated that the silencing of TRIM58 promoted the proliferation
of lung cancer cells (Fig. 3C). An
overexpression vector TRIM58-pcDNA3.1 was constructed in the
present study and an empty pcDNA3.1 vector was used as a negative
control. The expression level of pcDNA3.1-TRIM58 was significantly
increased compared with that of the vector (control group)
(Fig. 3B). TRIM58 overexpression
inhibited cell proliferation (Fig.
3D). Subsequently, wound-healing and transwell assays were used
to evaluate cell migration. Compared with the control group
(scramble siRNA), TRIM58 silencing potently accelerated the
migration of lung cancer cells (Fig. 3E,
G and I), whereas overexpression of TRIM58 exerted the opposite
effect (Fig. 3F, H and I). In
addition, flow cytometry analysis demonstrated that TRIM58
overexpression promoted the apoptosis of lung cancer cells compared
with empty pcDNA3.1 vector (Fig. 3J and
K). In summary, the overexpression of TRIM58, a potential tumor
suppressor gene inhibited cell proliferation and migration and
promoted cell apoptosis.
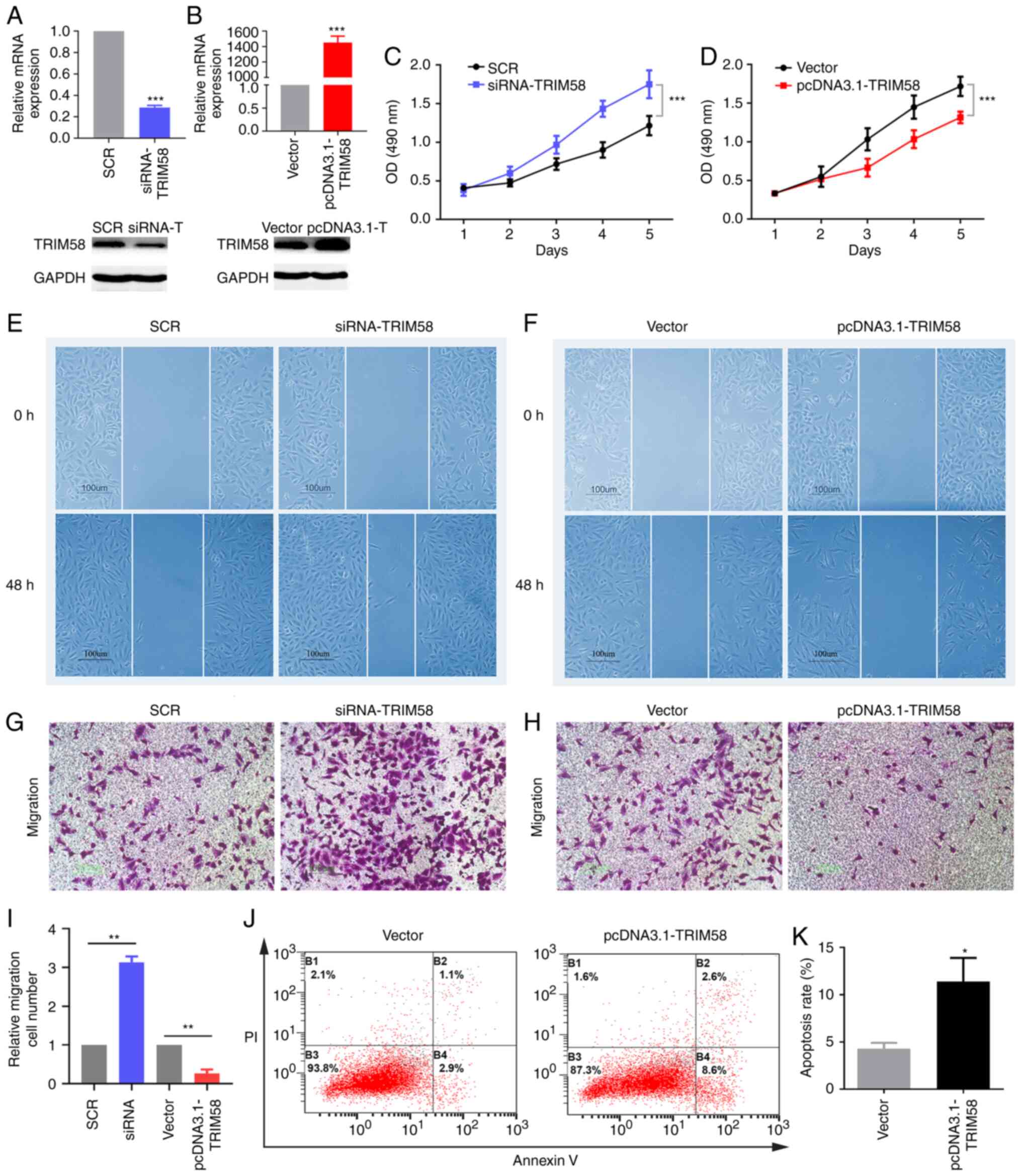 | Figure 3.TRIM58 inhibits the malignant
phenotypes of lung cancer cells. To evaluate the molecular
functions of TRIM58 in lung cancer, loss-of-function and
gain-of-function assays were conducted in A549 cell line. A series
of transfection experiments were performed. (A and B) siRNA and
plasmids effectively regulated the expression of TRIM58 in the A549
cell line. mRNA and protein expression of TRIM58 were detected by
RT-qPCR and western blotting, respectively. (C and D) MTS assay was
used to assess cell proliferation. Cell migration was assessed
using the (E and F) wound healing assay and (G, H and I) transwell
assay. (J and K) Flow cytometry was used to verify cell apoptosis.
*P<0.05, **P<0.01 and ***P<0.001. SCR, scrambled negative
control; si, small interfering; vector, pcDNA3.1; PI, propidium
iodide; TRIM58, tripartite motif containing 58; OD, optical
density. All these experiments are compared between the
experimental group and the control group. |
Identification of TRIM58-associated
signaling pathways in lung cancer
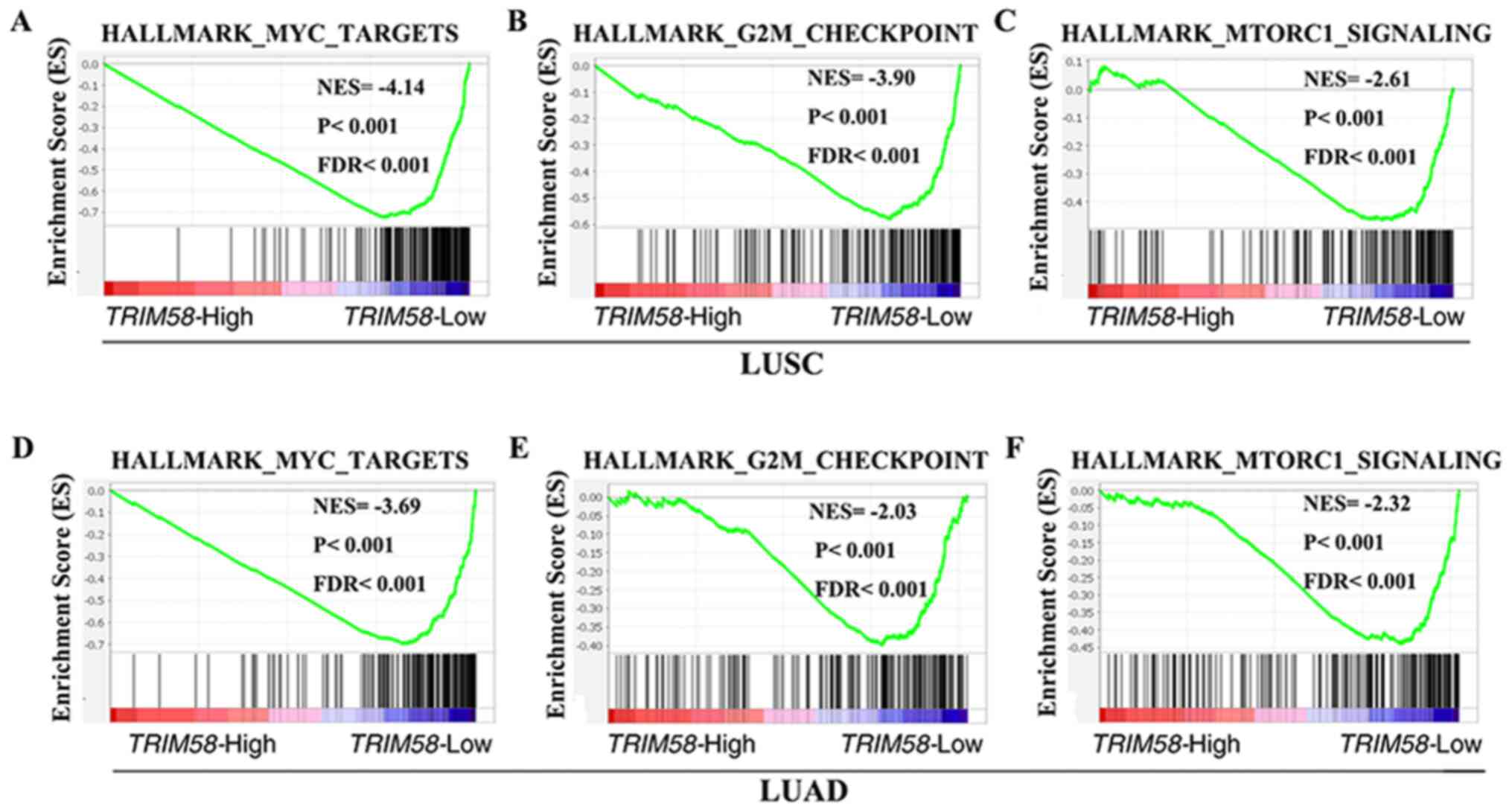
To explore the molecular mechanisms by which TRIM58
contributes to lung cancer progression, GSEA was performed using
mRNA expression data from the TCGA database. Several classic
mechanisms of carcinogenesis, such as MYC targets [P<0.001;
false discovery rate (FDR)<0.001; NESLUSC =−4.14;
P<0.001; FDR<0.001; NESLUAD=−3.69] (Fig. 4A and D) and G2M checkpoint-related
genes (P<0.001; FDR<0.001; NESLUSC=−3.90;
P<0.001; FDR<0.001; NESLUAD=−2.03) (Fig. 4B and E), were enriched in samples
with low TRIM58 expression. It was also observed that TRIM58 was
negatively correlated with the mTORC1 signaling pathway
(P<0.001; FDR<0.001; NESLUSC=−2.61; P<0.001;
FDR<0.001; NESLUAD=−2.32) (Fig. 4C and F), further indicating a tumor
suppressor role of TRIM58.
Protein interaction and co-expression
analysis
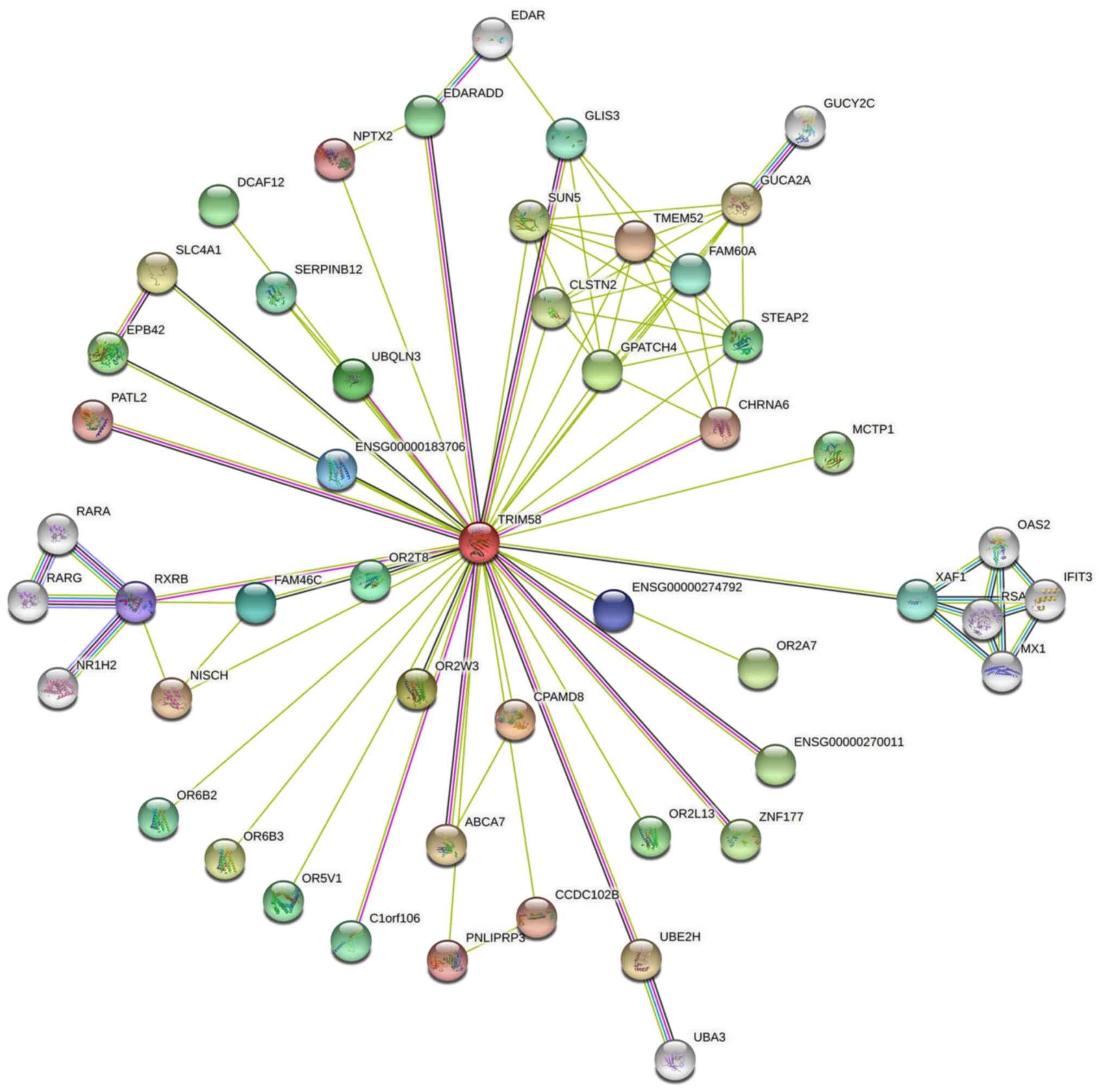
Protein-Protein Interaction Network analysis was
constructed using the STRING database (Fig. 5). A series of proteins interacting
with TRIM58 were identified, such as CPAMD8 (C3 and PZP-like
alpha-2-macroglobulin domain-containing protein 8), OR2W3
(Olfactory Receptor Family 2 Subfamily W Member 3), GPATCH4 (G
Patch Domain-Containing Protein 4), UBQLN3 (Ubiquilin 3), OR2T8
(Olfactory Receptor Family 2 Subfamily T Member 8), FAM46C (Family
with sequence similarity 46 member C) and RXRB (Retinoid X Receptor
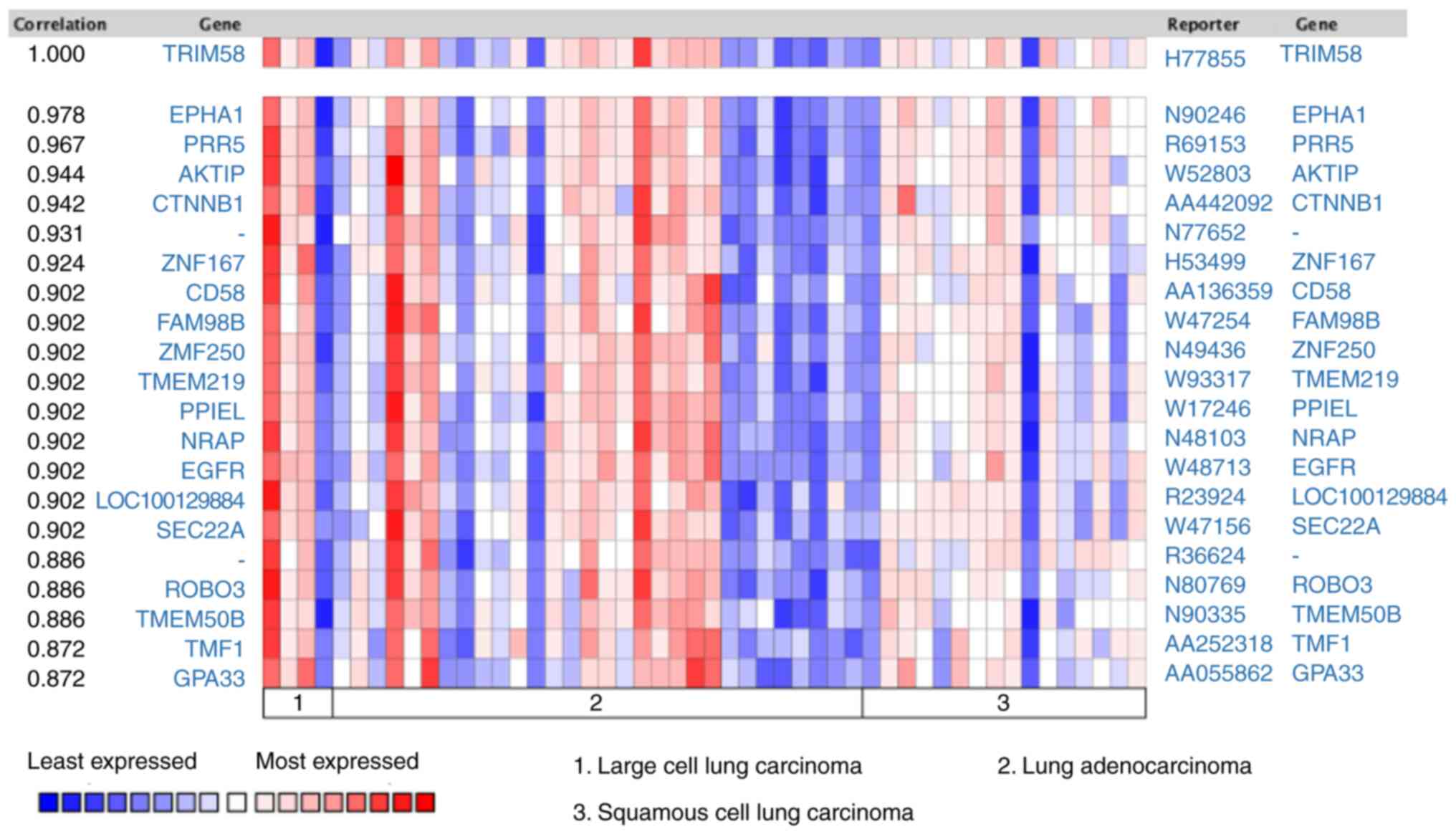
Beta). Subsequently, co-expression analysis was performed on lung
cancer samples from the Oncomine database. Tomida et al
(38) revealed that EPHA1 (EPH
Receptor A1) (r=0.978), PRR5 (Proline-Rich Protein 5) (r=0.967),
AKTIP (AKT Interacting Protein) (r=0.944), CTNNB1 (Catenin β-1)
(r=0.942), ZNF167 (Zinc Finger Protein 167) (r=0.924), CD58 (CD58
Antigen) (r=0.902), FAM98B (Family With Sequence Similarity 98
Member B) (r=0.902), ZNF250 (Zinc Finger Protein 250) (r=0.902),
TMEM219 (Transmembrane Protein 219) (r=0.902) were co-expressed
with TRIM58 (Fig. 6). These results
suggested that TRIM58 closely interacts with numerous functional
genes involved in lung cancer.
Discussion
Previous studies have shown that epigenetic changes
can be used as biomarkers for the detection of malignant tumors,
such as hypermethylation of GSTP1 (Glutathione S-Transferase Pi 1)
in prostate cancer (8,39). Abnormal hypermethylation in the
promoter region of tumor suppressor gene RASSF1A (RAS associated
domain family 1 A), repair gene MGMT, apoptosis-related genes EBF3
(Early B Cell Factor 3), cell cycle-related gene CDKN2B
(Cyclin-dependent kinase inhibitor 2B) and other important genes
often occurs in precancerous lesions or during the early
carcinogenesis of tumors, inhibiting transcriptional activity and
leading to tumor occurrence (40–42).
Additionally, abnormal genome-wide hypomethylation resulting in
genomic instability and oncogene activation can induce tumors
(43).
DNA methylation is an early event occurring in
tumors, providing a stable signal with high sensitivity and
specificity (44). In the present
study, high-throughput screening and independent validation
demonstrated that TRIM58 was hypermethylated and downregulated in
lung cancer compared to normal tissues. ROC analysis performed in
the present study revealed that TRIM58 had a strong predictive
value for the early diagnosis of lung cancer.
TRIM family proteins serve an important role in
tumorigenesis (17). There are few
reports of the molecular mechanism of TRIM58's regulatory role in
lung cancer. The morbidity and mortality of lung cancer is very
high and tumor metastasis is an important cause of treatment
failure and death (5). In
vitro and in vivo experiments (45) demonstrated that TRIM62 inhibits the
metastasis of cervical cancer by inhibiting the c-Jun/Slug
signaling pathway. Chen et al (46) demonstrated that TRIM62 as an
oncogene, negatively regulates TGF-β-mediated epithelial
mesenchymal transition, hence inhibiting tumor invasion and
metastasis. As a member of the same subfamily of TRIM62 (C-IV-1),
it was hypothesized that TRIM58 may regulate the malignant
phenotype of cancer. To further evaluate the molecular functions of
TRIM58 in lung cancer, loss-of-function and gain-of-function assays
were conducted in the present study in lung cancer cells (A549
cells). The results indicated that TRIM58 was a novel tumor
suppressor gene in lung cancer.
In addition, the present study attempted to explore
the signaling pathways related to TRIM58 in lung cancer to
understand the potential mechanisms by which TRM58 participates in
the regulation of tumor progression. Gene set enrichment analysis
revealed that TRIM58 expression was negatively correlated with MYC
targets, G2M checkpoints and the mTORC1 (mechanistic target of
rapamycin complex 1) signaling pathway. mTOR is a protein kinase
that can regulate a large number of cellular processes, such as
cell growth, proliferation and differentiation through the
PI3K/AKT/mTOR pathway (47). Among
mTOR proteins, mTORC1 is frequently activated in human cancers and
targeting mTORC1 signaling is a promising strategy for tumor
therapy (48). In addition, MYC and
G2M checkpoints are classic tumor-promoting signaling pathways
(49). Hence, the results of the
present study indicate that TRIM58 may serve an anticancer role by
inhibiting the signaling pathways of mTORC1.
In conclusion, through DNA methylation-based
profiling screening, the present study demonstrated that TRIM58
methylation has promise as a biomarker for the early diagnosis of
lung cancer. Cell experiments confirmed the role of TRIM58 as a
tumor suppressor gene in lung cancer and overexpression of TRIM58
inhibited the malignant phenotype of tumors. Gene set enrichment
analysis revealed that TRIM58 expression was negatively correlated
with the mTORC1 signaling pathway. Future studies are needed to
further explore the specific regulatory mechanisms to provide new
targets for the early diagnosis and effective treatment of lung
cancer.
Acknowledgements
The author would like to thank Professor Sheng
Deqiao (China Three Gorges University, Yichang, China) for guidance
and help with the experiments.
Funding
This work was supported by the Scientific Research
Project of Hunan Provincial Health Commission (grant no.
202103021291) and Natural Science Foundation of Hubei Province of
China (grant no. 2018CFB142).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author contributions
YXS performed the experiments, analyzed the data and
prepared the manuscript. YXS confirmed the authenticity of all the
raw data. The author has read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that he has no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cao M and Chen W: Epidemiology of lung
cancer in China. Thorac Cancer. 10:3–7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McIntyre A and Ganti AK: Lung cancer-A
global perspective. J Surg Oncol. 115:550–554. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liloglou T, Bediaga NG, Brown BR, Field JK
and Davies MP: Epigenetic biomarkers in lung cancer. Cancer Lett.
342:200–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wood SL, Pernemalm M, Crosbie PA and
Whetton AD: The role of the tumor-microenvironment in lung
cancer-metastasis and its relationship to potential therapeutic
targets. Cancer Treat Rev. 40:558–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Visconti R, Morra F, Guggino G and Celetti
A: The between now and then of lung cancer chemotherapy and
immunotherapy. Int J Mol Sci. 18:E13742017. View Article : Google Scholar
|
|
7
|
Shi YX, Yin JY, Shen Y, Zhang W, Zhou HH
and Liu ZQ: Genome-scale analysis identifies NEK2, DLGAP5 and ECT2
as promising diagnostic and prognostic biomarkers in human lung
cancer. Sci Rep. 7:80722017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Michalak EM, Burr ML, Bannister AJ and
Dawson MA: The roles of DNA, RNA and histone methylation in ageing
and cancer. Nat Rev Mol Cell Biol. 20:573–589. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seijo LM, Peled N, Ajona D, Boeri M, Field
JK, Sozzi G, Pio R, Zulueta JJ, Spira A, Massion PP, et al:
Biomarkers in lung cancer screening: achievements, promises, and
challenges. J Thorac Oncol. 14:343–357. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choo KB: Epigenetics in disease and
cancer. Malays J Pathol. 33:61–70. 2011.PubMed/NCBI
|
|
13
|
Schübeler D: Function and information
content of DNA methylation. Nature. 517:321–326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shinjo K and Kondo Y: Targeting cancer
epigenetics: Linking basic biology to clinical medicine. Adv Drug
Deliv Rev. 95:56–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jones PA, Issa JPJ and Baylin S: Targeting
the cancer epigenome for therapy. Nat Rev Genet. 17:630–641. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schiffmann I, Greve G, Jung M and Lübbert
M: Epigenetic therapy approaches in non-small cell lung cancer:
Update and perspectives. Epigenetics. 11:858–870. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hatakeyama S: TRIM family proteins: Roles
in autophagy, immunity, and carcinogenesis. Trends Biochem Sci.
42:297–311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hatakeyama S: TRIM proteins and cancer.
Nat Rev Cancer. 11:792–804. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Napolitano LM and Meroni G: TRIM family:
Pleiotropy and diversification through homomultimer and
heteromultimer formation. IUBMB Life. 64:64–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang S, Zhang Y, Huang J, Wong CC, Zhai J,
Li C, Wei G, Zhao L, Wang G, Wei H, et al: TRIM67 activates p53 to
suppress colorectal cancer initiation and progression. Cancer Res.
79:4086–4098. 2019.PubMed/NCBI
|
|
21
|
Cambiaghi V, Giuliani V, Lombardi S,
Marinelli C, Toffalorio F and Pelicci PG: TRIM proteins in cancer.
Adv Exp Med Biol. 770:77–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meroni G and Diez-Roux G: TRIM/RBCC, a
novel class of ‘single protein RING finger’ E3 ubiquitin ligases.
BioEssays. 27:1147–1157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reymond A, Meroni G, Fantozzi A, Merla G,
Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, et
al: The tripartite motif family identifies cell compartments. EMBO
J. 20:2140–2151. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Short KM and Cox TC: Subclassification of
the RBCC/TRIM superfamily reveals a novel motif necessary for
microtubule binding. J Biol Chem. 281:8970–8980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi YX, Wang Y, Li X, Zhang W, Zhou HH,
Yin JY and Liu ZQ: Genome-wide DNA methylation profiling reveals
novel epigenetic signatures in squamous cell lung cancer. BMC
Genomics. 18:9012017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robles AI, Arai E, Mathé EA, Okayama H,
Schetter AJ, Brown D, Petersen D, Bowman ED, Noro R, Welsh JA, et
al: An integrated prognostic classifier for stage I lung
adenocarcinoma based on mRNA, microRNA, and DNA methylation
biomarkers. J Thorac Oncol. 10:1037–1048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mansfield AS, Wang L, Cunningham JM, Jen
J, Kolbert CP, Sun Z and Yang P: DNA methylation and RNA expression
profiles in lung adenocarcinomas of never-smokers. Cancer Genet.
208:253–260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Selamat SA, Chung BS, Girard L, Zhang W,
Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al:
Genome-scale analysis of DNA methylation in lung adenocarcinoma and
integration with mRNA expression. Genome Res. 22:1197–1211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smyth GK: limma: Linear models for
microarray data. Bioinformatics and Computational Biology Solutions
Using R and Bioconductor. pp. 397–420. Springer; New York, NY:
2005, View Article : Google Scholar
|
|
32
|
Ghosh D: Incorporating the empirical null
hypothesis into the Benjamini-Hochberg procedure. Stat Appl Genet
Mol Biol. 11:/j/sagmb.2012.11.issue-4/1544-6115.1735/1544-6115.1735.xml2012.doi:
10.1515/1544-6115.1735. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goldman M, Craft B, Swatloski T, Cline M,
Morozova O, Diekhans M, Haussler D and Zhu J: The UCSC Cancer
Genomics Browser: Update 2015. Nucleic Acids Res. 43:D812–D817.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang WY, Hsu SD, Huang HY, Sun YM, Chou
CH, Weng SL and Huang HD: MethHC: A database of DNA methylation and
gene expression in human cancer. Nucleic Acids Res. 43:D856–D861.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe M and Hatakeyama S: TRIM proteins
and diseases. J Biochem. 161:135–144. 2017.PubMed/NCBI
|
|
37
|
Zhan W and Zhang S: TRIM proteins in lung
cancer: Mechanisms, biomarkers and therapeutic targets. Life Sci.
268:1189852021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tomida S, Koshikawa K, Yatabe Y, Harano T,
Ogura N, Mitsudomi T, Some M, Yanagisawa K, Takahashi T, Osada H,
et al: Gene expression-based, individualized outcome prediction for
surgically treated lung cancer patients. Oncogene. 23:5360–5370.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mari-Alexandre J, Diaz-Lagares A, Villalba
M, Juan O, Crujeiras AB, Calvo A and Sandoval J: Translating cancer
epigenomics into the clinic: Focus on lung cancer. Transl Res.
189:76–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Clark SJ and Melki J: DNA methylation and
gene silencing in cancer: Which is the guilty party? Oncogene.
21:5380–5387. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
You JS and Jones PA: Cancer genetics and
epigenetics: Two sides of the same coin? Cancer Cell. 22:9–20.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Diaz-Lagares A, Mendez-Gonzalez J, Hervas
D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga
LM, Zulueta J, et al: A novel epigenetic signature for early
diagnosis in lung cancer. Clin Cancer Res. 22:3361–3371. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu TY, Chen J, Shang CL, Shen HW, Huang
JM, Liang YC, Wang W, Zhao YH, Liu D, Shu M, et al: Tripartite
motif containing 62 is a novel prognostic marker and suppresses
tumor metastasis via c-Jun/Slug signaling-mediated
epithelial-mesenchymal transition in cervical cancer. J Exp Clin
Cancer Res. 35:1702016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen N, Balasenthil S, Reuther J and
Killary AM: DEAR1, a novel tumor suppressor that regulates cell
polarity and epithelial plasticity. Cancer Res. 74:5683–5689. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tamaddoni A, Mohammadi E, Sedaghat F,
Qujeq D and As'Habi A: The anticancer effects of curcumin via
targeting the mammalian target of rapamycin complex 1 (mTORC1)
signaling pathway. Pharmacol Res. 156:1047982020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lane HA and Breuleux M: Optimal targeting
of the mTORC1 kinase in human cancer. Curr Opin Cell Biol.
21:219–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tansey WP: Mammalian MYC proteins and
cancer. New J Sci. 2014.1–27. 2014.https://doi.org/10.1155/2014/757534 View Article : Google Scholar
|