Introduction
Prostate cancer (Pca) is the most common malignancy
affecting males and is the second leading cause of cancer-related
death among males in the US (1).
One consistent characteristic of this type of cancer is the
eventual progression to a hormonal refractory state. The
development of effective novel therapeutic strategies requires an
understanding of the mechanisms for the development of such a
refractory state. Targeting proliferative and survival pathways
provides a rationale for drug design and development for human
hormone refractory prostate cancer (HRPC). Prostate cancer cells
develop an enhanced redundancy in downstream survival signaling,
which is very important for the development and progression of HRPC
(2). Cyclooxygenase (COX)-2 is an
inducible enzyme stimulated by cytokines, growth factors, oncogenes
or tumor promoters during inflammation and malignancy. An increased
COX-2 expression is associated with decreased apoptosis, increased
tumor invasiveness, immunosuppression and angiogenesis.
Furthermore, an increased COX-2 expression correlates with poor
differentiation, increased tumor size, increased nodal and distant
disease and decreased patient survival in a variety of cancers
(3–7). The endothelin (ET) family is composed
of three isopeptides, ET-1, −2 and −3, which are potent mitogens
for several types of human tumors, including Pca. ET-1 and their
receptors are implicated in tumor progression through autocrine and
paracrine pathways (8,9).
ET-1 plays an important role in modulating COX-2
expression in various types of normal cells (10–13),
but the precise molecular mechanisms controlling these effects
remain undefined. Spinella et al reported that ET-1 appears
to lead to an increased COX-2 expression in human ovarian carcinoma
cells (14). However, the role of
ET-1 in the regulation of COX-2 in human HRPC cells has yet to be
investigated. The present study examined whether activation of the
endothelin A receptor (ETAR)/endothelin B receptor
(ETBR) by ET-1 leads to the up-regulation of COX-2
expression. Possible molecular mechanisms in the human PC3 cell
line were also investigated.
Materials and methods
Cell culture
The PC3 cell line (American Type Culture Collection,
Rockville, MD, USA) was cultured in F12 medium containing 10% fetal
bovine serum. The cells were serum-starved by incubation for 24 h
in serum-free F12 medium. Culture reagents were from Invitrogen
(Paisley, Scotland, UK). ET-1 (Merck, Darmstadt, Germany),
dissolved in deionized water, was added to the cell medium at the
indicated concentration and for the indicated time. BQ123
(ETAR antagonist) (1 μmol/l), BQ788 (ETBR
antagonist) (1 μmol/l), PD98059 (selective MEK inhibitor) (10
μmol/l), p38 SB203580 (p38 MAPK inhibitor) (5 μmol/l) and AG1478
[epidermal growth factor receptor (EGFR) antagonist) (0.1 μmol/l]
(Sigma, St. Louis, MO, USA) were all dissolved in 1% dimethyl
sulfoxide (DMSO). After their effects were studied, they were added
to the medium for 24 h with or without treatment with ET-1 (100
nmol/l). To remove any possible effect of the solvent DMSO on the
cells, the control group also contained 1% DMSO. Experiments were
repeated at least three times.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA from PC3 cells was extracted using TRIzol
reagent (Invitrogen Life Technologies, Burlington, Ontario,
Canada), according to the manufacturer’s instructions. The quality
of the RNA was verified by agarose gel electrophoresis using
ethidium bromide staining. For each PCR, 2 μg DNA-free total RNA
with oligo (deoxythymidine) primers and reverse transcriptase were
used. PCR was performed in 50-μl reactions containing 2.5 ng of
cDNA, 1 μl of each primer pair and 25 μl of Premix Taq (Takara,
Shiga, Japan). PCR was carried out in a T-gradient Biometra PCR
thermal cycler (Montreal Biotech Inc., Kirkland, Quebec, Canada) to
determine the annealing temperature for each set of paired primers.
The COX-2 primer pairs used were: 5′-CGAGGTGTATGTATGAGTGTG-3′
(forward) and 5′-TCTAGCCAGAGTTTCACCGTA-3′ (reverse), with the
length of the product being 582 bp. Thirty cycles of amplification
were performed under the following conditions: melting at 94°C for
30 sec, annealing at 55.5°C for 30 sec and extension at 72°C for 1
min. The PCR products were analyzed by electrophoresis on a 1%
agarose gel. Controls involved omitting reverse transcriptase, cDNA
or DNA polymerase and showed no reaction bands. Data were
normalized by β-actin RNA.
Western blot analysis
The PC3 cells were homogenized in a lysis buffer
containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 10 mM
NaF, 1 mM sodium orthovanadate, 1% Triton X-100, 0.5% sodium
deoxycholate, 1 mM phenylmethylsulfonyl fluoride and protease
inhibitor cocktail (Complete; Roche, Mannheim, Germany). The lysate
was then centrifuged at 12,000 × g for 20 min at 4°C. The total
protein concentration of each sample was analyzed using the BCA
Protein Assay kit (Pierce, Rockford, IL, USA). Equal amounts (40
μg) of protein were resolved by 5 and 10% SDS-PAGE and transferred
onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA).
Following transfer, membranes were blocked with 5% fat-free milk in
Tris-buffered saline plus 0.05% Tween-20 overnight at 4°C. The
membranes were then incubated with the primary antibody (goat
polyclonal COX-2 antibodies, diluted 1:500; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) for 2 h at room temperature.
After being washed in TBST (Tris-buffered saline Tween-ZO) three
times, the membranes were incubated with the peroxidase-linked
rabbit anti-goat IgG conjugates (Santa Cruz Biotechnology) for 1 h
at room temperature. Finally, they were washed again in TBST,
incubated in enhanced chemiluminescence reagents (Pierce) for 2
min, and exposed to X-Omat BT film (Eastman Kodak, Rochester, NY,
USA). The level of β-actin expression was used as the internal
control for equal loading. The Western blotting bands were scanned
and analyzed with a Bio-Rad image analysis system. For negative
controls, the primary antibody was omitted.
Statistical analysis
Most of the summarized densitometric data represent
the average from at least three experiments. Data were retrieved
and processed with the software SPSS13.0. In the experimental
section dealing with the ET-1 stimulation of COX-2 expression,
one-way ANOVA and the unpaired SNK-q-test were used when the
adjacent groups were compared. In the experimental section
involving the antagonists for each signaling pathway, one-way ANOVA
and the LSD test were used when the control and the remaining
groups were compared. Data were expressed as the means ± standard
error of means (SEM). P<0.05 was considered statistically
significant.
Results
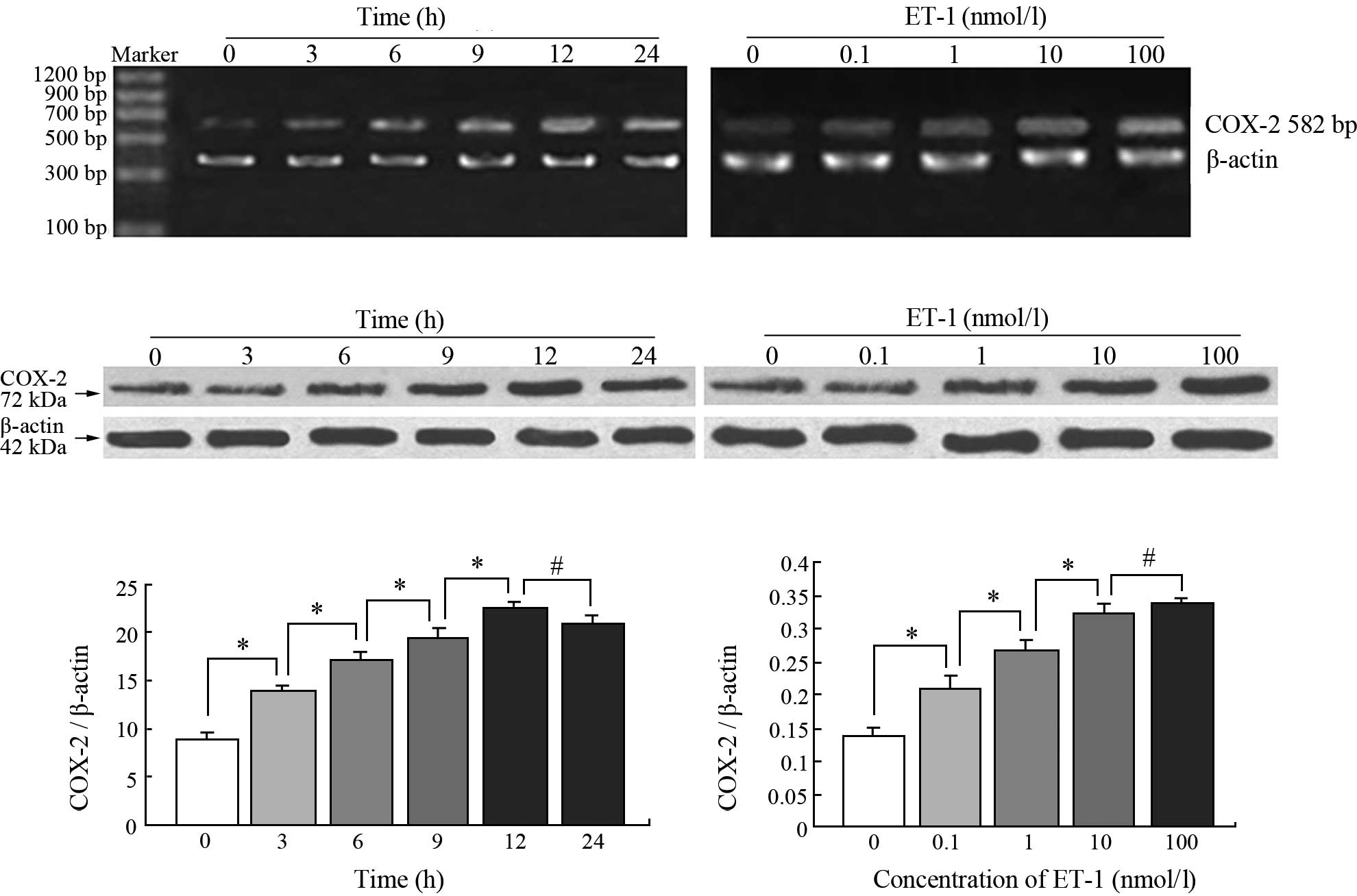
ET-1 stimulates COX-2 expression
The premise of whether ET-1 regulates COX-2
expression in the PC3 cell line was initially investigated. ET-1
markedly induced the time-dependent up-regulation of COX-2 mRNA in
the PC3 cells. RT-PCR analysis showed that the COX-2 mRNA levels
increased in the ET-1-treated cells compared with the control group
by 2-, 2.3-, 2.6-, 3- and 2.9-fold at 3, 6, 9, 12 and 24 h,
respectively (Fig. 1A). Moreover,
ET-1 treatment evoked a time-dependent increase in COX-2 protein
levels. Western blot analysis showed a low expression of COX-2
protein in the untreated PC3 cells, but a 1.5-fold increase after 3
h, and a 1.9-, 2.1-, 2.5- and 2.3-fold increase after 6, 9, 12 and
24 h of ET-1 stimulation, respectively (Fig. 1C and E). ET-1 also increased COX-2
mRNA and protein levels in a dose-dependent manner. Treatment of
PC3 cells with 0.1 and 1 nM ET-1 for 24 h showed 1.5 and 2-fold
increases in the COX-2 protein expression, respectively, which
reached maximum responses at 100 nM ET-1 (Fig. 1D). COX-2 mRNA levels increased
1.5-fold at 0.1 nM ET-1 and then increased gradually. The highest
level of COX-2 mRNA (2.4-fold compared with the control) was also
detected at 100 nM ET-1 (Fig. 1B and
F).
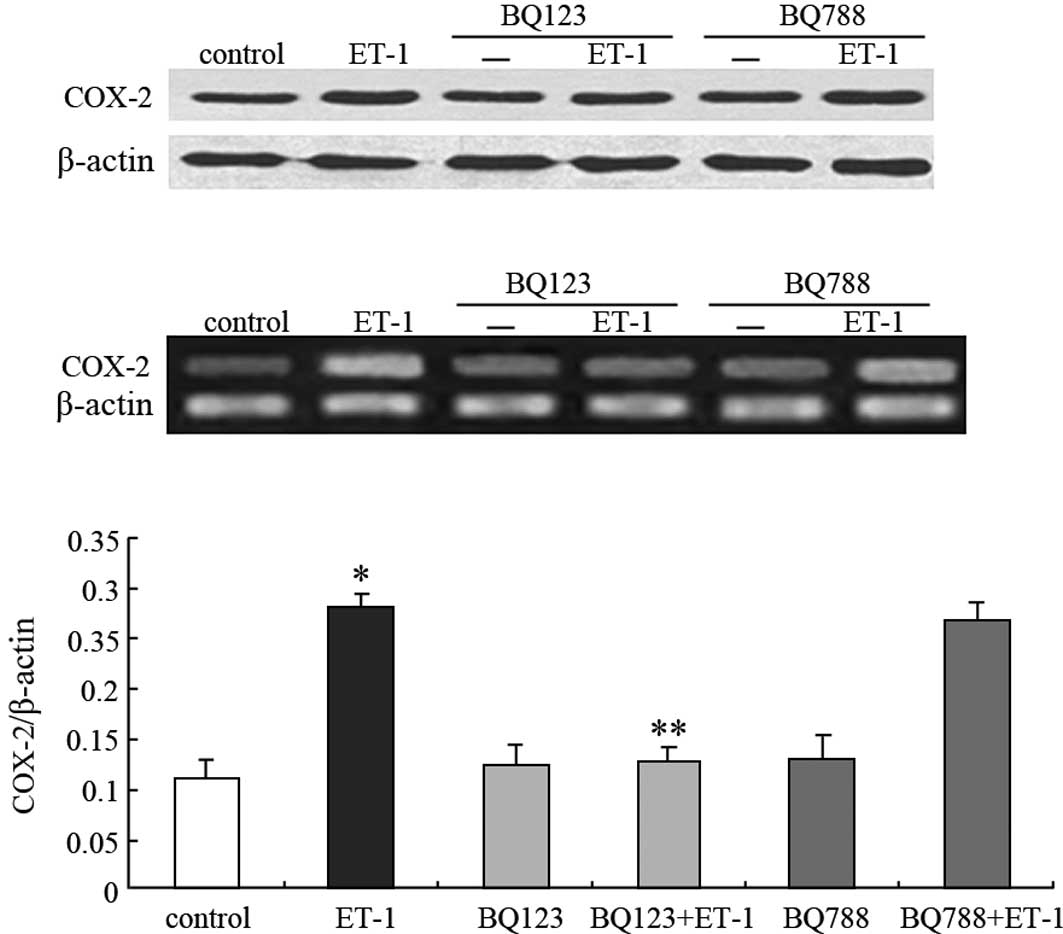
ET-1-induced COX-2 up-regulation is
mediated through ETAR
To investigate which receptor subtype mediates the
ET-1-induced up-regulation of COX-2 expression, selective
ETAR and ETBR antagonists, BQ123 and BQ788,
respectively, were used in the presence or absence of 100 nM ET-1.
As shown in Fig. 2B and C, BQ123
was able to completely block ET-1-induced COX-2 mRNA expression
(46% for ET-1, P<0.05; 118% for control, P>0.05), whereas
BQ788 did not (96% for ET-1, P>0.05; 248% for control,
P<0.05). This result was similar to that of the COX-2 protein
expression (Fig. 2A). Taken
together, these findings indicate that ET-1 acts through
ETAR to stimulate COX-2 expression in PC3 cells.
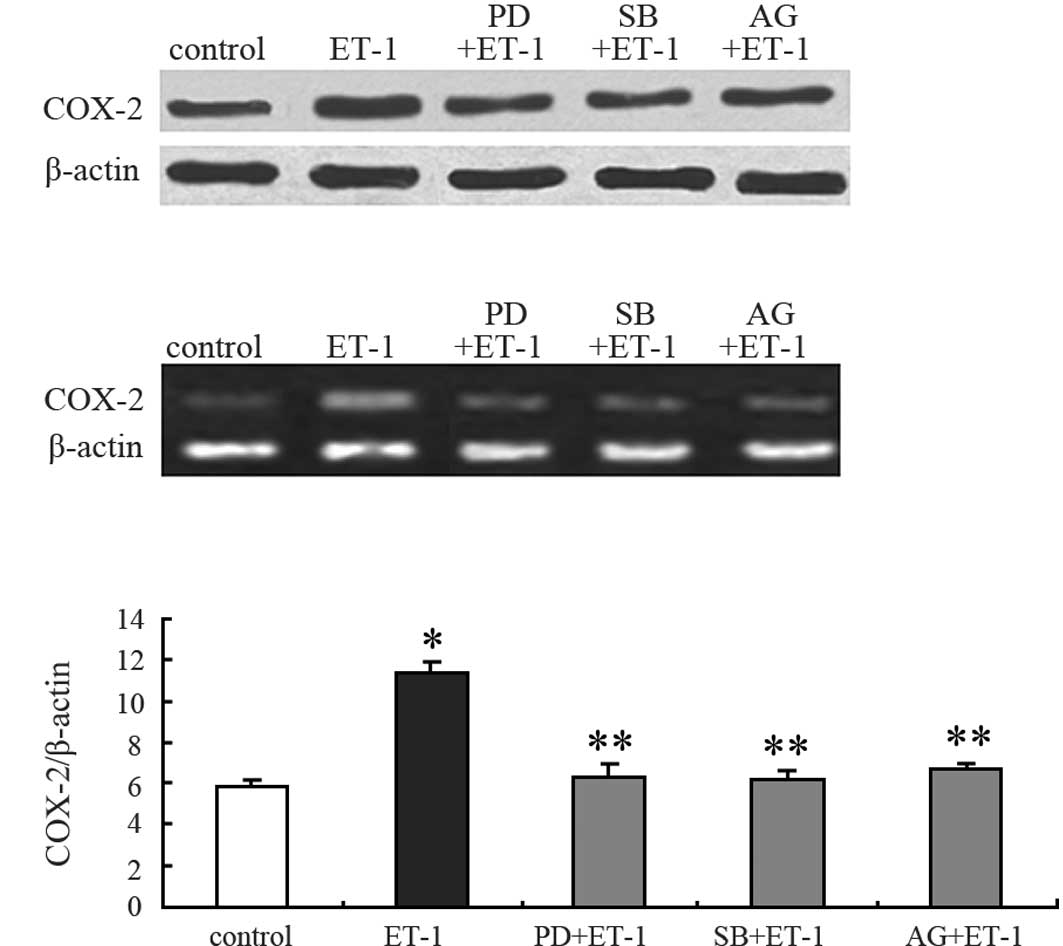
Signaling pathways are involved in
ET-1-stimulated COX-2 expression
To investigate the signaling pathways involved in
ET-1-induced COX-2 expression, 100 nmol/l ET-1 were added with
PD98059 (selective MEK inhibitor), SB20358 (p38 MAPK antagonist),
or AG1478 (specific EGFR antagonist) into the medium for 24 h. The
cell extracts were then analyzed for COX-2 expression by Western
blotting and RT-PCR. PD98059 (10 μM) and SB203580 (5 μM), which did
not affect the COX-2 protein basal levels, markedly inhibited
ET-1-stimulated COX-2 protein expression (Fig. 3A and C). Among downstream events
after ETAR activation, ET-1 resulted in the
transactivation of EGFR. Thus, the effect of AG1478 on
ETAR-mediated effects was examined. Treatment of PC3
cells with AG1478 (0.1 μM) markedly inhibited ET-1-induced COX-2
protein production (Fig. 3A and C),
indicating an involvement of EGFR in this mechanism. The trend of
COX-2 mRNA expression was similar to that of the protein expression
(Fig. 3B; data not shown). These
findings indicate that ET-1 acts through ETAR to induce
COX-2 production in PC3 cells and suggest that the transactivation
of EGFR, the activation of p38 MAPK-dependent and p42/44
MAPK-dependent pathways are involved in these mechanisms.
Discussion
Chemical carcinogenesis experiments and
epidemiological and clinical studies have collectively identified
COX-2 as an important molecule involved in the onset and
progression of a variety of malignancies (15). The development of selective
inhibitors of COX-2 clearly adds a novel potential pharmacological
target to cancer prevention and treatment. Subsequently, studies
aimed at identifying the metabolic pathways involved in COX-2
induction are significant from a biological as well as a clinical
point of view.
The ET-1/ETAR autocrine pathway plays a
key role in the development and progression of prostatic, ovarian
and cervical carcinomas (16).
Research has demonstrated that ET-1 increases the expression of
COX-2 in several cell types, such as human pulmonary epithelial
cells, human ovarian carcinoma cells, endothelial cells, mesangial
cells and macrophages (14,17–20).
As with previous observations, we found that ET-1 markedly induced
a time- and dose-dependent up-regulation of the COX-2 mRNA and
protein expression in PC3 cells. Notably, although COX-2 expression
reached a maximum response following treatment with 100 nM ET-1 for
24 h, no obvious differences were noted between the cell groups
treated with 10 and 100 nM ET-1. Thus, we determined that when ET-1
achieves a quantitative concentration, the combination of ET-1
ligands and their receptors reaches a saturation point, and the
dose-dependent relationship no longer exists.
ET-1 is known to activate the p42/44 MAPK pathway
through ETAR in ovarian carcinoma cell lines (21). Moreover, the inhibition of human
ovarian tumor growth in nude mice after treatment with the potent
ETAR-selective antagonist ABT-627 is associated with a
reduced COX-2 and vascular endothelial growth factor expression
(15). Therefore, we analyzed
whether these pathways are involved in ET-1-induced COX-2
expression in PC3 cells and found that the addition of a specific
ETAR antagonist, BQ123, blocked the ET-1-induced COX-2
expression. This finding showed that ETAR is a key
factor in the up-regulation of COX-2 by ET-1. Spinella et al
(22) and Rosanò et al
(23) previously demonstrated that
using the highly specific antagonist ABT-627, the in vivo
blockade of the ETAR autocrine pathway is associated
with an obvious reduction in microvessel density, VEGF expression,
matrix metalloproteinase-2, connexin 43 phosphorylation and
increased tumor apoptosis. This reduction indicates that the
anti-tumoral activity of this small molecule may also be due to the
inhibition of COX-2 activity (22,23).
Several signaling pathways, including p38 and p42/44
MAPK, have been implicated in the regulation of COX-2 expression
(24). ET-1 regulates COX-2
expression through p38 and p42/44 MAPK in vascular smooth muscle
cells (13) and through p42/44 MAPK
in osteoblast-like cells (11). The
activation of p38 MAPK was found to be involved in ET-1-stimulated
COX-2 expression in cultured feline esophageal smooth muscle cells
(25). Chen et al reported
that ET-1 treatment results in an increase in the phosphorylation
of both p38 and p42/44 MAPKs in peripheral lung microvascular
smooth muscle cells (13). Our
study demonstrated that the MEK pathway inhibitor, PD98059, as well
as the p38 MAPK inhibitor, SB203580, block the ET-1-induced COX-2
expression, indicating that ET-1-mediated effects are likely to be
dependent on the MAPK pathway. Moreover, the ET-1-induced COX-2
expression requires the ligand-independent activation of EGFR, as
demonstrated by the inhibitory effect exerted by the EGFR tyrosine
kinase inhibitor, AG1478, indicating that ET-1-induced effects are
also mediated by EGFR transactivation. Consistent with this
finding, Guo et al (24)
showed that, similar to ET-1, gastrin, another G protein-coupled
receptor agonist, stimulates COX-2 expression through multiple
signaling pathways, including EGFR transactivation in intestinal
epithelial cells, thus identifying a mechanism involved in the
initiation and progression of colorectal cancer.
Our study for the first time demonstrated that the
transactivation of the EGFR, p38 MAPK-dependent and p42/44
MAPK-dependent pathways are involved in the
ETAR-mediated regulation of COX-2 expression in PC3
cells. Although much research has been conducted in vitro,
the exact roles and the mechanisms of ET-1 and COX-2 in vivo
remain to be elucidated. However, blocking the activity of ET-1 and
COX-2 may have relevant implications in the prevention and
treatment of Pca in the future.
Acknowledgements
This study was supported by the Jiangsu Province Key
Laboratory of Human Functional Genomics (HFG007) and Nanjing
Medical University (09NJMUM074).
References
|
1
|
Nelson WG, De Marzo AM and Isaacs WB:
Prostate cancer. N Engl J Med. 349:366–381. 2003. View Article : Google Scholar
|
|
2
|
Uzgare AR and Isaacs JT: Prostate cancer:
potential targets of anti-proliferative and apoptotic signaling
pathways. Int J Biochem Cell Biol. 37:707–714. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuura H, Sakaue M, Subbaramaiah K, et
al: Regulation of cyclooxygenase-2 by interferon gamma and
transforming growth factor alpha in normal human epidermal
keratinocytes and squamous carcinoma cells: role of
mitogen-activated protein kinases. J Biol Chem. 274:29138–29148.
1999. View Article : Google Scholar
|
|
4
|
Ohneseit PA, Krebiehl G, Dittmann K,
Kehlbach R and Rodemann HP: Inhibition of cyclooxygenase-2 activity
by celecoxib does not lead to radiosensitization of human prostate
cancer cells in vitro. Radiother Oncol. 82:229–238. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jia RP, Xu LW, Su Q, Zhao JH, Li WC, Wang
F and Xu Z: Cyclooxygenase-2 expression is dependent upon epidermal
growth factor receptor expression or activation in androgen
independent prostate cancer. Asian J Androl. 10:758–764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saha D, Datta PK, Sheng H, Morrow JD, Wada
M, Moses HL and Beauchamp RD: Synergistic induction of
cyclooxygenase-2 by transforming growth factor-beta1 and epidermal
growth factor inhibits apoptosis in epithelial cells. Neoplasia.
1:508–517. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neeraja S, Ramakrishna B, Sreenath AS,
Reddy GV, Reddy PR and Reddanna P: Novel functional association of
rat testicular membrane-associated cytosolic glutathione S
transferases and cyclooxygenase in vitro. Asian J Androl.
7:171–178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nelson JB, Hedican SP, George DJ, Reddi
AH, Piantadosi S, Eisenberger MA and Simons JW: Identification of
endothelin-1 in the pathophysiology of metastatic adenocarcinoma of
the prostate. Nat Med. 1:994–999. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bagnato A, Rosanò L, Di Castro V, Albini
A, Salani D, Varmi M, Nicotra MR and Natali PG: Endothelin receptor
blockade inhibits proliferation of Kaposi’s sarcoma cells. Am J
Pathol. 158:841–847. 2001.
|
|
10
|
Pratt PF, Bokemeyer D, Foschi M, Sorokin A
and Dunn MJ: Alterations in subcellular localization of p38 MAPK
potentiates endothelin-stimulated COX-2 expression in glomerular
mesangial cells. J Biol Chem. 278:51928–51936. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Windischhofer W, Zach D, Fauler G,
Raspotnig G, Köfeler H and Leis HJ: Involvement of Rho and p38 MAPK
in endothelin-1-induced expression of PGHS-2 mRNA in
osteoblast-like cells. J Bone Miner Res. 17:1774–1784. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rebsamen MC, Capoccia R, Vallotton MB and
Lang U: Role of cyclooxygenase 2, p38 and p42/44 MAPK in the
secretion of prostacyclin induced by epidermal growth factor,
endothelin-1 and angiotensin II in rat ventricular cardiomyocytes.
J Mol Cell Cardiol. 35:81–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen D, Balyakina EV, Lawrence M,
Christman BW and Meyrick B: Cyclooxygenase is regulated by ET-1 and
MAPKs in peripheral lung microvascular smooth muscle cells. Am J
Physiol Lung Cell Mol Physiol. 284:L614–L621. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spinella F, Rosanò L, Di Castro V, Nicotra
MR, Natali PG and Bagnato A: Inhibition of cyclooxygenase-1 and −2
expression by targeting the endothelin A receptor in human ovarian
carcinoma cells. Clinical Cancer Res. 10:4670–4679. 2004.
|
|
15
|
Dannenberg AJ and Subbaramaiah K:
Targeting cyclooxygenase-2 in human neoplasia: rationale and
promise. Cancer Cell. 4:431–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nelson JB, Bagnato A, Battistini B and
Nisen P: The endothelin axis: emerging role in cancer. Nat Rev
Cancer. 3:110–116. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng H, Chen P, Cai Y, Chen Y, Wu QH, Li
Y, Zhou R and Fang X: Endothelin-1 increases expression of
cyclooxygenase-2 and production of interleukin-8 in hunan pulmonary
epithelial cells. Peptides. 29:419–424. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sugiyama T, Yoshimoto T, Sato R, Fukai N,
Ozawa N, Shichiri M and Hirata Y: Endothelin-1 induces
cyclooxygenase-2 expression and generation of reactive oxygen
species in endothelial cells. J Cardiovasc Pharmacol. 44(Suppl 1):
332–335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hughes AK, Padilla E, Kutchera WA, Michael
JR and Kohan DE: Endothelin-1 induction of cyclooxygenase-2
expression in rat mesangial cells. Kidney Int. 47:53–61. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimada K, Yonetani Y, Kita T, Suzumura A,
Takayanagi T and Nakashima T: Cyclooxygenase-2 expression by
endothelin-1-stimulated mouse resident peritoneal macrophages in
vitro. Eur J Pharmacol. 356:73–80. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bagnato A, Tecce R, Di Castro V and Catt
KJ: Activation of mitogenic signaling by endothelin 1 in ovarian
carcinoma cells. Cancer Res. 57:1306–1311. 1997.PubMed/NCBI
|
|
22
|
Spinella F, Rosanò L, Di Castro V, Nicotra
MR, Natali PG and Bagnato A: Endothelin-1 decreases gap junctional
intercellular communication by inducing phosphorylation of connexin
43 in human ovarian carcinoma cells. J Biol Chem. 278:41294–41301.
2003. View Article : Google Scholar
|
|
23
|
Rosanò L, Di Castro V, Spinella F, et al:
Therapeutic targeting of the endothelin A receptor in human ovarian
carcinoma. Cancer Res. 63:2447–2453. 2003.
|
|
24
|
Guo YS, Cheng JZ, Jin GF, Gutkind JS,
Hellmich MR and Townsend CM Jr: Gastrin stimulates cyclooxygenase-2
expression in intestinal epithelial cells through multiple
signaling pathways. Evidence for involvement of ERK5 kinase and
transactivation of the epidermal growth factor receptor. J Biol
Chem. 277:48755–48763. 2002. View Article : Google Scholar
|
|
25
|
Song HJ, Min YS, Shin CY, Jeong JH and
Sohn UD: Activation of p38 MAPK is involved in
endothelin-1-stimulated COX-2 expression in cultured feline
esophageal smooth muscle cells. Mol Cells. 22:44–50.
2006.PubMed/NCBI
|