Introduction
The ubiquitin system, which is composed of
ubiquitin-activating enzyme, ubiquitin-conjugating enzyme, and
ubiquitin ligase, regulates numerous cellular functions including
proteasomal degradation and signal transduction through the
generation of various ubiquitin chains (1–3).
Thus, dysfunctions in the ubiquitin system are associated with
multiple disorders (4). The linear
ubiquitin chain assembly complex (LUBAC), composed of RNF31 (also
known as HOIP), RBCK1 (HOIL-1L), and SHARPIN subunits, specifically
generates N-terminal M1-linked linear polyubiquitin chains (M1-Ub),
which activate the nuclear factor-kappa B (NF-κB) pathway,
affecting the development and activation of T cells and B cells
(5–8). Accordingly, dysregulation of M1-Ub
causes lymphoma (9–12), and two germline polymorphisms of
RNF31, Q584H and Q622L, are specifically enriched in patients with
activated B-cell-like subtype of diffuse large B-cell lymphoma
(ABC-DLBCL) (10). These
polymorphisms were reported to strengthen the interaction between
RNF31 and RBCK1, subsequently increasing the ligase activity of
LUBAC and NF-κB activation and causing ABC-DLBCL (10).
Because the role of dysregulated M1-Ub in other
tumors is unclear, we first aimed to clarify the prevalence of
RNF31 Q584 and Q622 polymorphisms in patients with lung cancer. We
identified two patients with a novel Q622H polymorphism.
Interestingly, one patient also had a history of ABC-DLBCL. We thus
speculated that the presence of ABC-DLBCL in the patient with RNF31
Q622H polymorphism was not a coincidence and that the Q622H may
have molecular functions similar to Q584H and Q622L. We further
evaluated NF-κB activation and the mutational background in lung
cancer and ABC-DLBCL with RNF31 Q622H polymorphism, and analyzed
the molecular effects of RNF31 Q622H on LUBAC activation.
Materials and methods
Patients and clinical course
We analyzed RNF31 polymorphisms in exon 10 of RNF31
in lung cancer patients who underwent surgery in our department
from 2003 to 2019. Details of the clinical courses of patients with
the RNF31 Q622H polymorphism are described in the Results
section.
PCR amplification and sanger
sequencing
The primers used to amplify RNF31 exon 10 were as
follows: forward 5′-CTGGGCTGGGTGCCTTTTCCTGTCAGG-3′ and reverse
5′-GAGTAATTCTTGGACCAGGTATCG-3′ (10). The PCR products were purified using
the ExoSAP-IT Kit (Thermo Fisher Scientific) and sequenced using
the BigDye sequencing system (Applied Biosystems).
Immunostaining
Immunohistochemistry was performed on
four-micrometer-thick formalin-fixed [10% neutral buffered formalin
for 24 to 48 h at room temperature (RT)], paraffin-embedded (FFPE)
tissue sections as previously described (13,14).
For RNF31 immunostaining (Fig.
S1), deparaffinized and rehydrated sections were treated with
0.3% hydrogen peroxide (H2O2) in methanol for
30 min at RT to block endogenous peroxidase activity. Antigen
retrieval was performed by autoclaving (5 min, citrate buffer pH
6.0). The sections were incubated with anti-RNF31/HOIP antibody
(ab187976; Abcam; 1:50 dilution) overnight at 4°C, followed by
incubation with a Histofine Simple Stain MAX PO (Nichirei), for 45
min at RT. The peroxidase reaction was carried out using 0.02%
3,3′-diaminobenzidine tetrahydrochloride and 0.01%
H2O2 in 0.05 M Tris-HCl (pH 7.4). The
immunoreaction was visualized with diaminobenzidine (DAB) and
briefly counterstained with hematoxylin. Negative control tissue
sections were stained as described above, except that the primary
antibody was omitted. For p65 immunostaining, antigen retrieval was
performed by autoclaving (10 min, citrate buffer pH 6.0). The
sections were incubated with anti-NF-κB p65 antibody (D14E12, Cell
Signaling Technology; 1:400 dilution), overnight at 4°C, followed
by immersing the sections in 3% solution of
H2O2 for 10 min at RT to block endogenous
peroxidase activity. Next, addition of a Histofine Simple Stain MAX
PO (Nichirei) was carried out for 30 min at RT. The immunoreaction
was visualized with DAB and briefly counterstained with hematoxylin
for 30 to 60 sec. Nuclear staining of NF-κB p65 was rated
positive.
Genomic DNA extraction
DNA from fresh frozen lung cancer tissue and
adjacent normal lung tissue were extracted using a DNeasy Tissue
Kit (QIAGEN). DNA from peripheral blood mononuclear cells was
extracted using a QIAamp DNA Blood Mini Kit (QIAGEN) and that from
ABC-DLBCL FFPE tissue was extracted from four micro-dissected
slides using a GeneRead DNA FFPE Kit (QIAGEN), all in accordance
with the manufacturer's instructions.
Genetic analysis
QIAseq DNA QuantiMIZE Kit (QIAGEN) was used to
qualify and quantify amplifiable FFPE DNA prior to the library
preparation. Ten nanograms (fresh frozen samples) or 100 ng (FFPE
samples) genomic DNA were subjected to genetic analysis using the
Human Comprehensive Cancer QIAseq DNA Panel (QIAGEN). For each
library, DNA concentrations and fragment sizes were measured using
the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and the
Bioanalyzer High Sensitivity DNA Kit (Agilent), respectively.
Paired-end sequencing was performed on a NextSeq 500 platform
(Illumina) for 151×2 cycles. The average number of read fragments
was 22,444,402 (range 17,939,522-30,178,462). Read fragments were
aligned to the hg19 assembly of the human genome (Genome Reference
Consortium Human Build 37, GRCh37) and variants were called using
the GeneGlobe v.2 smCounter (QIAGEN). The average coverage depth
and the percentage of target coverage at ≥20× were 3,573.19 (range
3,025.23-4,366.68) and 98.68% (range, 98.18%-99.03%), respectively.
VCFtools (v. 0.1.17) was used to filter out all variants other than
‘Pass’ variants with following command: ‘vcf-annotate-hard-filter’.
Variants were annotated using the ANNOVAR (http://annovar.openbioinformatics.org/en/latest/)
pipeline. Affectable mutation candidates were those with: 1) amino
acid changes or variants on splice site; 2) low minor allele
frequency in Japanese and east Asian populations (<0.01, HGVD2,
DBexome20161214; https://www.hgvd.genome.med.kyoto-u.ac.jp, gnomAD
exome EAS, v. 2.0.1); and 3) variant allele frequency >0.05. In
total, 15 variants were detected in at least one sample.
Furthermore, five variants with COSMIC v.70 (15) (cancer.sanger.ac.uk) registration
were selected.
Cell culture, transfection, and
luciferase assay
RNF31-knockout (KO) 293T cells were cultured in DMEM
containing 10% fetal bovine serum, 100 IU/ml penicillin G, and 100
µg/ml streptomycin at 37°C under 5% CO2. Transfection
experiments were performed using polyethyleneimine (PEI MAX;
Polysciences). Briefly, plasmid DNA and PEI [1:2 ratio of total DNA
(µg): PEI (µg)] were mixed in PBS, and incubated for 15 min at RT.
Then the DNA/PEI mixture was added to cells. For the luciferase
assay, a pGL4.32 [luc2P/NF-κB-RE/Hygro] vector and a pRL-TK Renilla
Luciferase control reporter vector (Promega) were co-transfected
into RNF31-KO 293T cells with a FLAG-RNF31 expression vector
(wild-type or mutant), RBCK1-myc, and HA-SHARPIN. At 24 h after
transfection, the cells were lysed, and the luciferase activity was
measured using a GloMax 20/20 luminometer (Promega) using the
Dual-Luciferase Reporter Assay System (Promega). The enzyme
activity of Renilla luciferase was used to normalize the firefly
luciferase enzyme activity.
Construction of RNF31-KO cells
A gRNA cloning vector and a pCAG-hCas9 vector were
obtained from Addgene. The nucleotide sequence
5′-TCAACCCTCAGGAAGCTCAGC-3′ in exon 2 of human RNF31 was selected
as the target. These plasmids and a puromycin-resistant vector
(pXS-Puro) were co-transfected into 293T cells (ATCC), and
puromycin-resistant cell clones were selected by limiting dilution.
Genome editing of RNF31 was screened by a BtsCI digestion assay
(New England BioLabs), and mutations were confirmed by sequencing.
RNF31 protein deficiency was confirmed by immunoblotting (Fig. S2).
Immunoprecipitation, SDS-PAGE, and
immunoblotting
FLAG-RNF31, RBCK1-myc, and HA-SHARPIN plasmids were
co-transfected into RNF31-KO 293T cells. At 24 h after
transfection, the cells were lysed with 50 mM Tris-HCl, pH 7.5, 150
mM NaCl, 1% Triton X-100, 2 mM PMSF, and complete protease
inhibitor cocktail (Sigma). The Bradford protein assay was
performed to determine the protein concentration. For
immunoprecipitation, 800 µg of the cell lysates was incubated with
1 µg of anti-FLAG antibody (F7425; Sigma-Aldrich) or normal rabbit
IgG (PM035; MBL) for 1 h at 4°C, and centrifuged at 20,000 g for 10
min at 4°C. The supernatants were incubated with Protein G agarose
beads (30 µl of the 50% slurry; GE Healthcare) for 1 h at 4°C with
gentle rotation. Then, beads were washed three times with 1 ml of
lysis solution, centrifuged at 1,500 g for 4 min at 4°C. The
samples were heated at 95°C for 5 min with SDS-PAGE sample buffer,
and separated by SDS-PAGE and transferred to PVDF membranes. For
SDS-PAGE, 2/15% gradient gel (Cosmobio) or 7.5% gel was used and
transferred to PVDF membranes. After blocking the membrane in
Tris-buffered saline containing 0.1% Tween-20 (TBS-T) with 5%
skim-milk for 2 h at RT, the membrane was incubated with the
appropriate primary antibodies diluted in TBS-T containing 5%
skim-milk at 4°C overnight. Then, the membranes were incubated with
anti-mouse IgG horseradish peroxidase-conjugated secondary antibody
(NA931V; 1:10,000; Cytiva) or anti-rat IgG horseradish
peroxidase-conjugated secondary antibody (NA935; 1:10,000; Cytiva)
diluted in TBS-T containing 5% skim-milk for 2 h at RT when using
non-HRP-conjugated primary antibodies. For detection, SuperSignal
West Pico PLUS (34577; Thermo Fisher Scientific) or Luminata Forte
(WBLUF0100; Millipore) was used.
Plasmids
The human cDNA open reading frame of RNF31 (16,17)
was amplified by reverse transcription PCR. Mutants of this cDNA
was prepared by the QuikChange method, and all nucleotide sequences
were verified. The cDNAs were ligated to the appropriate epitope
sequences and cloned into the pcDNA3.1 vector (Invitrogen).
Antibodies
The following antibodies were used for
immunoblotting analyses: DYKDDDDK (1E6, 015-22391; HRP-conjugate;
1:20,000; Wako), tubulin (CLT9002; 1:3,000; Cedarlane), β-actin
(sc-47778, 1:250; Santa Cruz Biotechnology), Myc (HRP-Conjugate,
M192-7; 1:20,000; MBL), HA (HRP-Conjugate, M180-7; 1:20,000; MBL),
HA (11867423001; 1:1,000; Roche), and linear ubiquitin (clone LUB9,
MABS451; 1:1,000; Millipore), RNF31/HOIP (ab125189; 1:1,000;
Abcam), RBCK1 (sc-49718, 1:250; Santa Cruz Biotechnology), SHARPIN
(14626-1-AP; 1:3,000; Proteintech).
Statistics
Data are shown as means ± SEM from at least three
experiments performed in triplicate. One-way ANOVA followed by a
post hoc Tukey HSD test or Student's t-test was performed using
KaleidaGraph software (Synergy Software, PA, USA). For all tests, a
P-value of less than 0.05 was considered statistically
significant.
Results
Analysis of RNF31 polymorphisms in
lung cancer patients
To identify the RNF31 Q584 and Q622 polymorphisms,
we sequenced exon 10 of RNF31 in 481 patients with lung
adenocarcinoma, 152 with squamous cell carcinoma, 16 with large
cell neuroendocrine carcinoma, 2 with adenosquamous carcinoma, and
22 with small cell lung cancer (Table
SI). Although the reported prevalence of Q584H and Q622L
polymorphisms in healthy individuals is 0.95% (GO Exome Sequencing
Project and 1000 Genome Project), no patient harbored these
polymorphisms in our cohort. However, two patients had a Q622H
polymorphism (Fig. 1A), which was
not identified in public databases (SNPnexus: http://www.snp-nexus.org/v4/). More interestingly, one
of these patients also had a history of ABC-DLBCL.
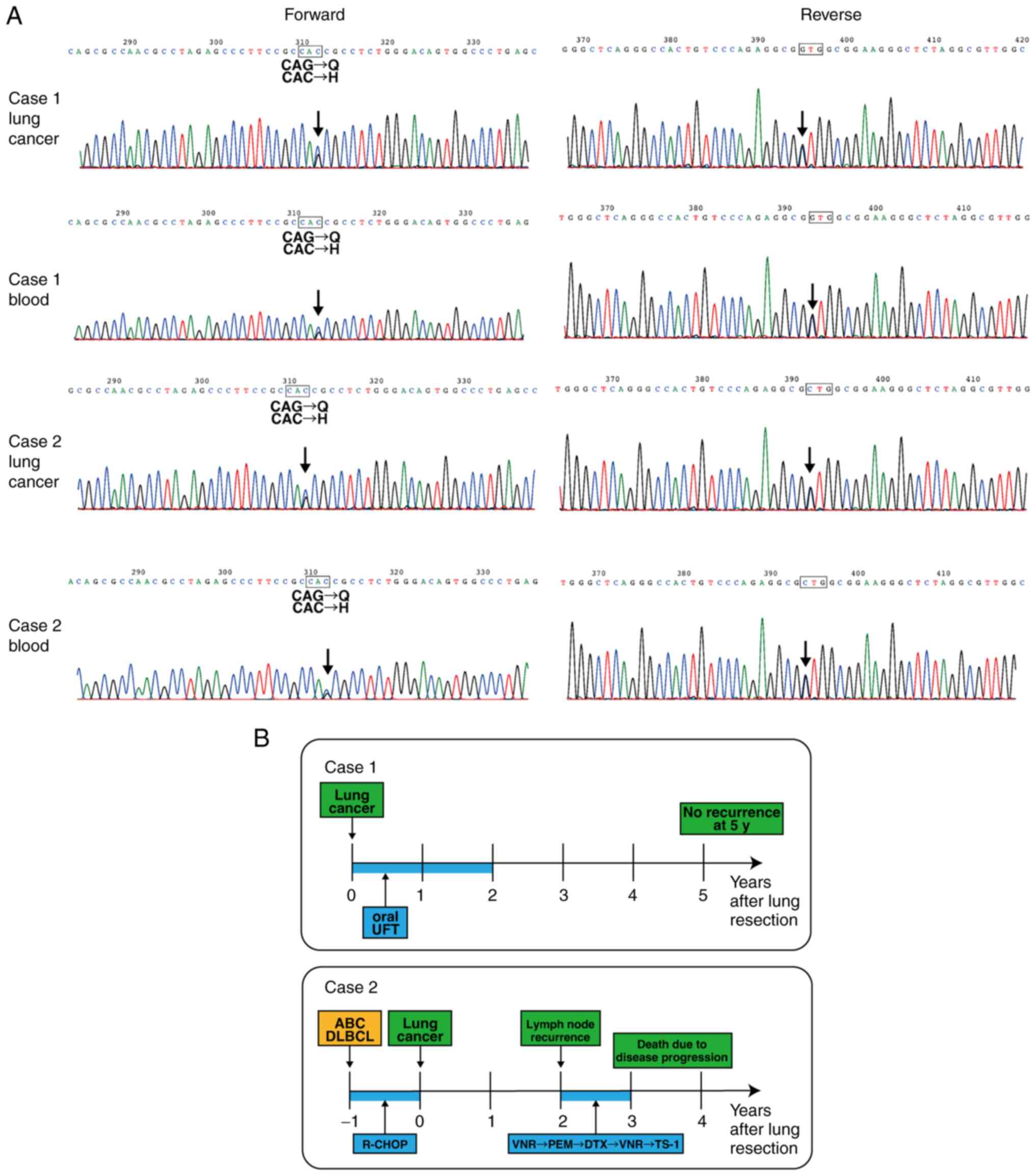 | Figure 1.RNF31 Q622H polymorphism in patients
with lung cancer and ABC-DLBCL. (A) DNA sequence electropherograms
of the region corresponding to the Q622H polymorphism in lung
cancer and blood. (B) Clinical course of the patients with the
Q622H polymorphism. UFT, uracil tegafur; ABC-DLBCL, activated B
cell-like subtype of diffuse large B-cell lymphoma; R-CHOP,
rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone;
VNR, vinorelbine; PEM, pemetrexed; DTX, docetaxel; TS-1,
tegafur/gimeracil/oteracil; RNF31, RING finger protein 31. |
Clinical course of patients with RNF31
Q622H polymorphism
The first case was a 71-year-old Japanese patient
who had undergone resection of lung adenocarcinoma, stage IB. The
patient received postoperative oral chemotherapy for 2 years and
had no recurrence at 5 years (Fig.
1B). The second case was a 78-year-old Japanese patient with a
history of ABC-DLBCL (Fig. 1B).
The patient underwent six cycles of R-CHOP therapy with radiation.
One year later, the patient was diagnosed with lung cancer and
underwent surgical resection. Pathological diagnosis was lung
adenocarcinoma, stage IA. Two years later, the patient developed
multiple lymph node metastases. The patient was administered
vinorelbine but further developed malignant pleuritis. The patient
then had pemetrexed as second-line, docetaxel as third-line,
vinorelbine rechallenge as fourth-line, and
tegafur/gimeracil/oteracil (TS-1) as fifth-line treatment. However,
the disease progressed, and the patient eventually died of
respiratory failure.
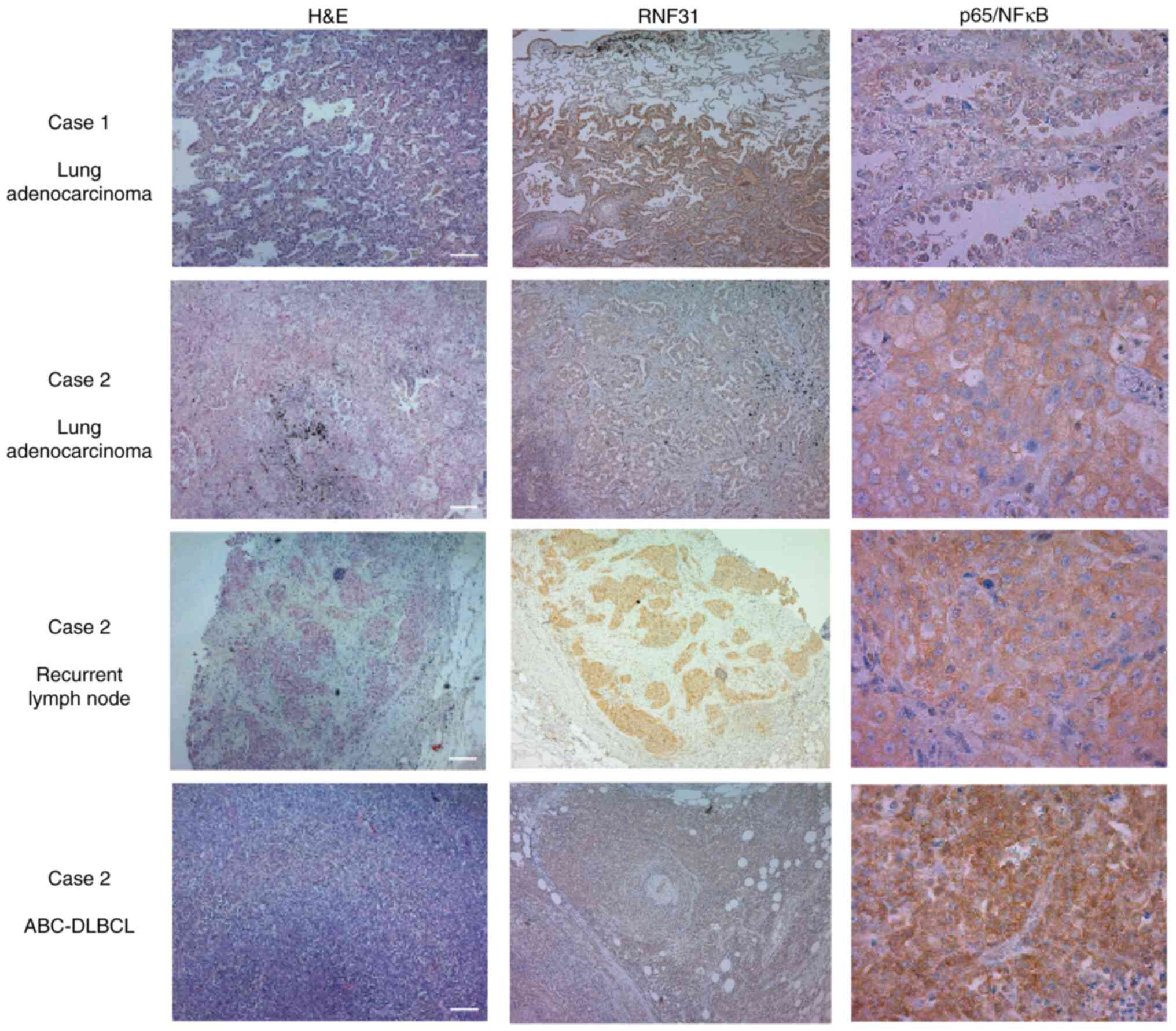
Histological analysis of lung cancer,
recurrent lymph nodes, and ABC-DLBCL
The previously reported RNF31 Q584H/Q622L
polymorphisms strengthen RNF31-RBCK1 binding, which subsequently
increase M1-Ub formation and NF-κB activation (10). We therefore analyzed whether the
RNF31 Q622H polymorphism had similar effects. Immunohistochemistry
showed that both patients strongly expressed RNF31 in tumor tissues
compared with the adjacent lung tissue (Fig. 2). NF-κB activation, evaluated by
nuclear localization of p65, was only observed in the ABC-DLBCL
specimen, and not in the lung cancer or recurrent lymph node
specimens.
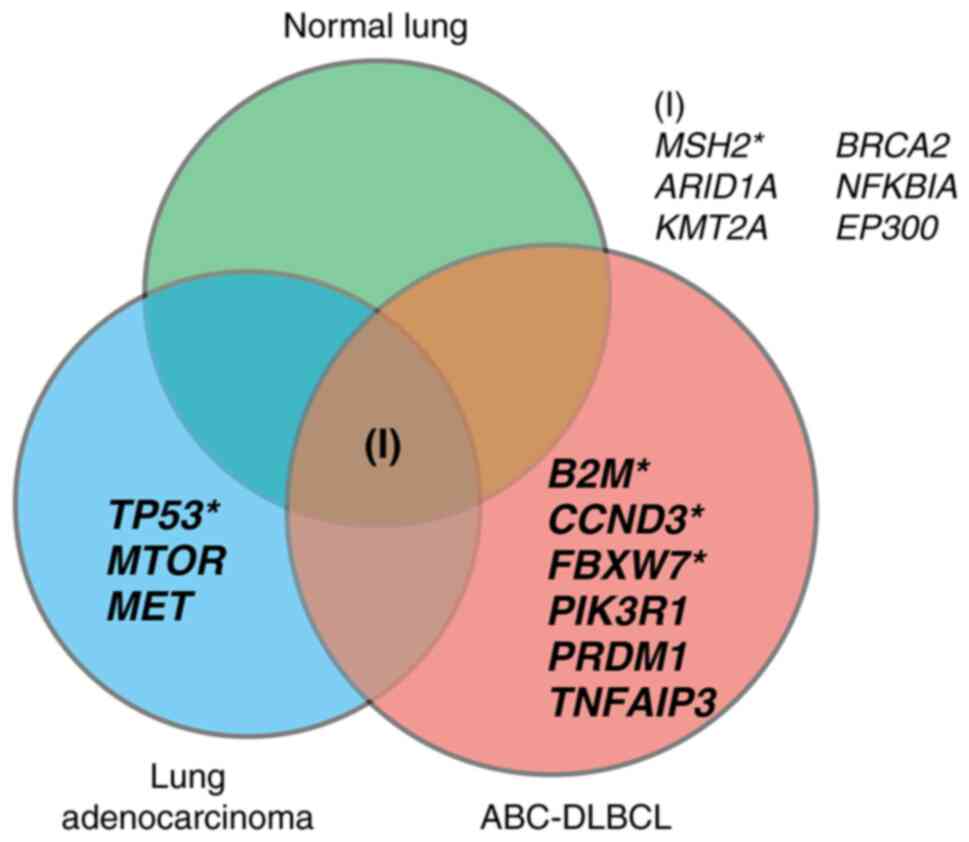
Mutational status of lung cancer and
ABC-DLBCL
Next, we examined co-mutations in the lung cancer
and ABC-DLBCL specimens by cancer panel analysis. Fifteen variants
were selected as candidate mutations. All samples harbored an MSH2
mutation (Fig. 3, Table SII) with similar variant allele
frequencies (0.59, 0.51, and 0.55 for lung cancer, ABC-DLBCL, and
normal lung, respectively). We also detected ARID1A, KMT2A, BRCA2,
NFKBIA, and EP300 mutations common to all samples. The lung cancer
sample was negative for EGFR, KRAS, BRAF, and NRAS mutations, but
we detected mutations in TP53, MET, and MTOR that were not detected
in the ABC-DLBCL lesion. In contrast, the ABC-DLBCL lesion harbored
multiple mutations, including B2M, FBXW7, CCND3, PIK3R1, PRDM1, and
TNFAIP3 mutations.
In vitro analysis of RNF31 Q622H
We next investigated the effect of RNF31 Q622H on
NF-κB activation, M1-Ub chain formation, and LUBAC complex
formation. We compared the NF-κB activation induced by
co-expression of RNF31 Q622H with RBCK1 and SHARPIN, in comparison
to RNF31 Q584H and Q622L. When wild-type RNF31 and each mutant were
overexpressed in 293T cells (Fig.
4A, lower panel), elevated NF-κB activation was detected in
samples with RNF31 Q584H and Q622L mutants, in contrast to RNF31
Q622H, which showed no difference with wild-type RNF31 (Fig. 4A, upper panel). We then compared
the formation of linear ubiquitin chains. In contrast to NF-κB
activation, the level of M1-Ub formation was similar amongst all
RNF31 mutants, with no significant increase in RNF31 Q584H and
Q622L mutants (Fig. 4B). We also
compared the binding ability of RNF31 mutants with RBCK1 and
SHARPIN by co-immunoprecipitation but found no significant
difference between RNF31 wild-type and RNF31 mutants (Fig. 4C).
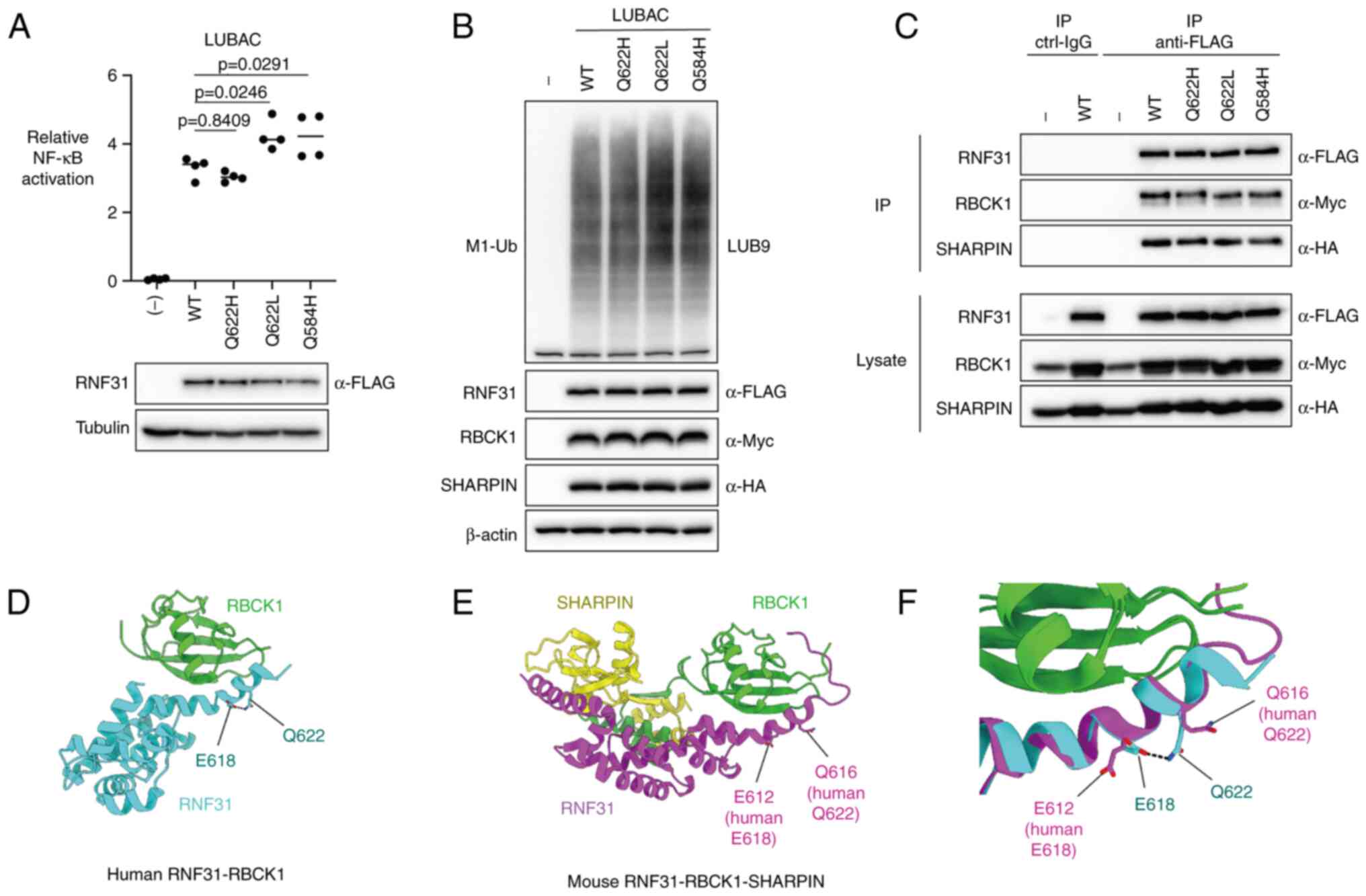 | Figure 4.In vitro assessment and predicted
crystal structure model of RNF31 Q622H polymorphism. (A) Activation
of NF-κB by RNF31 WT/Q622H/Q622L/Q584H. NF-κB activation by LUBAC
was evaluated using luciferase reporter assay (upper panel) in
RNF31-KO 293T cells. The expression of each protein was evaluated
using western blotting (lower panel). (B) Linear ubiquitin
formation by RNF31 WT/Q622H/Q622L/Q584H. The LUBAC expression
vectors were co-expressed in RNF31-KO 293T cells, and cell lysates
were immunoblotted with the indicated antibodies. (C) Effect of
RNF31 Q622H/Q622L/Q584H polymorphisms on RNF31, RBCK1 and SHARPIN
binding. FLAG-RNF31, RBCK1-myc, and HA-SHARPIN were overexpressed
in RNF31-KO 293T cells, as indicated. The cell lysates and
anti-FLAG immunoprecipitates were immunoblotted with the indicated
antibodies. (D) Crystal structure of human RNF31 UBA domain (cyan)
in complex with RBCK1 UBL domain (green) (PDB: 4dbg). (E) Mouse
RNF31 UBA domain (magenta) in ternary complex with RBCK1 (green)
and SHARPIN (yellow) (PDB: 5y3t). (F) Superposition of human and
mouse RNF31 UBA domains. Interaction between human E618 and Q622 is
shown in black dotted line. WT, wild-type; RNF31, RING finger
protein 31; LUBAC, linear ubiquitin chain assembly complex; KO,
knockout; RBCK1, RANBP2-type and C3HC4-type zinc finger containing
1; SHARPIN, SHANK-associated RH domain interactor; HA,
hemagglutinin. |
Structural prediction of RNF31
Q622H
We further analyzed the effect of RNF31 Q622H on
RNF31 and RBCK1 binding using the data from deposited crystal
structures of RNF31 (Fig. 4)
(18,19). We hypothesized that RNF31 Q622H
polymorphism could do one of the following: (1) directly affect the RNF31-RBCK1
interface; or (2) indirectly
affect RNF31-RBCK1 binding by changing high-layer structures, i.e.,
RNF31 Q622L polymorphism, which was proposed to affect the
electrostatic interaction between RNF31 Q622 and E618 residues
(10). In the crystal structures
of human RNF31-RBCK1 and murine RNF31-RBCK1-SHARPIN complexes, the
Q622 residue (or Q616 in mouse) is located in the
ubiquitin-associated (UBA) domain of RNF31, which interacts with
the ubiquitin-like (UBL) domain of RBCK1, leading to LUBAC
formation (Fig. 4D and E).
However, the side chain of the Q622 residue (Q616 in mouse) pointed
towards the outside of the molecule and not the binding cleft
between RNF31 and RBCK1. Therefore, the change in the Q622 residue
might not directly affect RNF31- RBCK1 binding. Next, to compare
the conformation of Q622 in the human and murine RNF31 structures,
the Cα atoms of the C-terminal helix of the human RNF31 UBA
(residues 610–624) in RNF31-RBCK1 was superposed onto the
equivalent region of murine RNF31-RBCK1-SHARPIN with a
root-mean-square deviation value of 0.857 (15 residues in total)
(Fig. 4F). Superposition of the
two structures showed that the human RNF31 Q622 formed an
electrostatic interaction with E618, whereas the side chain of
murine RNF31 Q616 and E612 pointed towards different directions
(Fig. 4F). This suggests that the
electrostatic interaction between murine E612 and Q616 (human E618
and Q622), which was previously proposed to affect the binding
affinity between RNF31 and RBCK1 (10), is not a crucial interaction in the
conformation of RNF31, at least in mice. Because this area is a
highly conserved region of RNF31 amongst species, we speculated
that the previously suggested human RNF31 E618 and Q622
electrostatic interaction is not crucial in humans either.
Discussion
Two germline polymorphisms of RNF31 were previously
reported as causative of ABC-DLBCL via enhanced binding between
RNF31 and RBCK1, resulting in elevated NF-κB activation (10). Because NF-κB is involved in the
carcinogenesis of various tumors (20), we searched for these NF-κB
activating RNF31 polymorphisms in lung cancer. Although no patient
examined had the RNF31 Q584H and Q622L polymorphisms, we identified
a novel Q622H germline polymorphism in two patients with lung
cancer, one of whom also had a history of ABC-DLBCL. Interestingly,
although we detected strong expression of RNF31 in both lung cancer
and ABC-DLBCL, NF-κB activation was only detected in the ABC-DLBCL
specimen, suggesting that RNF31 Q622H was not an activator of NF-κB
in these two cases of lung adenocarcinoma. Furthermore, in vitro
analysis did not show enhanced binding between RNF31 Q622H and
RBCK1, nor enhanced NF-κB activation or M1-Ub formation by Q622H,
in comparison to RNF31 wild-type. Curiously, structural prediction
failed to explain the role of Q622 in the binding between RNF31 and
RBCK1 because the Q622 residue was not directly involved in the
interaction of both molecules. We speculated that the reinforced
binding effect reported for the Q622L polymorphism might be caused
by a change in the higher layer structure and stability (10). We conclude that in contrast to the
RNF31 Q584H/Q622L polymorphisms, the Q622H polymorphism does not
show enhanced NF-κB activation, linear ubiquitin formation, and
RNF31-RBCK1 binding in comparison to RNF31 wild-type.
Studies have previously reported the clinical
features of patients with RNF31 and RBCK1 mutations (Table SIII) (21–24).
Mechanistically, the RNF31 L72P mutation destabilized RNF31 and
LUBAC expression, leading to impaired NF-κB activation (21). RBCK1 Q185X and L41fsX7 mutations
also cause impaired NF-κB activation (22). Therefore, in vitro, LUBAC
deficiency commonly leads to decreased NF-κB activation. However,
in vivo, a decrease in or deletion of LUBAC components results in
worse inflammation and subsequent carcinogenesis. For example, the
homozygous RNF31 L72P missense mutation results in multiorgan
autoinflammation, combined immunodeficiency, subclinical
amylopectinosis, and systemic lymphangiectasia (21). Similarly, patients with RBCK1
mutations also present immunodeficiency, autoinflammation, and
amylopectinosis. Such phenomena are also seen in the murine liver,
in which deletion of LUBAC causes increased inflammation, resulting
in hepatocarcinogenesis (25).
These results suggest that LUBAC is not a simple activator of
NF-κB, but rather a molecular rheostat that precisely modulates
immune response and prevents carcinogenesis (11–12).
In line with these findings, the patient with lung cancer and
ABC-DLBCL in this study also had interstitial lung disease and
pancreatitis, suggesting that the RNF31 Q622H polymorphism may have
promoted the development of these inflammatory diseases.
Critical signaling components are frequently mutated
in lymphoma and lung cancer. These alterations induce gain or loss
of function, overexpression, or deletion of genes. Previous reports
have shown that an increase in RNF31 expression itself was
insufficient to induce lymphoma (12). However, augmented LUBAC activity
overcomes cell death induced by DNA damage, thereby accelerating
the accumulation of somatic mutations. Thus far, the high number
and variety of mutations present in DLBCL make it difficult to
pinpoint which genetic lesions are driving the disease (26). In this study, mutation selection
led to six gene mutations in ABC-DLBCL, including a frameshift
mutation of TNFAIP3, which is a suppressor of linear ubiquitin
signaling and NF-κB activation. We detected none of the MYD88,
CARD11, CD79B, and CD79A mutations that have been previously
reported in ABC-DLBCL patients with RNF31 Q584H and Q622L
polymorphisms (10). These
findings suggest that although elevated NF-κB activation is a
common feature of ABC-DLBCL, the underlying mutational background
differs between patients with RNF31 Q584H/Q622L vs. Q622H
polymorphisms. Interestingly, mutations in the lung cancer specimen
included TP53 and MTOR mutations, as well as a MET mutation known
to activate MET in a fashion similar to MET exon 14 skipping
alterations (27,28).
A limitation of our study is that it was a
retrospective study from a single institute. The cohort was
therefore restricted to a Japanese population, and the prevalence
of RNF31 Q622H polymorphism was also low. A larger-scale analysis
is necessary to further clarify the role of RNF31 polymorphisms.
Furthermore, our in vitro analysis of RNF31 was mainly conducted by
overexpression and structural experiments. This might only depict
one aspect of the RNF31 polymorphism because its function might be
cell line- or context-dependent.
To the best of our knowledge, this is the first
study to analyze the RNF31 Q584 and Q622 polymorphisms in lung
cancer patients. The analysis revealed a previously unreported
RNF31 Q622H polymorphism in two patients with lung adenocarcinoma,
one patient having concurrent ABC-DLBCL. We had speculated that
RNF31 Q622H could have molecular effects similar to Q584H and
Q622L, which are RNF31 polymorphisms causing ABC-DLBCL. However,
RNF31 Q622H did not show enhanced LUBAC and NF-κB activation
compared with the wild-type, nor enhanced RNF31-RBCK1-SHARPIN
binding in vitro. The mutational background of RNF31 Q622H
ABC-DLBCL also differed from that of RNF31 Q584H/Q622L ABC-DLBCL.
Finally, there was a discrepancy between RNF31 expression and NF-κB
activation in tumor specimen, suggesting that the involvement of
the NF-κB pathway might be tissue-specific and the role of RNF31
could differ between lung cancer and ABC-DLBCL.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Takayuki Kosaka
(National Hospital Organization Takasaki General Medical Center,
Takasaki, Japan) and Dr Ryosuke Kaneko (Osaka University, Suita,
Japan) for expert advice; Dr Nobuhiko Kobayashi for advice on
ABC-DLBCL (Gunma University, Maebashi, Japan); Dr Kouki Hoshino, Ms
Ayako Morita, Mr Hayato Kawabata, Mr Tatsuya Yamazaki, Mr Yohei
Morishita, Ms Saori Umezawa, Ms Yoko Yokoyama, Ms Hiroko Matsuda,
Dr Yuichi Uosaki and Mr Yusuke Goto for technical support (Gunma
University, Maebashi, Japan).
Funding
This work was supported by KAKENHI Grant-in-Aid for Young
Scientists B (grant nos. 17K14982 and 17K15038) and KAKENHI
Grant-in-Aid for Young Scientists (grant no. 19K18203) Japan
Society for the Promotion of Science, Takeda Science Foundation,
and YOKOYAMA Foundation for Clinical Pharmacology (grant no.
YRY-2006). This work was the result of using research equipment
shared in the MEXT Project for promoting the public utilization of
advanced research infrastructure, program for supporting the
introduction of the new sharing system (grant no. JPMXS0420600120).
This work was also supported by the Fostering Health Professionals
for Changing Needs of Cancer by MEXT of Japan and Gunma University
Initiative for Advanced Research (GIAR).
Availability of materials and data
The datasets generated during and/or analyzed during
the current study are available from the corresponding author on
reasonable request but are not publicly available due to the fact
that the datasets have not been registered to a public database, in
accordance to the study protocol.
Authors' contributions
SNa, DO and FT conceived and designed the study.
SNa, RM, RKI, DO, KK, SNo, YS and AS acquired data. SNa, RM, RKI,
DO, KK, FT, SNo, YS, AS, TY and KS made substantial contributions
to data analysis and interpretation. SNa, RM, RKI, DO, KK, FT, SNo,
YS, AS, TY and KS wrote, reviewed and/or revised the manuscript.
SNa, DO, RKI, FT, KK, SNo, AS, TY and KS provided material support.
SNa, RKI, FT, SNo, TY and KS supervised the study. SNa, DO and RKI
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the ethics committee of
Gunma University Faculty of Medicine (approval nos. 1344 and
HS2021-219). Because of the retrospective design of the study, the
requirement for written informed consent from each patient was
waived.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yau R and Rape M: The increasing
complexity of the ubiquitin code. Nat Cell Biol. 18:579–586. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swatek KN and Komander D: Ubiquitin
modifications. Cell Res. 26:399–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grumati P and Dikic I: Ubiquitin signaling
and autophagy. J Biol Chem. 293:5404–5413. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Popovic D, Vucic D and Dikic I:
Ubiquitination in disease pathogenesis and treatment. Nat Med.
20:1242–1253. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sasaki Y, Sano S, Nakahara M, Murata S,
Kometani K, Aiba Y, Sakamoto S, Watanabe Y, Tanaka K, Kurosaki T
and Iwai K: Defective immune responses in mice lacking
LUBAC-mediated linear ubiquitination in B cells. EMBO J.
32:2463–2476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Teh CE, Lalaoui N, Jain R, Policheni AN,
Heinlein M, Alvarez-Diaz S, Sheridan JM, Rieser E, Deuser S,
Darding M, et al: Linear ubiquitin chain assembly complex
coordinates late thymic T-cell differentiation and regulatory
T-cell homeostasis. Nat Commun. 7:133532016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Redecke V, Chaturvedi V, Kuriakose J and
Häcker H: SHARPIN controls the development of regulatory T cells.
Immunology. 148:216–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klein T, Fung SY, Renner F, Blank MA,
Dufour A, Kang S, Bolger-Munro M, Scurll JM, Priatel JJ, Schweigler
P, et al: The paracaspase MALT1 cleaves HOIL1 reducing linear
ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat
Commun. 6:87772015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song Z, Wei W, Xiao W, Al-Saleem ED,
Nejati R, Chen L, Yin J, Fabrizio J, Petrus MN, Waldmann TA and
Yang Y: Essential role of the linear ubiquitin chain assembly
complex and TAK1 kinase in A20 mutant Hodgkin lymphoma. Proc Natl
Acad Sci USA. 117:28980–28991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Y, Schmitz R, Mitala J, Whiting A,
Xiao W, Ceribelli M, Wright GW, Zhao H, Yang Y, Xu W, et al:
Essential role of the linear ubiquitin chain assembly complex in
lymphoma revealed by rare germline polymorphisms. Cancer Discov.
4:480–493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dubois SM, Alexia C, Wu Y, Leclair HM,
Leveau C, Schol E, Fest T, Tarte K, Chen ZJ, Gavard J and Bidère N:
A catalytic-independent role for the LUBAC in NF-κB activation upon
antigen receptor engagement and in lymphoma cells. Blood.
123:2199–2203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jo T, Nishikori M, Kogure Y, Arima H,
Sasaki K, Sasaki Y, Nakagawa T, Iwai F, Momose S, Shiraishi A, et
al: LUBAC accelerates B-cell lymphomagenesis by conferring
resistance to genotoxic stress on B cells. Blood. 136:684–697.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yokobori T, Bao P, Fukuchi M, Altan B,
Ozawa D, Rokudai S, Bai T, Kumakura Y, Honjo H, Hara K, et al:
Nuclear PROX1 is associated with hypoxia-inducible factor 1α
expression and cancer progression in esophageal squamous cell
carcinoma. Ann Surg Oncol. 22 (Suppl 3):S1566–S1573. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sasaki A, Hirato J, Hirose T, Fukuoka K,
Kanemura Y, Hashimoto N, Kodama Y, Ichimura K, Sakamoto H and
Nishikawa R: Review of ependymomas: Assessment of consensus in
pathological diagnosis and correlations with genetic profiles and
outcome. Brain Tumor Pathol. 36:92–101. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tate JG, Bamford S, Jubb HC, Sondka Z,
Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E,
et al: COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 47(D1): D941–D947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tokunaga F, Nakagawa T, Nakahara M, Saeki
Y, Taniguchi M, Sakata S, Tanaka K, Nakano H and Iwai K: SHARPIN is
a component of the NF-κB-activating linear ubiquitin chain assembly
complex. Nature. 471:633–636. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tokunaga F, Sakata SI, Saeki Y, Satomi Y,
Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et
al: Involvement of linear polyubiquitylation of NEMO in NF-kappaB
activation. Nat Cell Biol. 11:123–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujita H, Tokunaga A, Shimizu S, Whiting
AL, Aguilar-Alonso F, Takagi K, Walinda E, Sasaki Y, Shimokawa T,
Mizushima T, et al: Cooperative domain formation by homologous
motifs in HOIL-1L and SHARPIN plays A crucial role in LUBAC
stabilization. Cell Rep. 23:1192–1204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yagi H, Ishimoto K, Hiromoto T, Fujita H,
Mizushima T, Uekusa Y, Yagi-Utsumi M, Kurimoto E, Noda M, Uchiyama
S, et al: A non-canonical UBA-UBL interaction forms the
linear-ubiquitin-chain assembly complex. EMBO Rep. 13:462–468.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karin M: NF-kappaB as a critical link
between inflammation and cancer. Cold Spring Harb Perspect Biol.
1:a0001412009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boisson B, Laplantine E, Dobbs K, Cobat A,
Tarantino N, Hazen M, Lidov HG, Hopkins G, Du L, Belkadi A, et al:
Human HOIP and LUBAC deficiency underlies autoinflammation,
immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med.
212:939–951. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boisson B, Laplantine E, Prando C, Giliani
S, Israelsson E, Xu Z, Abhyankar A, Israël L, Trevejo-Nunez G,
Bogunovic D, et al: Immunodeficiency, autoinflammation and
amylopectinosis in humans with inherited HOIL-1 and LUBAC
deficiency. Nat Immunol. 13:1178–1186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nilsson J, Schoser B, Laforet P, Kalev O,
Lindberg C, Romero NB, Dávila López M, Akman HO, Wahbi K, Iglseder
S, et al: Polyglucosan body myopathy caused by defective ubiquitin
ligase RBCK1. Ann Neurol. 74:914–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oda H, Beck DB, Kuehn HS, Sampaio Moura N,
Hoffmann P, Ibarra M, Stoddard J, Tsai WL, Gutierrez-Cruz G, Gadina
M, et al: Second case of HOIP deficiency expands clinical features
and defines inflammatory transcriptome regulated by LUBAC. Front
Immunol. 10:4792019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimizu Y, Peltzer N, Sevko A, Lafont E,
Sarr A, Draberova H and Walczak H: The linear ubiquitin chain
assembly complex acts as a liver tumor suppressor and inhibits
hepatocyte apoptosis and hepatitis. Hepatology. 65:1963–1978. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pomerantz JL: HOIP for targeting diffuse
large B-cell lymphoma. Blood. 136:646–647. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Awad MM, Oxnard GR, Jackman DM, Savukoski
DO, Hall D, Shivdasani P, Heng JC, Dahlberg SE, Jänne PA, Verma S,
et al: MET exon 14 mutations in non-small-cell lung cancer are
associated with advanced age and stage-dependent MET genomic
amplification and c-Met overexpression. J Clin Oncol. 34:721–730.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frampton GM, Ali SM, Rosenzweig M,
Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D,
et al: Activation of MET via diverse exon 14 splicing alterations
occurs in multiple tumor types and confers clinical sensitivity to
MET inhibitors. Cancer Discov. 5:850–859. 2015. View Article : Google Scholar : PubMed/NCBI
|