Introduction
Follicular lymphoma (FL) represents 20% of all non-Hodgkin lymphoma (NHL) cases, with over 8,000 new cases diagnosed in the USA each year (1). Treatment for FL includes successive lines of therapy that unfortunately result in progressively shorter periods of disease-free survival followed by the development of either chemo-refractory disease, large cell transformation or death from treatment-related toxicities (2). CD20 is a B-cell-specific antigen expressed on malignant FL cells. As such, targeting CD20 is a viable therapeutic strategy and plays a central role in the treatment of FL. This strategy includes the monoclonal anti-CD20 antibody, rituximab (RTX), alone or in combination with chemotherapy for patients who have advanced-stage disease. The mechanisms of action by which RTX kills FL cells include complement-dependent cytotoxicity (CDC) (3), antibody-dependent cellular cytotoxicity (ADCC) (4,5)-herein referred to as ‘effector function’ mechanisms-as well as CD20 signaling effects (6). The pathobiology of FL is complex and presents changes at the genome and the epigenome levels. These changes are thought to lead to tumor-microenvironment interactions via factors secreted by stromal or epithelial cells in response to lymphoma cell-produced cytokines (7). Previous studies have established CA125 as a tumor-microenvironment factor that can be detected and found elevated (above normal 35 U/ml levels) in the serum of up to 40% of FL patients and other NHL patients. High serum level of CA125 is used as an indicator of tumor abdominal involvement, advanced staging, relapse rates and survival in NHL patients (8–12). Up to 37% of FL patients have elevated CA125 serum levels (11); however, despite its prevalence in FL and other cancers, the role of CA125 in tumor biology and immunity has been only recently elucidated. Previous clinical evidence suggested that high levels of serum CA125 have a negative impact on the effectiveness of the experimental therapeutic antibodies farletuzumab and amatuximab (13–15). Similar to RTX, the modes of action of farletuzumab and amatuximab include ADCC and CDC (16–18). In addition, CA125 inhibits CDC by blocking C1q from binding to farletuzumab (19). Key evidence showing that ADCC plays an important role in the clinical effect of RTX was provided by the analysis of Fc gamma RIIIa (CD16a) polymorphisms, where patients carrying CD16a 158V/V genotype (associated with high affinity Fc gamma receptor) showed significantly improved objective response rates compared with FL patients carrying at least one 158F allele (associated with low affinity Fc gamma receptor) (20). The role of CD16a gene locus polymorphisms on ADCC has been further elucidated by an independent study (21). Considering that ADCC and CDC play a major role in RTX mode of action and that CA125 mediates the suppression of ADCC and CDC elicited by some investigational antibodies, it is compelling to note that high CA125 serum levels are correlated with reduced responses to RTX treatment in FL patients (22). In a study including 82 FL patients, RTX-containing regimen showed a superior 5-year progression-free survival in FL patients having CA125 level below 35 U/ml [85.9%; 95% confidence interval (CI) 0.75–0.97] compared with patients with CA125 above 35 U/ml (54.5%; 95% CI 0.36–0.73; P=0.004) (22). This result suggested that similarly to farletuzumab and amatuximab, RTX clinical activity may also be limited by CA125 immunosuppressive effects.
In a previous study, we demonstrated that CA125 binds to RTX, reducing its C1q binding and consequently its CDC activity (19). The present study built upon that initial observation and performed additional cellular and molecular assays to study RTX and CA125 interaction, as well as generating a library of RTX variants using a proprietary technology called Block-Removed Immunoglobulin Technology (BRITE) that combines randomized amino acid substitutions and high-throughput functional screenings to identify CA125-refractory RTX variants.
Materials and methods
Cell lines
Daudi and Ramos (used as target cells), and Jurkat cell lines were purchased from the American Type Culture Collection (ATCC). Cells were cultured in complete RPMI (cRPMI) medium consisting of RPMI (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), placed at 37°C in a humidified incubator containing 5% CO2 and passaged twice weekly. Jurkat-Luc effector cells were purchased from Promega Corporation, cultured in cRPMI and placed at 37°C in a humidified incubator containing 5% CO2. CA125-positive OVCAR3 cells (13) (Fig. 3E) were purchased from ATCC and grown in cRPMI. To generate CA125- and CD20-positive target cells, OVCAR3 cells were grown in cRPMI and transfected using Lipofectamine (Thermo Fisher Scientific, Inc.) with 10 µg CD20 cDNA expression plasmid (pUNO1-hCD20a; InvivoGen). Transfected cells were selected using 30 µg/ml blasticidin.
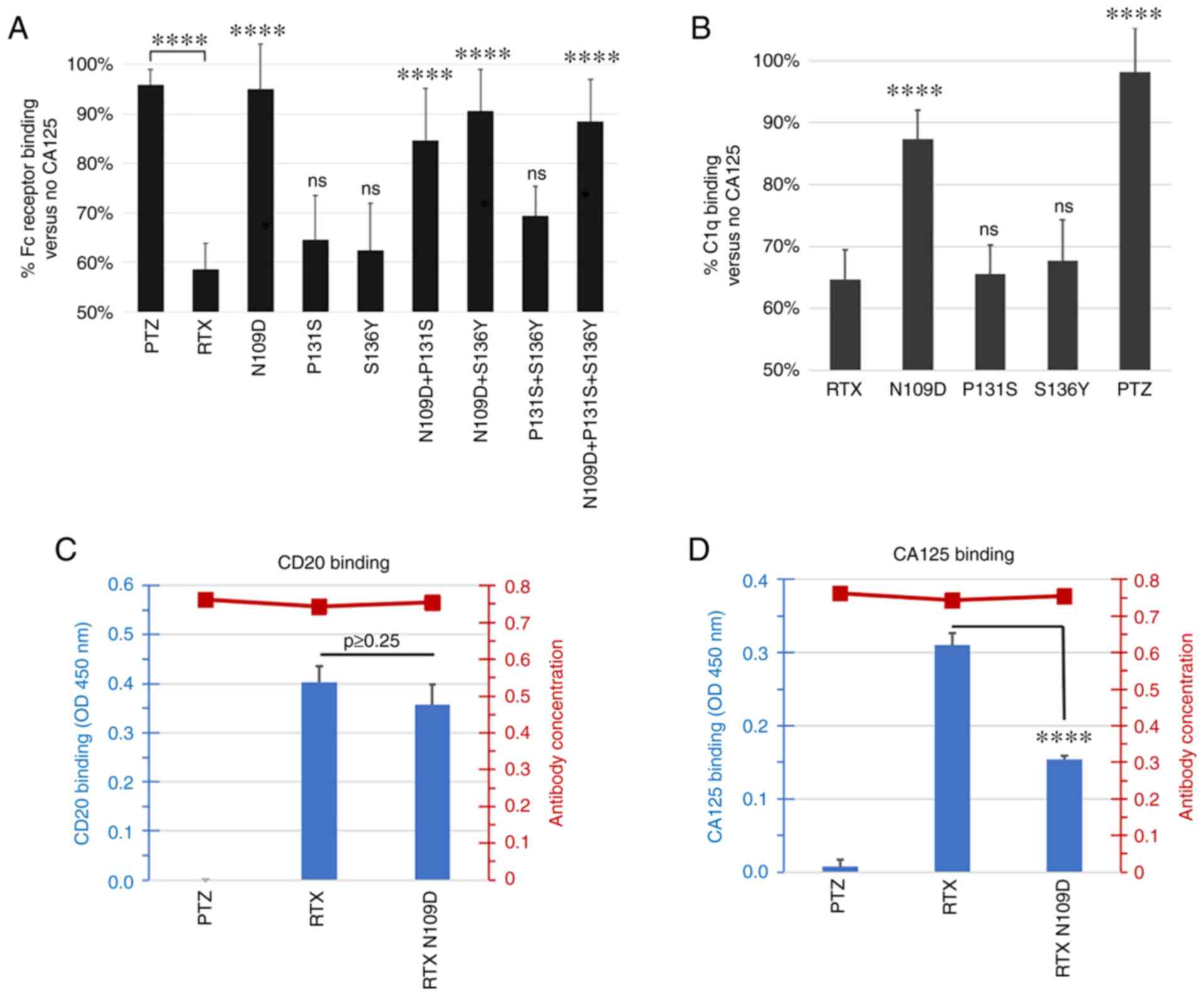 |
Figure 3.
Characterization of RTX-166 variant mutations. (A) Single or double mutations N109D, P131S, S136Y, N109D + P131S, N109D + S136Y and P131S + S136Y, as well as RTX-166 (N109D + P131S + S136Y) were compared for Fc Receptor binding in the presence of CA125. Data were plotted as a percentage of binding with CA125 vs. no CA125. Any variant carrying at least the N109D mutation appeared to have higher binding to Fc receptor compared with parent RTX in the presence of CA125. The summary of P-value following ANOVA and Tukey's post hoc test comparing RTX variants vs. RTX is shown over the individual RTX variants bars. RTX was also compared to PTZ. (B) Single mutations N109D, P131S and S136Y were compared for C1q binding in the presence of CA125. Data are plotted as percentage of binding with CA125 vs. no CA125. This analysis revealed that RTX N109D variant was the least affected by the presence of CA125 and retained ~90% of C1q binding compared with <65% for parent RTX. The summary of P-value following ANOVA and Tukey's post hoc test comparing PTZ and RTX variants vs. RTX is shown over the individual bars. (C) Antibody binding to CD20 peptide was measured using ELISA assay. RTX N109D CD20 binding was comparable to RTX and the difference was not statistically significant (ANOVA and Tukey's post hoc test; P>0.25). (D) Antibody binding to CA125 was evaluated using an ELISA assay. Binding of RTX N109D to CA125 was found to be reduced by 50% compared with RTX and the difference was statistically significant (P<0.00009). The secondary Y axis on the right side of each graph in (C) and (D) panel shows the amount of detected coated biotinylated antibody used in the assay. In all experiments, PTZ was used as control as a CA125-refractory control antibody. ****P<0.0001. CA125, cancer antigen 125; ns, non-significant; PTZ, pertuzumab; RTX, rituximab.
|
CD16a/Fc receptor activation assay
CD16a activation assays were conducted using Jurkat-Luc effector cells, which are part of the ADCC Reporter Bioassay Core Kit, according to the manufacturer's instructions (Promega Corporation). Briefly, 2×104 target cells were seeded overnight in black opaque 96-well plates in triplicate in assay buffer (RPMI + L-glutamine + 1% ultra-low Ig serum; Gibco; Thermo Fisher Scientific, Inc.). The following day, 1×105 Jurkat-Luc effector cells were seeded into each well in assay buffer for an effector:target ratio of 5:1. Rituximab, rituximab variants and pertuzumab antibody as well as effector and target cells were incubated for 16 h at 37°C in a humidified incubator containing 5% CO2. Plates were equilibrated at room temperature for 10 min. Bio-Glo reagent (50 µl; Promega Corporation) was eventually added to each well. Luminescence was read using a Varioskan LUX plate reader at 560 nm emission and using SkanIt™ version 4.1 (Thermo Fisher Scientific, Inc.).
Antibody-dependent cellular cytotoxicity
IL-2-stimulated (10 ng/ml) human peripheral blood mononuclear cells (PBMCs) isolated from healthy donors (PPA Research; catalog. no. 15-00006) were cultured in cRPMI and were placed at 37°C in a humidified incubator containing 5% CO2. The following day, Daudi target cells (2×104) were seeded in clear U-bottom 96-well plates in triplicate in assay buffer [PFAB; phenol-free RPMI (Quality Biological; catalog no. 112-040-101) containing 2 mM L-glutamine and 1% ultra-low Ig serum]. PBMCs were washed in PFAB and added to target wells at effector:target ratios as indicated in figure legends. Rituximab, rituximab variants and pertuzumab antibodies, and CA125 (25 KU/ml) were added and incubated at 37°C in 5% CO2 for 6 h. Subsequently, 10 µl of 10% Triton X-100 was added for 2 min to a subset of wells containing only target and effector cells to mimic complete cell lysis allowing the calculation of maximum killing (see formula below). Plates were centrifuged at 500 × g for 2 min at room temperature. Supernatants (50 µl) were transferred to clear flat bottom 96-well plates. Lactate dehydrogenase reagent (100 µl; Promega Corporation) was added to the wells and plates were incubated 15 min in the dark at room temperature. Absorbance at 492 nm was obtained using a Varioskan LUX plate reader and SkanIt™ version 4.1 (Thermo Fisher Scientific, Inc.) and % cytotoxicity was calculated as follows: % cytotoxicity=[experimental well-(Effector + Target cells only well)]/[Triton X-100 well-(Effector + Target cells only well)] × 100%.
CA125 binding competition
ELISA plate (Santa Cruz Biotechnology, Inc.) wells were coated with 1,000 units of CA125 (Lee Bio) in 0.05 M carbonate buffer overnight at room temperature. Wells were washed with PBS and blocked with 5% bovine serum albumin solution (BSA; MilliporeSigma) for 1 h at room temperature. RTX was biotinylated using EZ-link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, Inc.) and added at 2.5 µg/ml with or without recombinant complement factor C3B (Sigma-Aldrich; Merck KGaA) or Protein L (Pierce; Thermo Fisher Scientific, Inc.) at 5 µg/ml in PBS buffer containing 5% BSA and incubated for 1 h at room temperature. After washing at room temperature using PBS, bound RTX was detected using 0.3 µg/ml of HRP-conjugated streptavidin (Jakson ImmunoResearch; catalog no. 016-030-084) for 1 h at room temperature. After washing using PBS, reactions were developed for up to 10 min by adding 75 µl TMB substrate (Pierce; Thermo Fisher Scientific, Inc.) and stopped by adding 75 µl 0.1 N H2SO4. Absorbance was read at 450 nm on a Varioskan plate reader and using SkanIt™ version 4.1 (Thermo Fisher Scientific, Inc.).
Competition of CD20 binding in the presence of CA125
ELISA plates were coated overnight at 4°C with diluted antigen in 0.05 M carbonate buffer at pH 9.5. Coated antigen was CD20/MS4A1 linear peptide (amino acid 142–184 of CD20 extracellular domain; Alpha Diagnostic; catalog no. cd20-rp1l-p) at 10 µg/ml. Plates were blocked with 0.05 M PBS at pH 7.2 containing 5% BSA for 1 h at room temperature then washed twice with PBS. Biotinylated RTX (by EZ-link Sulfo-NHS-LC-Biotin; Thermo Fisher Scientific, Inc.) or negative control human serum albumin (HSA; Sigma-Aldrich; Merck KGaA) were added to the plate in PBS at 5 µg/ml for 1 h at room temperature. Competition was carried out by adding 10 µg/ml unlabeled RTX (2-fold excess) or 30 KU/ml of CA125. Wells were washed three times with PBS followed by addition of streptavidin-HRP (Jakson ImmunoResearch; catalog no. 016-030-084) in PBS/BSA for 1 h at room temperature. After washing, reactions were developed for up to 10 min by adding 75 µl of TMB substrate (Pierce; Thermo Fisher Scientific, Inc.) and stopped by adding 75 µl 0.1 N H2SO4. Absorbance was read at 450 nm on a Varioskan plate reader and using SkanIt™ version 4.1 (Thermo Fisher Scientific, Inc.).
Generation and production of RTX variants
Parent RTX sequences were obtained from Biogen's patent EP2000149B1. Heavy chain (HC) and light chain (LC) cDNAs were cloned into expression vectors. A library of RTX HC mutants was engineered by using an error-prone PCR-based random mutagenesis technique. A pair of primers was designed that framed a region that started with residue ala95, located on the N-terminus of RTX HC complementarity determining region 3 (CDR3), and ended with pro235, located within the hinge sequence (CysProProCys). RTX HC cDNA sequence was cloned downstream of a CMV promoter in a DNA plasmid named CMV-RTX-HC. This vector, along with RTX LC cDNA cloned downstream of a CMV promoter in a plasmid named CMV-RTX-LC, were used to produce the parent version of RTX antibody. The CMV-RTX-HC plasmid was then used as the template for the PCR-based random mutagenesis method. The primers used were as follows: RTX-mut forward, 5′-CGCCGTGTATTACTGTGCT-3′ and reverse, 5′-CACGGTGGGCATGTGTGAGTTT-3′.
PCR reactions were set up using Mutazyme II kit (Agilent) according to the manufacturer's instructions. The PCR product was then loaded on 1.5% agarose gel to separate residual primers, dNTP and buffer by electrophoresis. A gel fragment containing only the amplified 423 bp DNA segment was excised and further purified by gel extraction using EZ-10 Spin Column (Bio Basic). The purified DNA was used as megaprimer for the subsequent PCR reaction using EZClone enzyme mix (Agilent) according to the manufacturer's instructions. To digest wild type methylated DNA strands and maintain newly synthesized, unmethylated DNA strands, 1 µl (10 Units) of Dpn I endonuclease was added to PCR product and incubated for 2 h at 37°C. Digested sample (2 µl) was used to transform XL-Gold E. coli cells by heat-shock. Transformed cells were plated on multiple agar plates containing 50 µg/ml of ampicillin, since CMV-RTX-HC plasmid DNA contains the beta lactamase resistance gene. More than 1,000 colonies were formed, each containing putative mutants of RTX HC at the targeted region. More than 200 random colonies were inoculated each in 2 ml of LB medium containing 50 µg/ml of ampicillin and grown at 37°C by shaking at 250 RPM. Plasmid DNA was extracted from overnight cultures by alkaline lysis and DNA binding through NucleoSpin columns (Takara Bio USA, Inc.).
Purified HC plasmids along with CMV-RTX-LC were used for transfections to produce putative mutated version of RTX variants. Briefly, 1 µg each of HC and LC plasmids were mixed with 25 µl of polyethylenimine (Polysciences, Inc.) and added to FreeStyle™ 293-F cells (Thermo Fisher Scientific, Inc.) after 20 min. Cells were grown in T25 flasks under shaking conditions at 37°C in 5% CO2 in serum-free BalanCD medium (Irvine Scientific). After one week, conditioned media were collected and cleared of cells and cell debris by centrifugation at >10,000 × g at 20°C for 15 min and used for CD20 and CA125 binding screenings.
As a control, pertuzumab (PTZ) was used throughout these studies. PTZ sequences were obtained from Genentech's patent US20110117097. HC and LC cDNAs were cloned into expression vectors.
For production of lead RTX variants used for validation screenings as well as PTZ, transfections were scaled up to 80 ml culture, scaling up the amount of plasmids and polyethylenimine proportionally. Conditioned media were cleared of cells and cell debris by centrifugation at >10,000 × g, filtered through 0.45 mm membrane and further purified by protein A affinity column using Pierce Protein A binding and elution buffers (Thermo Fisher Scientific, Inc.). Purity of antibodies was assessed by SDS-PAGE and Coomassie staining and quantitated by spectrophotometry by measuring 280 nm absorbance using a NanoDrop instrument (Thermo Fisher Scientific, Inc.). Purity was typically >95%.
CD20 and CA125 binding ELISA
ELISA plates were coated overnight at 4°C with diluted antigens in 0.05 M carbonate buffer, pH 9.5. Coated antigens were CA125 (15 KU/ml; Lee Bio) or human CD20/MS4A1 linear peptide (amino acid 142–184 of CD20 extracellular domain) at 10 µg/ml. Plates were blocked with 0.05 M PBS at pH 7.2 containing 5% BSA for 1 h at room temperature then washed twice with PBS. Biotinylated antibodies (by EZ-link Sulfo-NHS-LC-Biotin; Thermo Fisher Scientific, Inc.) or negative control HSA were added to the plate in PBS at 5 µg/ml for 1 h at room temperature. Wells were washed three times with PBS followed by addition of streptavidin-HRP in PBS/BSA for 1 h at room temperature. After washing, reactions were developed for up to 10 min by adding 75 µl of TMB substrate (Pierce; Thermo Fisher Scientific, Inc.) and stopped by adding 75 µl 0.1 N H2SO4. Absorbance was read at 450 nm on a Varioskan plate reader and using SkanIt™ version 4.1 (Thermo Fisher Scientific, Inc.).
Immunostaining of CD20-positive and CD20-negative cells using 488-conjugated antibodies to determine affinity
The assessment of antibody equilibrium dissociation constant (Kd) was carried out using a cell-based fluorescent assay involving the immunostaining of antigen-positive (Ramos) and antigen-negative (Jurkat) cell lines. RTX and RTX-N109D were labeled using Alexa Fluor™ 488 Antibody Labeling kit (Thermo Fisher Scientific Inc.) following the manufacturer's instructions. Cells were stained with a 2-fold concentration titration of the 488-labeled antibody ranging from 0.78 to 100 nM. Briefly, cells were grown in suspension in cRPMI. Confluent cultures with viability >85% were centrifuged at 300 × g for 6 min at room temperature and cell pellets were resuspended in 4 ml of Animal-Free Blocker (Vector Laboratories, Inc.; cat. no. SP-5035) in a 50-ml tube to wash the cells. Cells were centrifuged again as aforementioned and resuspended in Animal-Free Blocker at 10 million/ml. Samples were incubated for 20 min on ice to block any non-specific binding sites. Cells (200 µl/2 million cells/well) were then transferred in V-bottom, polypropylene 96-well microplates (Corning; cat. no. 3363) and centrifuged in a benchtop centrifuge at 600 × g for 2 min at room temperature. Supernatants were aspirated and the cells were resuspended in 90 µl/well of Animal-Free Blocker. Each condition was tested in duplicate wells. Diluted 488-labeled antibody (10 µl/well) was added to the corresponding well and samples were incubated for 1 h on ice with mixing every 20 min. Cells were then washed four times using 100 µl/well of Animal-Free Blocker and centrifuged at 600 × g for 2 min at room temperature after each wash. After the last wash, cells were fixed with 100 µl/sample of 10% Formalin (LabChem; cat. no. LC146702) for 10 min at room temperature. Samples were centrifuged again as aforementioned and cells were resuspended in 100 µl/well of Animal-Free Blocker and transferred into a black microplate (Greiner Bio-One; cat. no. 655086). The fluorescence was read at 494 nm excitation/519 nm emission for each sample using a Varioskan LUX plate reader. The non-specific background binding to antigen-negative cells was subtracted from the binding signal from the antigen-positive cells. Subtracted values were plotted using a nonlinear regression analysis for ‘saturation binding with one site’ in GraphPad Prism version 9 (GraphPad Software, Inc.). This model determined the Kd as being the ligand concentration that binds to half the receptor sites at equilibrium.
Complement-mediated cytotoxicity (CDC)
Target cells (25,000 cell/well) were seeded in triplicates in 96-well microplates in RPMI supplemented with 7.5% FBS. The following day, medium was replaced with RPMI containing 1% FBS, 5 µg/ml antibody and 10% rabbit complement (Bio-Rad Laboratories, Inc.), with or without 30 KU/ml CA125. After a 2-h incubation, CellTiter-Glo® (Promega Corporation) solution was added and samples were processed according to the manufacturer's instructions. The percentage of CDC was calculated as [1-(experimental well/cells + complement)] ×100%.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software (GraphPad Software, Inc.). All experiments were performed in triplicate and data are presented as the means ± standard error of the mean. In experiments involving >2 groups, data were analyzed using one-way ANOVA followed by Tukey's multiple comparison test. Otherwise, 2-group analysis was carried out using unpaired two-tailed t-test.
Results
CA125 reduces RTX-mediated ADCC via inhibition of CD16/Fc receptor interaction
We have previously reported that CA125 interacts with RTX and reduces its interaction with the complement factor C1q, leading to reduced CDC activity (19). Because CA125 has been reported to also inhibit ADCC activity (13,15), we hypothesized that CA125 could also affect RTX-mediated ADCC and thus investigated this hypothesis. The results demonstrated that CA125 significantly reduced RTX's ADCC activity against the CD20-positive Daudi cells (Fig. 1A). CA125 also inhibited the RTX-dependent activation of the Fc-gamma receptor FCGR3A/CD16a (‘Fc receptor’; Fig. 1B), confirming the mechanism of action previously reported for the reduced ADCC activity of farletuzumab, whereby the binding of CA125 to this antibody reduces its engagement with both Fc receptor as well as C1q (13,15,19). Furthermore, CA125 did not decrease RTX binding to CD20, thus ruling out reduced binding to the target cells as a mechanism of decreased ADCC and CDC (Fig. 1C). Taken together, these findings suggested that CA125 may bind to RTX and inhibit C1q as well as Fc receptor binding, leading to the suppression of ADCC and CDC activity.
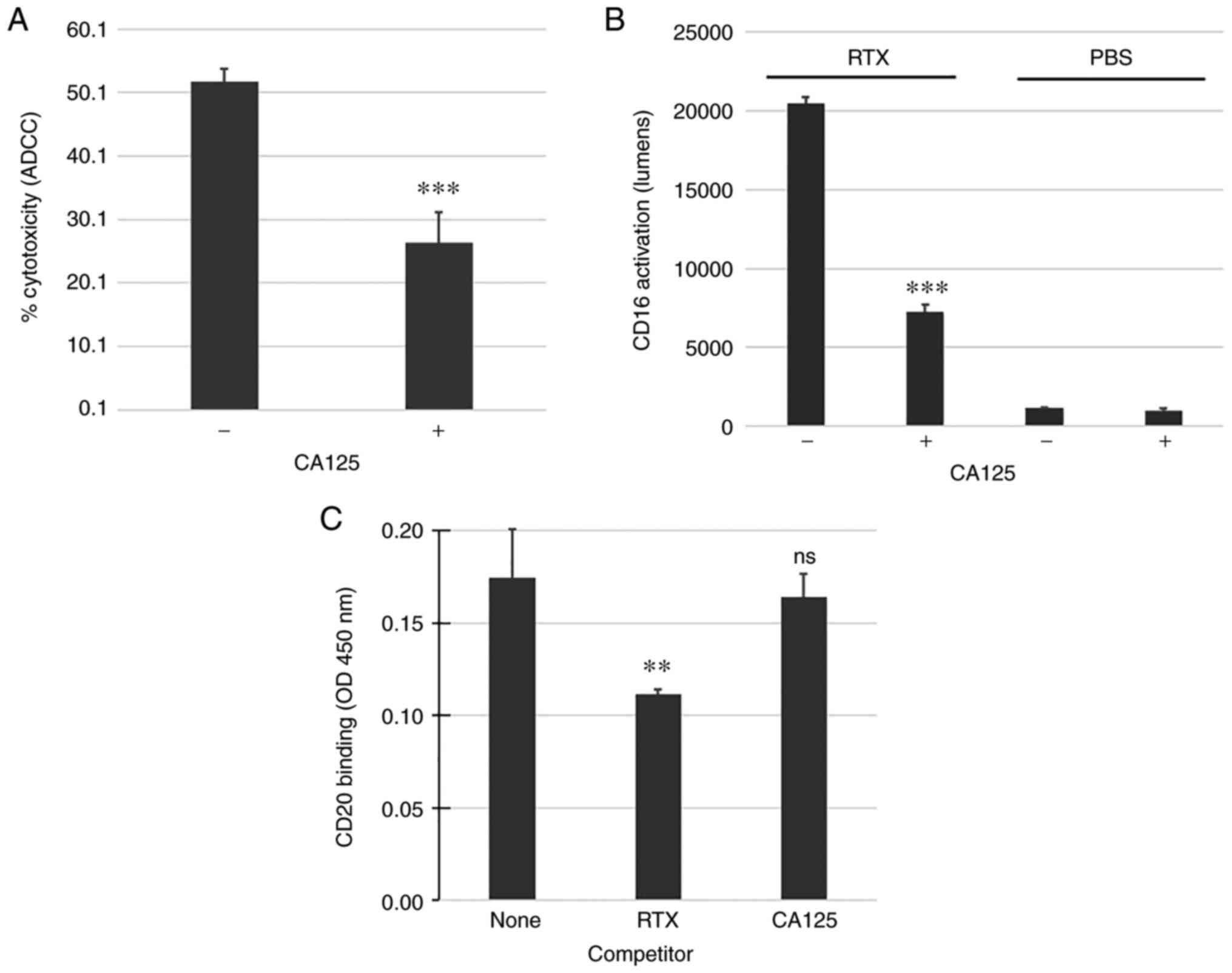 |
Figure 1.
CA125 inhibits RTX-mediated ADCC and Fc receptor/CD16a activation. (A) CD20-positive Daudi cells were targeted with RTX (30 µg/ml) and human peripheral blood mononuclear cells (effector:target ratio of 10:1). CA125 (50,000 U/ml) was added in some reactions and was shown to be immunosuppressive against RTX-mediated ADCC compared with reactions without CA125. (B) CD16a activation mediated by RTX was significantly inhibited by CA125. (C) CA125 did not affect RTX binding to CD20. A 2-fold excess of unlabeled RTX was used as a competitor control and was shown to reduce CD20 binding of biotinylated RTX while CA125 had no effect. **P<0.01 and ***P<0.001. ADCC, antibody-dependent cellular cytotoxicity; CA125, cancer antigen 125; ns, non-significant; RTX, rituximab.
|
Identification of the CA125/RTX binding region
To improve RTX's CDC and ADCC activities in the presence of CA125, we generated a variant of RTX with reduced sensitivity to CA125′s immunosuppressive effects. To determine where RTX interacts with CA125 and knowing that CA125 did not bind to PTZ (13), hybrids of PTZ and RTX were generated by swapping the LC and HC of these two antibodies to determine whether one chain was responsible for the interaction with CA125. CA125 binding to these hybrid antibodies was measured and the results indicated that only the hybrid comprising the RTX HC and PTZ LC retained CA125 binding, while the hybrid comprising the PTZ HC and RTX LC had lost this property (Fig. 2A). These data supported the hypothesis that the CA125 binding region may be located within RTX HC. In addition, the present study demonstrated that the RTX/CA125 interaction could be competed by the complement protein C3B (according to methods from Data S1), which binds to the constant region 1 (CH1) domain of the HC, but not by Protein L, which binds to the LC variable domain, further supporting the hypothesis that the putative CA125 binding site is located within the RTX HC (Fig. 2B). Because the PTZ and RTX antibodies produced in our laboratory share identical HC constant regions, these results suggested that the CA125 binding site could be within the variable region. Furthermore, in view of the competitive binding exerted by the CH1-binding C3B protein, we hypothesized that the CA125 binding interface could span across both the variable region and the CH1 domain. Therefore, we broadened the region targeted for mutagenesis by including the amino acid sequence encompassing the HC CDR3 (H-CDR3), the HC framework 4 (H-FWR4) as well as the first domain of CH1.
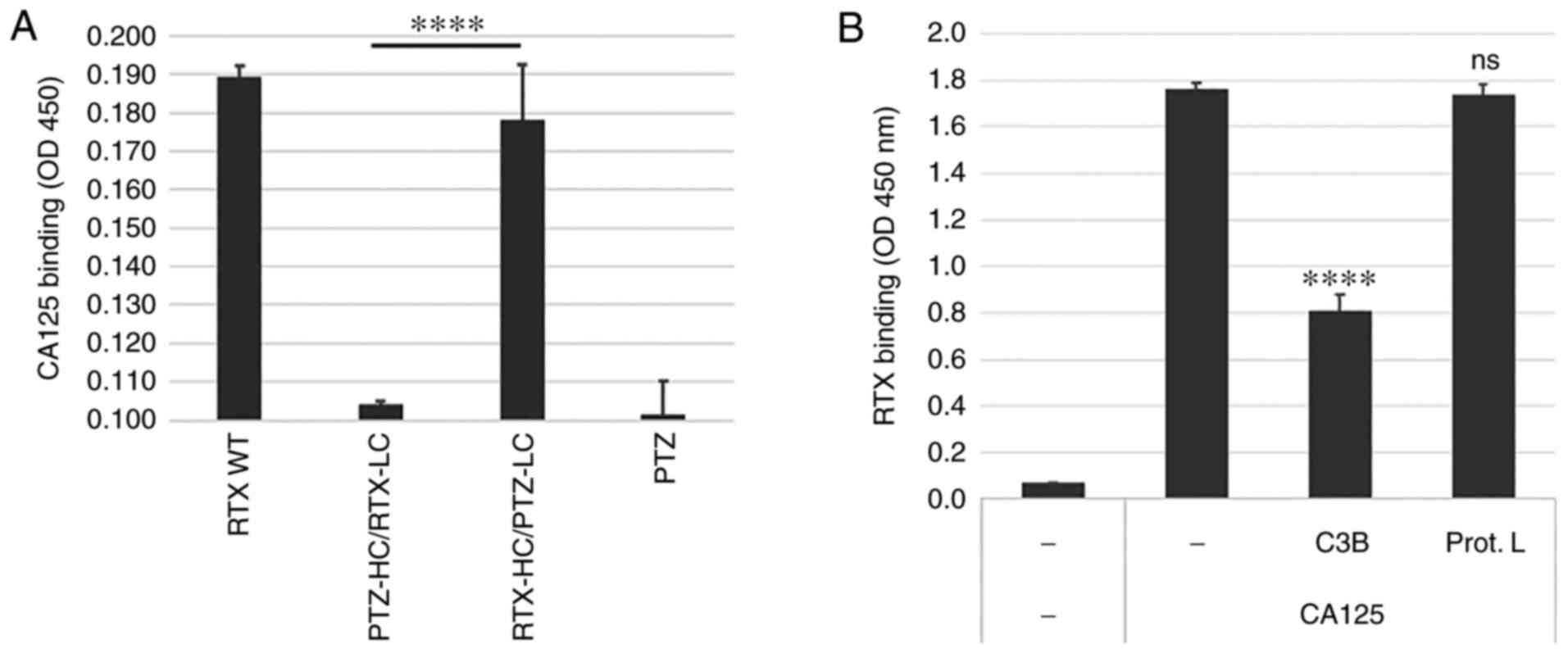 |
Figure 2.
RTX/CA125 binding interface is localized in the RTX HC. (A) Parent RTX (RTX WT), PTZ and their chain swap hybrids (PTZ-HC/RTX-LC and RTX-HC/PTZ-LC) were compared for CA125 binding activity. All antibodies were produced in 293 cells and affinity-purified. Only the hybrid comprising the RTX HC and PTZ LC retained CA125 binding. (B) CH1-binding protein complement factor C3B but not the LC-binding protein L could compete RTX binding to CA125, suggesting that RTX/CA125 binding interface may be localized in the RTX HC within the region encompassing H-CDR3, H-FWR4 and CH1. ****P<0.0001. CA125, cancer antigen 125; CH1, heavy constant region; HC, heavy chain; H-CDR3, heavy chain of complementarity determining region 3; H-FWR4, heavy chain framework 4; LC, light chain; ns, non-significant; PTZ, pertuzumab; RTX, rituximab; WT, wild type.
|
Generation and production of RTX variants library
To target the amino acid sequence encompassing H-CDR3, H-FWR4 and CH1, a pair of primers was designed that framed a sequence starting with residue ala95, located on the N-terminus of H-CDR3 and ending with pro235, located within the hinge sequence (cys-pro-pro-cys). A total of 204 variants of RTX HC were therefore generated. Expression vectors containing each variant HC were paired with parent RTX LC and transfected in 293 cells to produce each RTX variant. The RTX variants were subsequently tested for CD20 as well as CA125 binding and compared to parent RTX. A total of 12 random RTX HC variant clones were sequenced to determine the mutation frequency. The number of mutated bases in each clone ranged from 0 to 13, with an average number of mutations per RTX HC clone of 4.75, thus demonstrating a sufficient degree of mutation frequency within the library (Fig. S1A). The different possible base changes were ranked based on the number of occurrences and the data showed a high degree of base change diversity (Fig. S1B). In addition, only two clones had either one or two deleted bases, leading to open reading frame shift and predicted truncation of the HC, suggesting that ~90% of the library included full-length HC variants.
Screening of RTX variants library and identification of leads with reduced sensitivity to CA125
The outcome of the RTX variant library screening is summarized in Table SI.
A total of 204 RTX variants (RTX-001 to RTX-204) were produced and screened by measuring CD20 as well as CA125 binding levels using ELISA-based assays. Primary leads were ranked and selected based on CD20 binding activity being comparable to parent RTX as well as having decreased binding to CA125. A total of 30 primary leads were re-tested (secondary ELISA screening) and 11 secondary hits were advanced to confirmatory tests. The top five tertiary leads were selected and compared head-to-head to select a final lead. Among these top RTX variant leads, RTX-166 showed consistent CA125-refractory property (Figs. S2 and S3) and was further studied.
Sequence analyses demonstrated that the RTX-166 variant had three amino acid changes (N109D, P131S and S136Y). To study the contribution of each mutation to the observed phenotype, additional RTX variants were engineered, each having one of the three amino acid changes found in the RTX-166 variant as well as combinations of double mutations. The analyses of these new variants (N109D, P131S, S136Y, N109D + P131S, N109D + S136Y and P131S + S136Y) suggested that the presence of the N109D mutation was correlated with reduced sensitivity to CA125-mediated suppression of Fc receptor (Fig. 3A) as well as C1q binding (Fig. 3B). Upon further testing, the N109D variant (referred to as RTX N109D) was shown to bind CD20 at levels comparable to RTX (Fig. 3C) and, importantly, its binding to CA125 was significantly reduced by 50% (Fig. 3D). The nucleotide as well as the amino acid sequence alignments between RTX and RTX N109D immediately surrounding the N109D change are shown in Fig. S4A. Taken together, these results indicated that the mutation N109D may be necessary and sufficient to reduce RTX sensitivity to CA125. It was therefore crucial to determine whether RTX N109D could perform better than RTX in the presence of immunosuppressive CA125.
Functional characterization of RTX N109D variant
As the residue N109 lies within H-CDR3, further characterization was needed to unequivocally determine whether this mutation had any impact on CD20 binding or on the effector function mediated by RTX N109D compared with parent RTX. First, we further assessed RTX N109D binding to CD20. Similarly to RTX, RTX N109D bound to CD20-positive Ramos cells in a dose-dependent manner, while it did not bind to CD20-negative Jurkat cells, confirming retention of target specificity (Fig. 4A and B). In addition, the measured affinity/Kd for RTX and RTX N109D were 20.6 and 30.5 nM, respectively (Fig. 4C and D). While a small drop in RTX N109D affinity was expected and consistent with previous CD20 binding data using a different assay (Fig. 3C), RTX N109D showed CD20-specific binding and affinity comparable to parent RTX. Since CA125 inhibits C1q binding to RTX, leading to reduced CDC activity (19), we investigated whether this CA125 effect was attenuated when using RTX N109D. Our previous analysis already indicated that C1q binding to RTX N109D was in fact less suppressed by CA125 compared with RTX (Fig. 3B). This effect on C1q binding resulted in enhanced CDC activity mediated by RTX N109D compared with RTX using target cells expressing immunosuppressive CA125 as well as CD20 (Fig. 5A), while no significant killing was noted when using CA125-positive but CD20-negative cells (Fig. 5B).
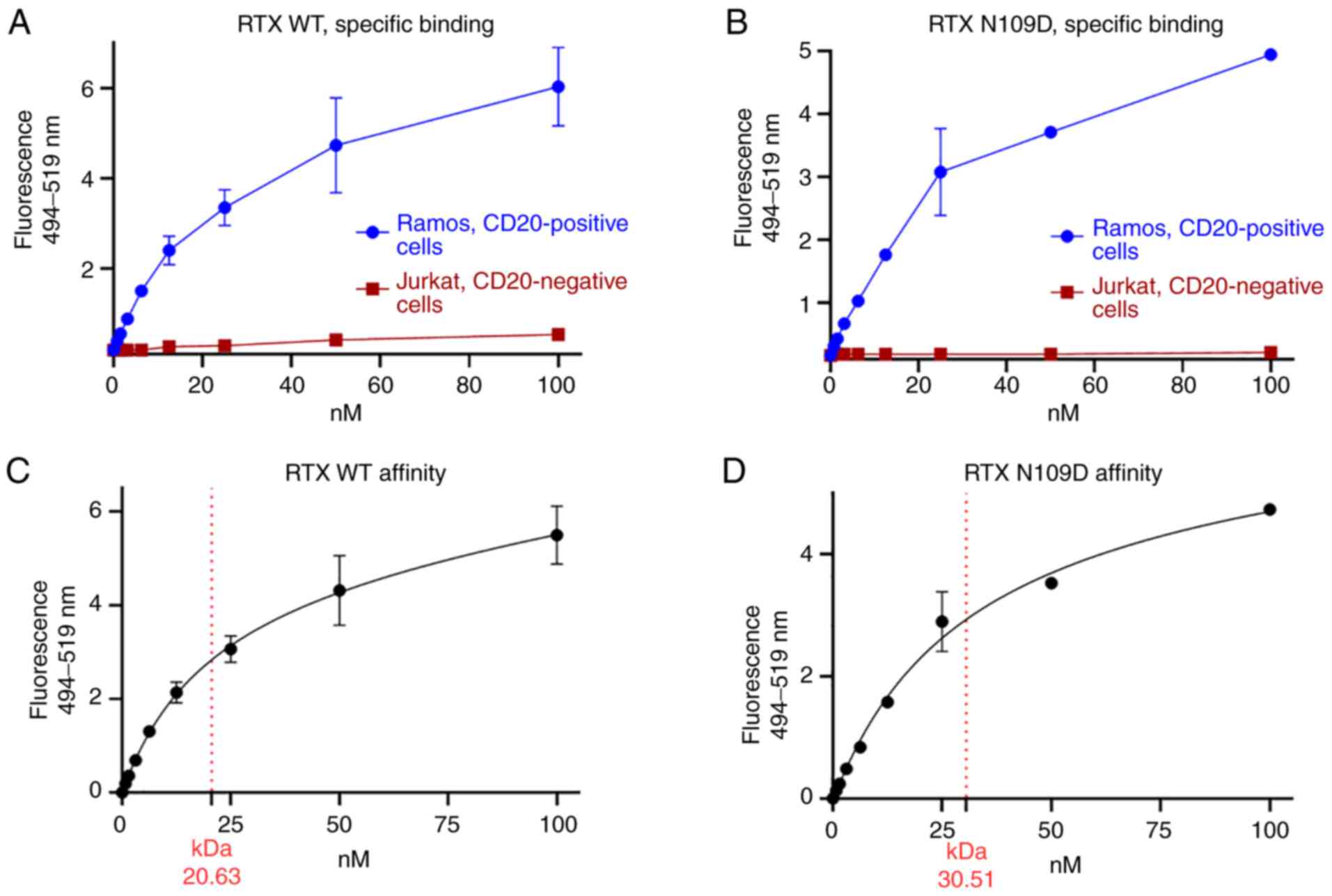 |
Figure 4.
Characterization of RTX N109D CD20 binding. Parent (WT) RTX (A and C) and RTX N109D (B and D) were directly conjugated to AlexaFluor 488 dye and used to stain live cells at concentration ranging from 0.78 to 100 nM. Specific binding to CD20-positive and negative cells were measured. (C and D) Extrapolation of Kd as the concentration of antibody bound to half the receptor sites at equilibrium by using a nonlinear regression analysis of one-site saturation binding. RTX, rituximab; WT, wild type.
|
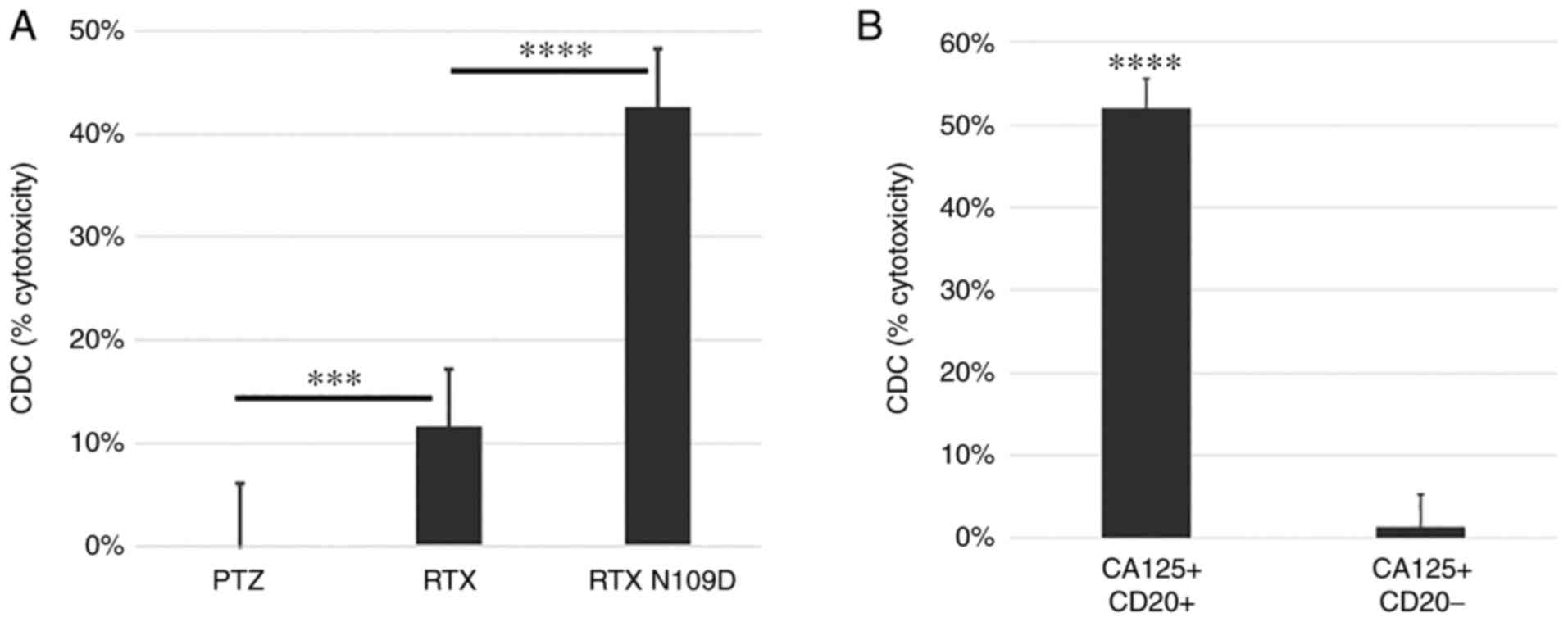 |
Figure 5.
Comparison of complement-mediated killing mediated by RTX N109D vs. parent RTX. (A) CD20-positive cancer cells expressing CA125 were targeted with antibodies in the presence of complement. This representative experiment showed that RTX N109D had superior CDC activity against CA125-positive cells compared with parent RTX. (B) CD20-positive and CD20-negative cancer cells expressing CA125 were targeted with RTX N109D in the presence of complement. This representative experiment showed that RTX N109D specifically killed only CD20-positive cells. ***P<0.001 and ****P<0.0001. CA125, cancer antigen 125; CDC, complement-dependent cytotoxicity; PTZ, pertuzumab; RTX, rituximab.
|
Because CA125 inhibited RTX binding to Fc receptor as well as its signaling activation (Fig. 1B), which led to reduced ADCC activity (Fig. 1A), we investigated whether this immunosuppressive effect was lessened when using RTX N109D. The results demonstrated that RTX N109D could bind to Fc receptor as well as activate its signaling at significantly higher levels compared with parent RTX in target cells that expressed CA125, while no significant difference was noted when using CA125-negative cells (Fig. 6A and B). An important consequence of the enhanced Fc receptor interaction and activation was that RTX N109D was able to mediate a more robust Fc receptor-dependent cytotoxic activity (ADCC) against target cells in the presence of the immunosuppressive CA125 compared with parent RTX (Fig. 6C and D). Taken together, these findings suggested that RTX N109D may perform better than parent RTX in the presence of the immunosuppressive CA125.
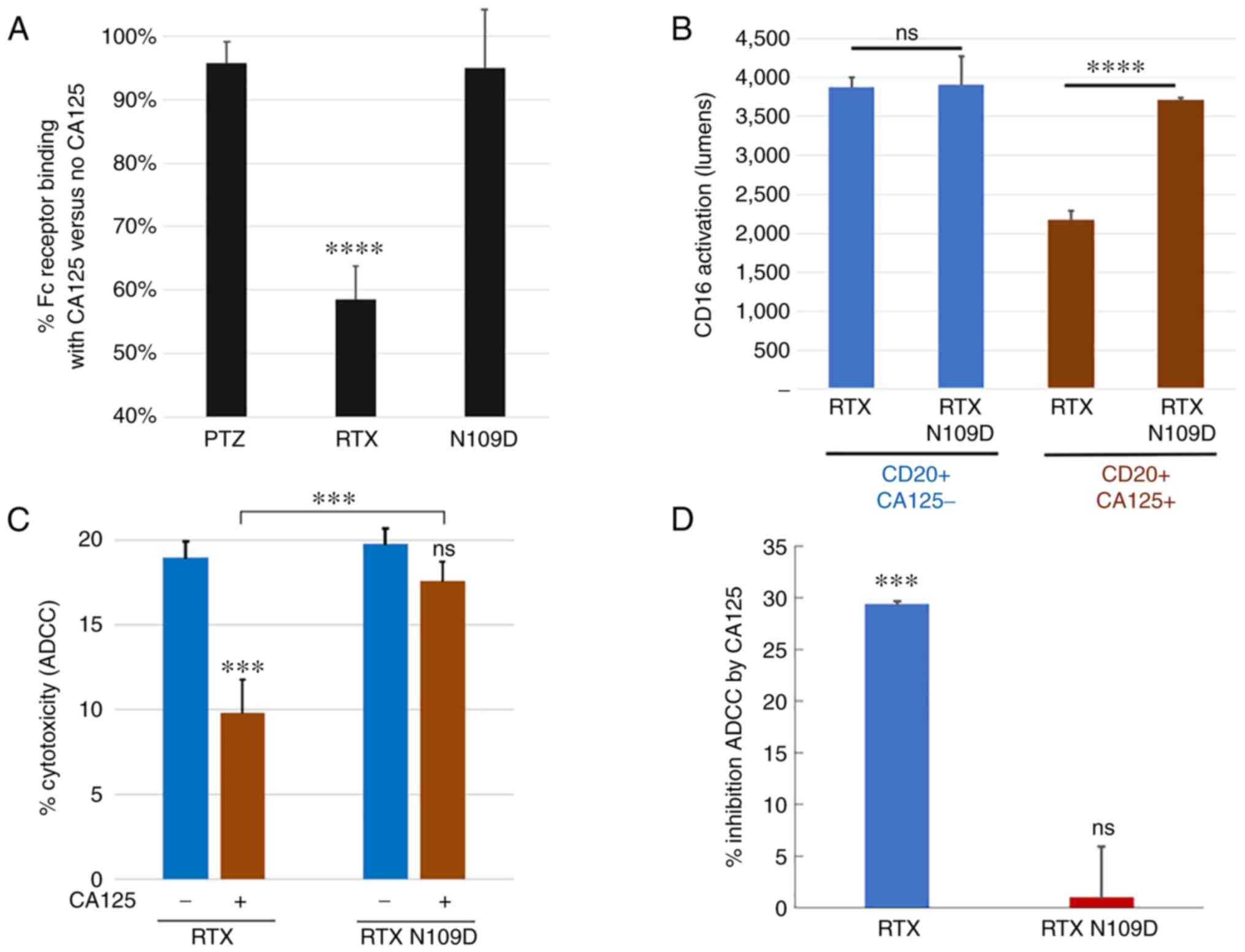 |
Figure 6.
Comparison of CD16a/Fc receptor-related activities mediated by RTX N109D vs. parent RTX. (A) RTX N109D retained >90% Fc receptor binding in the presence of CA125 compared with <60% for RTX. (B) CD16a receptor activation mediated by RTX N109D in the presence of CA125 was significantly higher than parent RTX. (C) Human PBMCs-mediated killing of Daudi target cells (effector:target cells ratio of 5:1) with or without CA125 (25 KU/ml). (D) Human PBMCs-mediated killing of Ramos target cells (effector:target cells ratio of 20:1). Data are presented as % of inhibition of cytotoxicity mediated by the addition of CA125. ADCC mediated by RTX N109D in the presence of CA125 was significantly higher than parent RTX. ***P<0.001 and ****P<0.0001. ADCC, antibody-dependent cellular cytotoxicity; CA125, cancer antigen 125; ns, non-significant; PBMC, peripheral blood mononuclear cell; RTX, rituximab.
|
Discussion
The present study identified CA125 as an immunosuppressive factor of humoral immunity (ADCC and CDC) mediated by RTX. This finding may have potential implications in the treatment of NHL cases, particularly FL cases where CA125 is elevated and RTX is part of the standard-of-care. Because of its effects on anti-cancer humoral immunity, we have named CA125 as ‘humoral immuno-oncology factor 1’ (HIO-1).
In the present study, we generated a library of RTX variants that was screened using a set of assays where the immunosuppressive effects mediated by CA125 were recreated in vitro. We identified RTX N109D (internally referred to as NAV-006) as a variant of RTX as being less sensitive to CA125-mediated immunosuppressive effects. Collectively, this approach is referred to as BRITE. The BRITE platform provides an opportunity to overcome immunosuppression affecting some therapeutic antibodies that are sensitive to CA125/HIO-1 as well as other HIO factors that we have recently discovered as having similar immunosuppressive effects (unpublished data).
Elucidation of the molecular mechanism by which the N109D change could affect the CA125/RTX interface in the context of C1q and Fc gamma receptor/CD16a binding will require additional structural analyses, including crystallography. One possible scenario would be that the N109D change in the Fab region may lead to allosteric changes in the CH2 domain of the Fc region where the canonical binding sites for both C1q and Fc gamma receptor are located (23,24). This hypothetical mechanism would be reminiscent of the allosteric effects involving Fab and Fc regions triggered by antigen binding that modulate binding of the antibody's Fc region to low-affinity Fc receptors including CD16a (25). Furthermore, it is known that different constant regions can affect the binding affinity of antibodies having the same variable region (26,27). Altogether, this evidence indicates the existence of dynamic structural influences between Fab and Fc regions that can occur in both directions. In the present study, we hypothesized that the binding interface between CA125 and RTX may contain multiple contact points within the region spanning the H-CDR3 and CH1 domains, and that the N109D change may lie in one of these points. The possibility that the CH1 region may also be involved is underscored by the observation that the RTX/CA125 interaction could be partially competed by the complement protein C3B that binds to the CH1 domain (Fig. 2B).
According to several CDR sequence definitions, the residue N109 is located at the C-terminus of the H-CDR3 region. Specifically, based on the ‘Contact’ definition, N109 represents the very last residue within H-CDR3 (Table SII) and its contribution to the antigen binding interface may be minimal, providing a possible explanation for the little impact of the N109D change on CD20 binding affinity. That N109 is not essential for H-CDR3 conformation for antigen binding is suggested by the notion that the unique conformations within the CDR3 loop are provided principally by 4-residue fragments enriched with tyrosine and glycine residues, represented in the RTX H-CDR3 by the ‘Tyr-Tyr-Gly-Gly’ sequence (28). Crystallography analysis supported the key role of these residues in the CD20/RTX binding interface (29). Interestingly, potential circumstantial evidence may be that CA125-refractory PTZ has D109 in its CDR3 instead of N109 as in RTX (Fig. S4B), which would also indicate that a naturally occurring residue could be substituted for another naturally occurring amino acid during mutagenesis.
Whenever mutations are introduced in a protein-based therapeutic agent, an increase in immunogenicity vs. the parent protein is a possible drawback. Introducing mutations to enhance activity of an antibody that is already on the high-end of the immunogenicity spectrum is not advisable. Because RTX has been reported to have a good immunogenic profile in the clinics (30), the risk of heightened immunogenicity appears to be minimal since RTX N109D is 99.9% identical to the parent RTX's whole IgG sequence. Furthermore, preliminary in-silico analysis designed to uncover any potential immunogenicity mediated by T-cell epitopes using well-established algorithms (31) demonstrated that the RTX N109D has equivalent or even reduced immunogenicity score associated with peptides within the H-CDR3/FWR4 region compared with parent RTX (Fig. S5).
Over the past few years, we have identified HIO factors that are produced by tumor cells or tumor-associated cells that have immunosuppressive effects on humoral immunity mediated by certain therapeutic antibodies, including RTX. It is therefore crucial to investigate the underlying mechanisms of HIO in order to counteract their effects. This study presented the BRITE platform as a solution to improve existing therapeutic agents that use CDC and ADCC mechanisms, so that their therapeutic benefits could be further extended to patient populations whose cancers express HIO factors diminishing humoral immunity.
Supplementary Material
Supporting Data
Acknowledgements
This abstract was presented at the AACR Annual Meeting 2021, April 10–15, 2021, and May 17–21, 2021 in Philadelphia, PA, and was published as abstract no. 1622 in Cancer Res 81 (Suppl 13): 2021.
Funding
This study was supported by the National Cancer Institute of the National Institutes of Health (grant no. R03CA241784). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
This study was supported by the National Cancer Institute of the National Institutes of Health (grant no. R03CA241784). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
LG, NN and JK analyzed and interpreted all data. ELISA assays were performed by NN. RTX variants were designed by LG. RTX and variant affinity were determined by LG. ADCC and CDC assays were performed by JK. LG authored and revised the manuscript, and all authors read and approved the final manuscript. LG, NN and JK confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Human peripheral blood mononuclear cells isolated from healthy donors used for ADCC assays were purchased from a commercial source (PPA Research). Because these human specimens were not collected specifically for our study and since we had no access to the subject identifiers linked to the specimens, our study was exempted from Federal regulations for the Protection of Human Subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
National Cancer Institute, . Surveillance, Epidemiology, and End Results (SEER) Program: Cancer Stat Facts: NHL — Follicular Lymphoma. https://seer.cancer.gov/statfacts/html/follicular.htmlOctober 27–2021
|
|
2
|
Gallagher CJ, Gregory WM, Jones AE, Stansfeld AG, Richards MA, Dhaliwal HS, Malpas JS and Lister TA: Follicular lymphoma: Prognostic factors for response and survival. J Clin Oncol. 4:1470–1480. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Manches O, Lui G, Chaperot L, Gressin R, Molens JP, Jacob MC, Sotto JJ, Leroux D, Bensa JC and Plumas J: In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 101:949–954. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clynes RA, Towers TL, Presta LG and Ravetch JV: Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 6:443–446. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P and Watier H: Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 99:754–758. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seyfizadeh N, Seyfizadeh N, Hasenkamp J and Huerta-Yepez S: A molecular perspective on rituximab: A monoclonal antibody for B cell non Hodgkin lymphoma and other affections. Crit Rev Oncol Hematol. 97:275–290. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scott DW and Gascoyne RD: The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 14:517–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dilek I, Ayakta H, Demir C, Meral C and Ozturk M: CA 125 levels in patients with non-Hodgkin lymphoma and other hematologic malignancies. Clin Lab Haematol. 27:51–55. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fehm T, Beck E, Valerius T, Gramatzki M and Jäger W: CA 125 elevations in patients with malignant lymphomas. Tumour Biol. 19:283–289. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lazzarino M, Orlandi E, Klersy C, Astori C, Brusamolino E, Corso A, Bellio L, Gargantini L, Morra E and Bernasconi C: Serum CA 125 is of clinical value in the staging and follow-up of patients with non-Hodgkin's lymphoma: Correlation with tumor parameters and disease activity. Cancer. 82:576–582. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gutiérrez A, Martínez-Serra J, Barceló B, Sampol A, Viñas L, González G, Bea MD, Amat JC, Martín J, Ramos R, et al: Prognostic value of serum CA125 levels in diffuse large B-cell lymphoma: Potential role of a new sex- and age-adjusted reference value. Int J Lab Hematol. 32:582–589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Memar B, Aledavood A, Shahidsales S, Ahadi M, Farzadnia M, Raziee HR, Noori S, Tayebi-Meybodi N, Amouian S and Mohtashami S: The prognostic role of tumor marker CA-125 in B-cell non-Hodgkin's lymphoma. Iran J Cancer Prev. 8:42–46. 2015.PubMed/NCBI
|
|
13
|
Kline JB, Kennedy RP, Albone E, Chao Q, Fernando S, McDonough JM, Rybinski K, Wang W, Somers EB, Schweizer C, et al: Tumor antigen CA125 suppresses antibody-dependent cellular cytotoxicity (ADCC) via direct antibody binding and suppressed Fc-γ receptor engagement. Oncotarget. 8:52045–52060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vergote I, Armstrong D, Scambia G, Teneriello M, Sehouli J, Schweizer C, Weil SC, Bamias A, Fujiwara K, Ochiai K, et al: A randomized, double-blind, placebo-controlled, phase III study to assess efficacy and safety of weekly farletuzumab in combination with carboplatin and taxane in patients with ovarian cancer in first platinum-sensitive relapse. J Clin Oncol. 34:2271–2278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nicolaides NC, Schweizer C, Somers EB, Wang W, Fernando S, Ross EN, Grasso L, Hassan R and Kline JB: CA125 suppresses amatuximab immune-effector function and elevated serum levels are associated with reduced clinical response in first line mesothelioma patients. Cancer Biol Ther. 19:622–630. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin J, Spidel JL, Maddage CJ, Rybinski KA, Kennedy RP, Krauthauser CL, Park YC, Albone EF, Jacob S, Goserud MT, et al: The antitumor activity of the human FOLR1-specific monoclonal antibody, farletuzumab, in an ovarian cancer mouse model is mediated by antibody-dependent cellular cytotoxicity. Cancer Biol Ther. 14:1032–1038. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ebel W, Routhier EL, Foley B, Jacob S, McDonough JM, Patel RK, Turchin HA, Chao Q, Kline JB, Old LJ, et al: Preclinical evaluation of MORAb-003, a humanized monoclonal antibody antagonizing folate receptor-alpha. Cancer Immun. 7:62007.PubMed/NCBI
|
|
18
|
Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, Chao Q, Jacob S, Turchin H, Gibbs L, et al: Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 7:202007.PubMed/NCBI
|
|
19
|
Kline JB, Fernando S, Ross EN, Grasso L and Nicolaides NC: Tumor-shed antigen CA125 blocks complement-mediated killing via suppression of C1q-antibody binding. Eur J Immunol. 48:1872–1882. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weng WK and Levy R: Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 21:3940–3947. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, et al: Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 110:2561–2564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Procházka V, Faber E, Raida L, Kapitáňová Z, Langová K, Indrák K and Papajík T: High serum carbohydrate antigen-125 (CA-125) level predicts poor outcome in patients with follicular lymphoma independently of the FLIPI score. Int J Hematol. 96:58–64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Painter RH, Foster DB, Gardner B and Hughes-Jones NC: Functional affinity constants of subfragments of immunoglobulin G for Clq. Mol Immunol. 19:127–131. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chappel MS, Isenman DE, Everett M, Xu YY, Dorrington KJ and Klein MH: Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc Natl Acad Sci USA. 88:9036–9040. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orlandi C, Deredge D, Ray K, Gohain N, Tolbert W, DeVico AL, Wintrode P, Pazgier M and Lewis GK: Antigen-induced allosteric changes in a human IgG1 Fc increase low-affinity Fcγ receptor binding. Structure. 28:516–527.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dam TK, Torres M, Brewer CF and Casadevall A: Isothermal titration calorimetry reveals differential binding thermodynamics of variable region-identical antibodies differing in constant region for a univalent ligand. J Biol Chem. 283:31366–31370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pritsch O, Magnac C, Dumas G, Bouvet JP, Alzari P and Dighiero G: Can isotype switch modulate antigen-binding affinity and influence clonal selection? Eur J Immunol. 30:3387–3395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Regep C, Georges G, Shi J, Popovic B and Deane CM: The H3 loop of antibodies shows unique structural characteristics. Proteins. 85:1311–1318. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rougé L, Chiang N, Steffek M, Kugel C, Croll TI, Tam C, Estevez A, Arthur CP, Koth CM, Ciferri C, et al: Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. 367:1224–1230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Denoel A, Dieude P, Chollet-Martin S and Grootenboer-Mignot S: SAT0181 immunogenicity of rituximab in patients with rheumatoid arthritis: A kinetic analysis. Ann Rheum Dis. 74 (Suppl≈2):S720.2–720. 2015. View Article : Google Scholar
|
|
31
|
Dhanda SK, Karosiene E, Edwards L, Grifoni A, Paul S, Andreatta M, Weiskopf D, Sidney J, Nielsen M, Peters B and Sette A: Predicting HLA CD4 immunogenicity in human populations. Front Immunol. 9:13692018. View Article : Google Scholar : PubMed/NCBI
|














