Introduction
Primary pulmonary myxoid sarcoma (PPMS) is a rare
malignant tumor arising within the lung, originally described by
Nicholson et al (1) in 1999.
In 2011, Thway et al (2)
introduced the term based on its distinctive genetic profile. A
characteristic feature of PPMS is the EWSR1-CREB1 gene fusion,
which aids in its identification and differentiation from other
sarcomas. The World Health Organization formally recognized PPMS as
a distinct interlobar tumor entity in 2015, marking its
classification among rare pulmonary neoplasms (3). To date, fewer than 40 cases have been
reported to the best of our knowledge (4).
Due to its scarcity, the understanding of PPMS
clinical presentation, biological behavior and optimal management
strategies remains limited. Histologically, PPMS is characterized
by a unique myxoid stroma and spindle-shaped cells, which can
resemble other soft tissue sarcomas and contribute to potential
diagnostic challenges (5). The
clinical presentation of PPMS is typically non-specific, and the
lack of clear symptoms often results in incidental detection
(5). Additionally, the pathogenesis
of PPMS remains poorly understood, with few molecular studies
exploring its underlying genetic alterations (2,6).
However, recent advances, such as the identification of EWSR1 gene
rearrangement through fluorescence in situ hybridization
(FISH) testing, reverse transcription-PCR (RT-PCR) or sequencing,
have begun to shed light on the molecular characteristics of this
tumor.
The current study presents the case of a 41-year-old
male diagnosed with PPMS that was initially suspected to be
pleomorphic adenoma based on frozen section analysis. The diagnosis
was confirmed through histological examination and
immunohistochemical staining, along with FISH analysis revealing
EWSR1 rearrangement. This case highlights the importance of
increasing awareness and advancing diagnostic precision to better
understand and manage this rare tumor.
Case report
A 41-year-old male was admitted to Jeonbuk National
University Hospital (Jeonju, South Korea) in April 2024, following
the incidental discovery of a mass in the left lower lobe during a
routine health check-up. The patient had no significant past
medical history, and his physical examination was otherwise
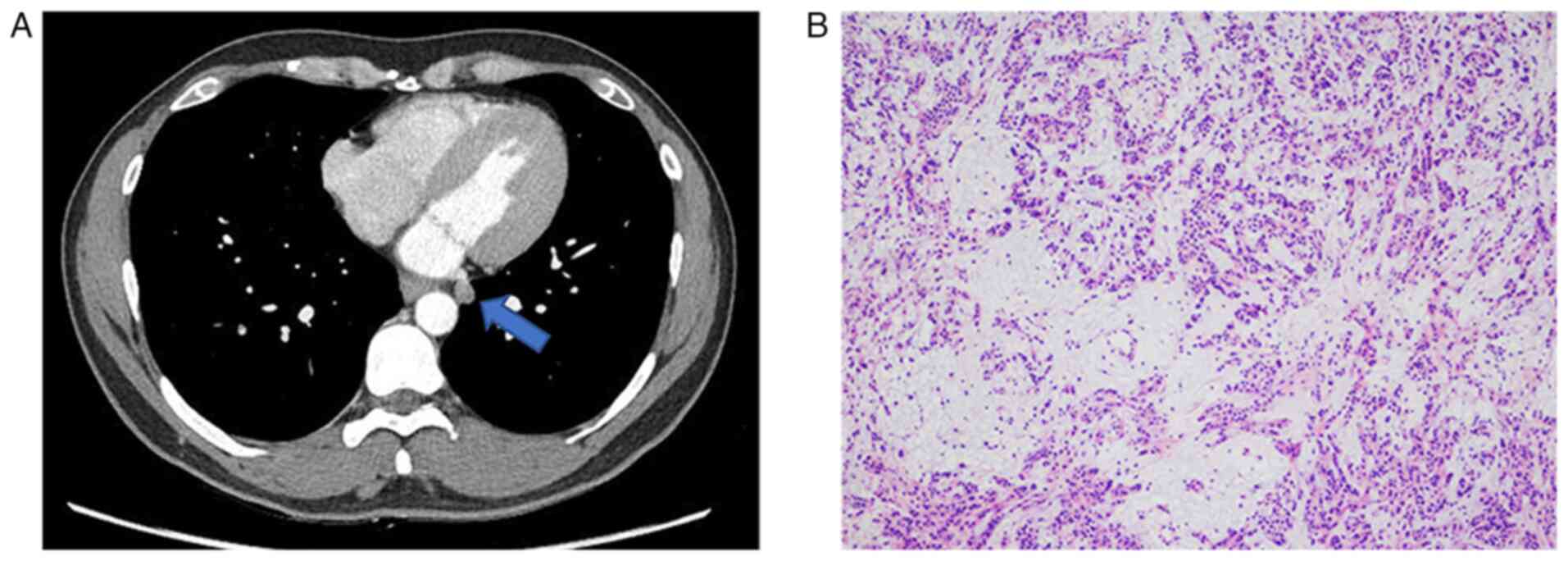
unremarkable. A contrast-enhanced computed tomography (CT) scan
performed at Jeonbuk National University Hospital revealed a small
pulmonary nodule, ~1 cm in size, located in the mediobasal segment
of the left lower lobe (Fig. 1A).
The nodule had relatively well-defined margins, though some areas
appeared irregular, raising suspicion for a potentially malignant
lesion.
Due to its deep-seated location, a percutaneous
transthoracic needle biopsy was considered challenging. Therefore,
to obtain a definitive diagnosis, the patient underwent a wedge
resection of the left lower lobe mass. Intraoperative frozen
section analysis was performed using standard intraoperative
pathological procedures. Tissue samples were rapidly frozen at
−20°C using a cryostat. Sections were cut at a thickness of 5 µm
and mounted on glass slides. Staining was performed with
haematoxylin and eosin (H&E); slides were stained in
haematoxylin at room temperature for 5 min, rinsed in water and
counterstained with eosin at room temperature for 1 min. All
sections were imaged using a light microscope. Frozen section
analysis suggested a high likelihood of pleomorphic adenoma;
however, the final diagnosis was deferred pending permanent
histopathological analysis (Fig.
1B). For permanent histopathological analysis, tissue specimens
were fixed in 10% neutral buffered formalin at room temperature for
24 h. After fixation, samples were processed and embedded in
paraffin. Sections were cut at a thickness of 4 µm using a
microtome and mounted on glass slides.
H&E staining was performed with haematoxylin
staining for 3 minutes at room temperature, followed by eosin
staining for 7 min utes at room temperature. All stained sections
were examined usingnder a light microscope. Examination of the
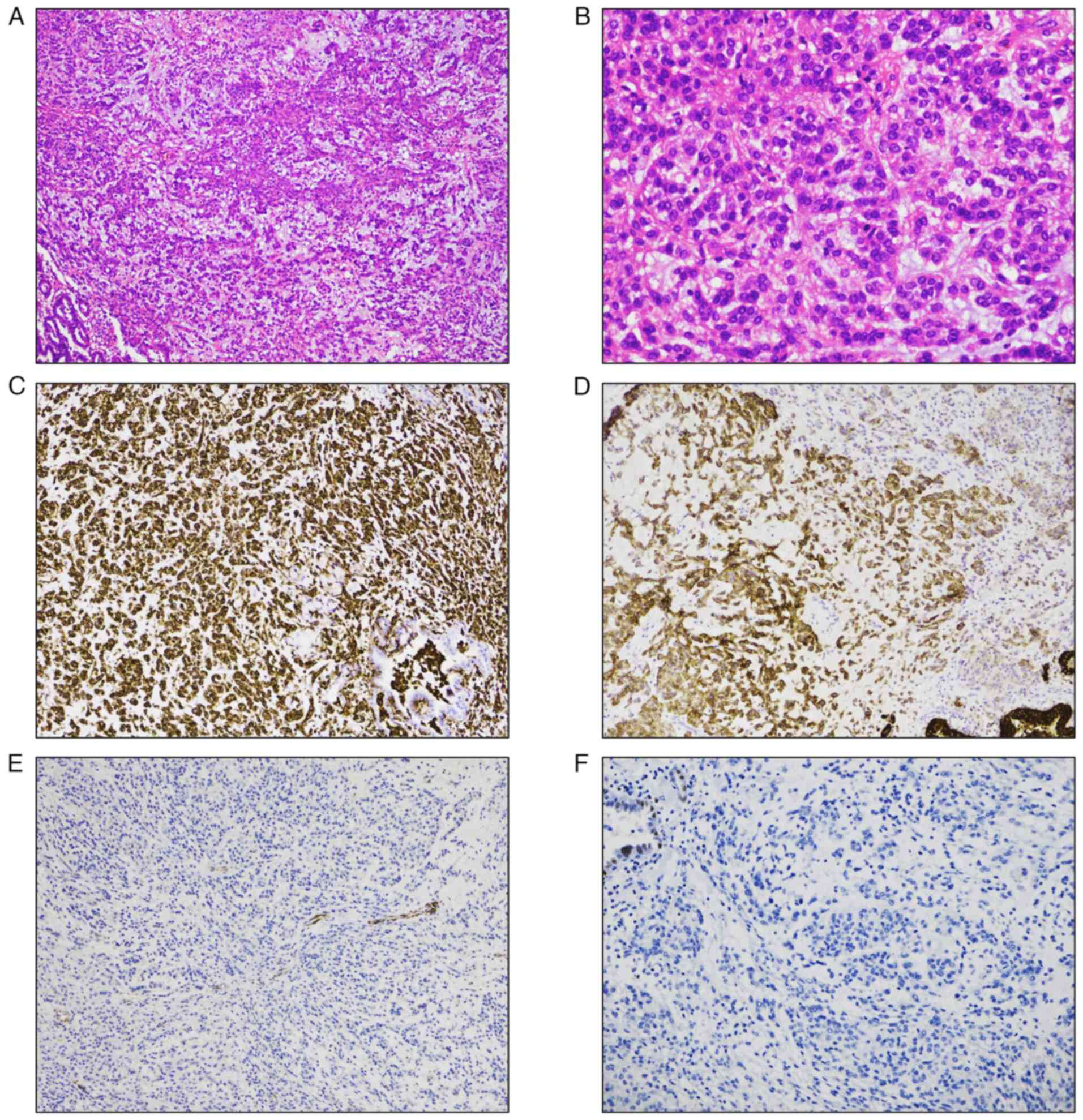
permanent sections revealed distinctive features, including an
abundant myxoid matrix with embedded spindle, stellate and
rounded/epithelioid cells arranged in a reticular pattern,
exhibiting mild cellular atypia and rare mitotic figures. Notably,
giant or bizarre tumor cells, which have been reported in some
previous cases, were not observed in the present case (Fig. 2A and B).
Immunohistochemical (IHC) staining was performed to
further aid in diagnosis. Staining was performed using an automated
immunostainer (BenchMark ULTRA; Ventana Medical Systems, Inc.)
according to the manufacturer's protocol. Tissue sections were
fixed in 10% neutral buffered formalin at room temperature for 24
h, processed routinely, and embedded in paraffin. Sections were cut
at a thickness of 4 µm and mounted on glass slides. All primary
antibodies used were ready-to-use products provided by the
manufacturer (Roche); therefore, no dilution was required. All
staining steps, including deparaffinization, antigen retrieval,
blocking, antibody incubation, and detection, were carried out
automatically under pre-optimized and standardized conditions
according to the manufacturer's instructions.
Hematoxylin was applied as a counterstain for 5
minutes at room temperature. All stained slides were examined using
a light microscope. The tumor cells were positive for vimentin and
epithelial membrane antigen but negative for myoepithelial cell
markers, including calponin, high-molecular-weight cytokeratin and
p63 (Fig. 2C-F). This pattern
effectively ruled out pleomorphic adenoma and other myoepithelial
tumors. Other markers, such as CD68, anaplastic lymphoma kinase
(ALK), insulinoma-associated 1 (INSM1) and synaptophysin were
negative. Given these histological and immunohistochemical
findings, which aligned with characteristics of PPMS, the
possibility of PPMS was considered.
To further confirm the diagnosis, fluorescence in
situ hybridization (FISH) analysis was performed on formalin-fixed,
paraffin-embedded (FFPE) tissue sections. A commercially available
break-apart probe targeting the EWSR1 gene (Vysis LSI
EWSR1 Break Apart Rearrangement Probe; Abbott
Pharmaceutical) was used in accordance with the manufacturer's
protocol. This probe is specifically designed to detect
EWSR1 gene rearrangement but does not identify the specific
fusion partner. All procedures, including hybridization and washing
steps, were carried out in accordance with the manufacturer's
instructions. Fluorescent signals were evaluated using a
fluorescence microscope, and at ≥100 nuclei were assessed by a
trained cytogeneticist. A result was considered positive when
>15% of the examined nuclei demonstrated a split signal pattern,
consistent with EWSR1 rearrangement. In this case, the FISH
analysis demonstrated the presence of an EWSR1 gene
rearrangement. Following this, RNA was extracted from the specimen
to specifically verify the presence of the EWSR1-CREB1 fusion.
Total RNA was extracted from FFPE tumor tissue using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Reverse
transcription was performed using the SuperScript™ IV First-Strand
Synthesis System (Thermo Fisher Scientific, Inc.) following the
manufacturer's standard protocol. PCR amplification was carried out
using primers designed to target exon 7 of EWSR1
(5′-TCCTACAGCCAAGCTCCAAGTC-3′) and exon 7 of CREB1
(5′-GTACCCCATCGGTACCATTGT-3′). The PCR thermocycling conditions
were as follows: initial denaturation at 95°C for 5 min, followed
by 35 cycles of denaturation at 95°C for 30 sec, annealing at 60°C
for 30 sec and extension at 72°C for 30 sec, with a final extension
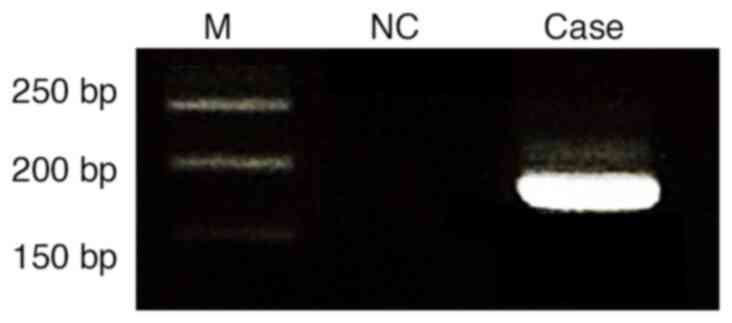
at 72°C for 5 min. PCR products were analyzed by electrophoresis on
a 2% agarose gel stained with ethidium bromide and visualized under
UV illumination. A distinct band corresponding to the
EWSR1-CREB1 fusion transcript was observed (Fig. 3). Based on the histopathological
findings, immunohistochemical profile and molecular analysis, a
final diagnosis of primary pulmonary myxoid sarcoma was
established. The patient did not receive adjuvant therapy
postoperatively and has been monitored with imaging studies every 3
months. As of 10 months after surgery, there is no evidence of
recurrence or metastasis. Due to the rarity of PPMS and its
potential for misdiagnosis, further studies are needed to improve
our understanding of its biological behavior, treatment responses,
and prognostic factors.
Discussion
PPMS is a rare neoplasm, and as such, comprehensive
epidemiological data are limited (5). The tumor most commonly affects
middle-aged adults, with a median age of 49 years (range, 26–80
years) and a slight female predominance (5). Due to its rarity, precise incidence
rates have not been established in population-based studies
(5). The etiology of PPMS remains
unclear, with no definitive causative factors identified. Unlike a
number of other malignancies, PPMS has not been associated with
smoking or linked to known inherited cancer predisposition
syndromes (5). From a prognostic
standpoint, surgical resection remains the mainstay of treatment,
with ~90% of patients remaining disease-free following surgery
(5). Nonetheless, a small subset of
cases demonstrates metastatic progression, with documented spread
to the lung, kidney or brain, and rare instances of lethal outcomes
have been reported (5).
PPMS is a challenging tumor to diagnose, given its
histological overlap with other soft tissue sarcomas and
myoepithelial tumors. The present case exemplifies the diagnostic
complexities associated with PPMS, which often lead to its initial
misidentification. In the present patient, the intraoperative
frozen section analysis suggested pleomorphic adenoma, a
differential diagnosis often considered due to the myxoid nature of
the lesion. However, definitive histopathology,
immunohistochemistry and molecular testing ultimately confirmed the
diagnosis of PPMS. The current case highlights the critical role of
an integrated approach that combines histological,
immunohistochemical and molecular analyses to achieve an accurate
diagnosis.
The diagnostic process in the present case
illustrates the challenges involved in diagnosing PPMS. The initial
frozen section suggested pleomorphic adenoma, a diagnosis that was
ultimately excluded based on the permanent histopathological and
immunohistochemical findings. Histologically, PPMS is characterized
by an abundant myxoid matrix with embedded tumor cells exhibiting
spindle, stellate or rounded/epithelioid morphology, typically
arranged in a reticular or cord-like pattern (5). The tumor cells display mild to
moderate atypia, with low mitotic activity, though variability
exists among cases (5). These
features can closely resemble those of other soft tissue tumors,
making accurate differentiation essential (5). Immunohistochemistry plays a crucial
role in differentiating PPMS from other myoepithelial tumors
(7). In the present case, the tumor
cells stained positive for vimentin, a mesenchymal marker, but were
negative for markers of myoepithelial differentiation, such as
calponin, high-molecular-weight cytokeratin and p63, which helped
rule out pleomorphic adenoma and similar entities.
A key diagnostic feature of PPMS is the EWSR1-CREB1
gene fusion, detectable by FISH or molecular techniques such as
RT-PCR and sequencing (7). In the
current case, the diagnosis was confirmed by identifying the EWSR1
rearrangement through FISH analysis, along with supportive
immunohistochemical staining. Additionally, RT-PCR was conducted
using primers designed to target exon 7 of EWSR1 and exon 7 of
CREB1, and the amplification results confirmed the presence of the
EWSR1-CREB1 fusion, further supporting the diagnosis. This
highlights the crucial role of molecular testing in distinguishing
PPMS from other sarcomas. The EWSR1-CREB1 fusion, although not
entirely specific to PPMS, is highly characteristic and has been
instrumental in defining this rare entity (8). A limitation of the present case report
is the inability to confirm the EWSR1-CREB1 fusion using RNA
sequencing. While the diagnosis was established through FISH and
RT-PCR, RNA-sequencing would have provided a more definitive and
comprehensive molecular validation of the fusion event.
However, several other tumors with similar
histological characteristics also show EWSR1 rearrangements, which
can complicate the diagnosis of PPMS (9,10).
Extraskeletal myxoid chondrosarcoma (EMC) is one tumor with
overlapping features, as it is characterized by multinodular growth
and a rich myxoid matrix, similar to PPMS (11). It contains spindle-shaped to ovoid
cells embedded in cords within the matrix. EMC is characterized by
the presence of an EWSR1-NR4A3 fusion (11). Therefore, testing solely for EWSR1
rearrangement may not reliably distinguish EMC from PPMS.
Immunohistochemical staining can aid in differentiating these two
entities. EMC may show positive staining for neuroendocrine markers
such as INSM1 and synaptophysin, reflecting a neural
differentiation profile not typically seen in PPMS. By contrast,
PPMS generally does not express INSM1 or synaptophysin, which
serves as an important clue in distinguishing it from EMC (12). Angiomatoid fibrous histiocytoma
(AFH) is another tumor with overlapping histological features,
including a myxoid stroma and spindle cell morphology (13). Similar to PPMS, AFH can harbor EWSR1
rearrangements, most commonly resulting in EWSR1-CREB1 or
EWSR1-ATF1 fusions, which can complicate the differential diagnosis
(13). Immunohistochemistry offers
additional diagnostic insight, as AFH often shows positive staining
for markers such as CD68 and ALK, which are typically absent in
PPMS (14). These
immunohistochemical differences provide important clues for
distinguishing AFH from PPMS. In the present case,
immunohistochemical staining demonstrated negative results for
synaptophysin, INSM1, CD68 and ALK. These findings further support
the diagnosis of PPMS.
The treatment of PPMS poses significant challenges
due to its rarity and limited clinical data. Due to the lack of
established treatment guidelines, management is largely based on
case reports and small case series. Surgical resection is generally
considered the primary treatment for PPMS, especially for localized
tumors, as complete resection offers the best chance for prolonged
survival (6). In cases where the
tumor is confined to the lung, lobectomy or wedge resection with
negative margins is often pursued to achieve local control and
reduce the risk of recurrence (4,15). For
patients with inoperable or metastatic PPMS, systemic therapies,
such as chemotherapy, may be considered, although the response
rates in PPMS are not well documented (7). Sarcoma-oriented chemotherapy regimens,
including agents such as doxorubicin and ifosfamide, have been used
in some cases, but their efficacy remains uncertain (2,16). Due
to the slow growth of PPMS in some patients, close surveillance may
be preferred in select cases where aggressive treatment may not
provide significant benefit (6).
Emerging molecular insights into PPMS, particularly the
identification of EWSR1-CREB1 fusions, may pave the way for
targeted therapies in the future (6). While no targeted therapies specific to
PPMS are available, identifying molecular characteristics could
open possibilities for the development of novel therapies aimed at
these fusion proteins. Future research and collaboration among
institutions will be essential to improve understanding of the
biology of PPMS and to establish more standardized treatment
protocols for this rare tumor.
In conclusion, the present case highlights the
critical role of comprehensive diagnostic techniques, including
histological, immunohistochemical and molecular approaches, in
identifying PPMS. Increased awareness of PPMS among clinicians and
pathologists is essential for avoiding misdiagnosis, given its
resemblance to other myxoid neoplasms. Further research and
accumulation of case data are necessary to enhance our
understanding of PPMS and establish standardized treatment
protocols, improving patient outcomes for this rare malignancy.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JHK and KMK conceptualized the study and wrote the
original manuscript. KMK searched the literature and obtained
case-related data. KMK and JHK analyzed data and relevant
literature. KMK reviewed and edited the final draft. KMK and JHK
confirm the authenticity of all the raw data. Both authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of Jeonbuk National University Hospital (IRB no. 2024-09-008)
and was conducted according to the Declaration of Helsinki.
Patient consent for publication
The patient provide written informed consent for the
publication of his images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nicholson AG, Baandrup U, Florio R,
Sheppard MN and Fisher C: Malignant myxoid endobronchial tumour: A
report of two cases with a unique histological pattern.
Histopathology. 35:313–318. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thway K, Nicholson AG, Lawson K, Gonzalez
D, Rice A, Balzer B, Swansbury J, Min T, Thompson L, Adu-Poku K, et
al: Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: A new
tumor entity. Am J Surg Pathol. 35:1722–1732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Travis WD, Brambilla E, Burke AP, Marx A
and Nicholson AG: Introduction to the 2015 World Health
Organization classification of tumors of the lung, pleura, thymus,
and heart. J Thorac Oncol. 10:1240–1242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu T, Wu L, Ye H, Luo S and Wang J:
Primary pulmonary myxoid sarcoma in the interlobar fissure of the
left lung lobe: A case report. BMC Pulm Med. 24:3132024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
World Health Organization (WHO), . WHO
Classification of Tumours Editorial Board Thoracic Tumours.
International Agency for Research on Cancer; Lyon: 2021
|
|
6
|
Chen Z, Yang Y, Chen R, Ng CS and Shi H:
Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: A case
report and review of the literature. Diagn Pathol. 15:1–10. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miao X, Chen J, Yang L and Lu H: Primary
pulmonary myxoid sarcoma with EWSR1:: CREB1 fusion: A literature
review. J Cancer Res Clin Oncol. 150:1082024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koelsche C, Tavernar L, Neumann O, Heußel
CP, Eberhardt R, Winter H, Stenzinger A and Mechtersheimer G:
Primary pulmonary myxoid sarcoma with an unusual gene fusion
between exon 7 of EWSR1 and exon 5 of CREB1. Virchows Arch.
476:787–791. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thway K and Fisher C: Mesenchymal tumors
with EWSR1 gene rearrangements. Surg Pathol Clin. 12:165–190. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Flucke U, van Noesel MM, Siozopoulou V,
Creytens D, Tops BBJ, van Gorp JM and Hiemcke–Jiwa LS: EWSR1-the
most common rearranged gene in soft tissue lesions, which also
occurs in different bone lesions: an updated review. Diagnostics
(Basel). 11:10932021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Finos L, Righi A, Frisoni T, Gambarotti M,
Ghinelli C, Benini S, Vanel D and Picci P: Primary extraskeletal
myxoid chondrosarcoma of bone: Report of three cases and review of
the literature. Pathol Res Pract. 213:461–466. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giner F, López-Guerrero JA, Machado I,
Rubio–Martínez LA, Espino M, Navarro S, Agra–Pujol C, Ferrández A
and Llombart–Bosch A: Extraskeletal myxoid chondrosarcoma: p53 and
Ki-67 offer prognostic value for clinical outcome-an
immunohistochemical and molecular analysis of 31 cases. Virchows
Arch. 482:407–417. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rossi S, Szuhai K, Ijszenga M, Tanke HJ,
Zanatta L, Sciot R, Fletcher CDM, Tos APD and Hogendoorn PCW:
EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous
histiocytoma. Clin Cancer Res. 13:7322–7328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ponmar M, Badrinath T, Ramachandran A,
Kurian JJ, Gaikwad P, Thomas BP, Kandagaddala M and Prabhu AJ: Case
series of angiomatoid fibrous histiocytoma (AFH)-A
clinico-radiological and pathological conundrum. Indian J Surg
Oncol. 16:1–12. 2024.PubMed/NCBI
|
|
15
|
Hutchings H, Schwarze E, Ahsan B, Cox J
and Okereke I: Primary pulmonary myxoid sarcoma: A case report and
review of the literature. Glob J Perioper Med. 7:1–3. 2023.
View Article : Google Scholar
|
|
16
|
Niu SY, Sun L, Hsu ST, Hwang SF, Liu CK,
Shih YH, Lu TF, Chen YF, Lai LC, Chang PL and Lu CH: Efficacy and
toxicities of doxorubicin plus ifosfamide in the second-line
treatment of uterine leiomyosarcoma. Front Oncol. 13:12825962023.
View Article : Google Scholar : PubMed/NCBI
|