Introduction
Ovarian cancer is the most frequent cause of death
from gynaecological cancer in western countries (1). At diagnosis, epithelial ovarian cancer
is currently treated by cytoreductive surgery followed by
platinum-based chemotherapy. While treatment of ovarian cancer with
platinum-based agents has been established for decades, these still
remain the most active substances for this entity (2). Accordingly, primary resistance to
platinum-based adjuvant therapy is associated with a worse
disease-free and overall survival (3). However, virtually all patients
eventually develop secondary resistance to platinum based agents
and compounds used for second or third line treatment display
substantially less anti-cancer activity as compared to platinum
(4). Thus, there is a definite need
for new treatment strategies which overcome platinum-resistance.
This would have an immediate impact on the clinical management of
patients with advanced ovarian cancer. Accordingly, a better
understanding of mechanisms responsible for platinum-resistance is
of paramount importance.
The serine/threonine kinase AKT/PKB-pathway is a
promising target for cancer therapy, as it is a main nodal point
where extracellular and intracellular oncogenic signals are
integrated. Alterations of the AKT-pathway have been detected in
several human malignancies including ovarian cancer (5). AKT has a broad range of downstream
effectors that regulate cell processes such as cell growth, cell
cycle progression, survival, migration, and angiogenesis (6). Due to the key role of AKT in malignant
transformation numerous inhibitors of the AKT-pathway have been
developed, and are currently in various stages of clinical
development (7).
In human specimens of ovarian cancer AKT was found
to be activated in 68% (5) and
PI3K, an upstream component of the AKT-pathway, was found to be
mutated in 12% of the cases (8).
Recent evidence by our group and others has shown that
overactivation of the AKT-pathway may be associated with
platinum-resistance (9–12). The present study evaluates the
effect of overexpression and downregulation of AKT on sensitivity
to cisplatin in a platinum-sensitive parental human ovarian cancer
cell line and the corresponding platinum-resistant cell line.
Materials and methods
Plasmid construction
The plasmid pFLAG-CMV-2-human-WT-Akt1 (13,14)
was kindly provided by Dr Naoya Fujita. The DNA-sequence coding for
human Akt-1 was amplified by PCR using pFLAG-CMV-2-human-WT-Akt1 as
template with the following primers: 5′-CGGTGGGAGGATCCTATAAGC
AGAGC-3′ and 5′-CCATCCCTCCAAGATATCGTC-3′. The resulting PCR product
was purified with QIAquick PCR purification kit (Quiagen, Hilden,
Germany) and than digested with BamHI and EcoRV (New
England Biolabs, Frankfurt, Germany). The Akt-1 coding sequence was
cloned into BamHI and EcoRV digested pcDNA(+) and
pcDNA(−) (Invitrogen, Karlsruhe, Germany), respectively, resulting
in plasmids coding for the Akt-1 sense (AKT+) or
antisense (AKT−) cDNA. For verification the resulting
plasmids were sequenced (Entelechon, Regensburg, Germany).
Cell culture and stable transfection
A2780 and A2780cis cell lines (both are p53 and KRAS
wild-type cell lines) were obtained from ECACC (Salisbury, UK). The
cisplatin-resistant A2780cis cell line has been developed by
chronic exposure of the parental cisplatin-sensitive A2780 cell
line to increasing concentrations of cisplatin (15). Cells were cultured in RPMI,
supplemented with 10% FCS, 100 U per ml penicillin G, and 100 mg/ml
streptomycin (Invitrogen). Transfection of A2780 and A2780cis cells
was performed with TransIT-LT1 (Mirus, Madison, WI, USA) according
to the manufacturer′s instructions. Briefly, 24 h after seeding
cells in 6-well plates the cells were washed with PBS and 1000 μl
serum-free medium was added to each well. Then, in each well a
mixture of 100 μl serum-free medium, 3 μl transfection reagent and
1 μg plasmid was added. After 6-h transfection was stopped by
replacing the transfection medium with serum-containing medium.
Forty-eight hours after transfection selection of transfected cells
was started with 200 μg G-418/ml (Invitrogen).
Preparation of cell lysates and western
blotting
Preparation of cell lysates was performed as
previously described (16).
Membranes were probed overnight with: anti-phospho-Akt (no. 2118),
antibody from Epitomics (Burlingame, CA, USA). The secondary
antibody horseradish peroxidase (HRP)-conjugated anti-rabbit IgG
was from Cell Signaling (Frankfurt, Germany). The chemiluminescent
HRP substrate solution (Millipore, Schwalbach, Germany) was used
for detection.
MTT assay
To quantify the cytotoxicity of
cis-diamminedichloroplatinum(II) (cisplatin) viability of
cells was measured with a non-radioactive cell counting assay as
previously described (17).
Experiments were performed 6-fold, and at least three independent
experiments were performed for each cell line.
Clonogenicity assay
Cells were cultured in 96-well flat-bottom plates,
in humidified 37°C and 5% CO2 atmosphere. Cell density
was initially adjusted to 2.5×103 cells/ml in a final
volume of 200 μl/well. Twenty-four hours after seeding cells were
treated with different concentrations of cisplatin as indicated and
the 96-well plates were incubated for 7 days. Then the supernatant
was discarded and the cells were washed with PBS before 50 μl
crystal violet solution [0.5% (w/v) in methanol (Roth, Karlsruhe,
Germany)] was added in each well. Plates were incubated under
shaking for 10 min at room temperature. After washing three times
with tap water the plates were air dried at room temperature.
Formed cell colonies were counted under a microscope (Leica DMIL,
Wetzla, Germany). Three independent experiments were performed, and
each experiment was carried out in triplicate.
Flow cytometry
For cell cycle analysis, cells were treated with
cisplatin as indicated, harvested, fixed and permeabilized
overnight in ice-cold 70% ethanol (Merck, Darmstadt, Germany). The
cells were washed twice with PBS. RNA was digested with RNase A
(Invitrogen). DNA was stained with PI (50 μg/ml). Fluorescence was
recorded in a FACSCalibur (Becton-Dickinson, Heidelberg, Germany).
Instrument settings were adjusted to move the
G0/G1 peak to 200 relative fluorescence
units. Cells to the left of this peak have a DNA content <2n,
indicative of cell death. Aggregated cells were gated out. A total
of 20,000 events per condition were recorded.
Results
Generation of cell lines with stable
AKT-overexpression and downregulation
Four stable ovarian cell cultures were established
by transfection of platinum-resistant A2780cis and parental A2780
cells with either an AKT-1 or an AKT-1-antisense expression vector.
AKT-1-overexpressing (A2780AKT+ and
A2780cisAKT+) and underexpressing (A2780AKT−
and A2780cisAKT−) cells were selected from pooled
populations of transfected cells, in order to avoid clonal
variations. First of all, the established cell lines and the
parental cells were analysed on protein level by western blotting
with AKT-1-specific antibody (Fig.
1). The observed AKT-1 expression corresponded to the expected
variations: the cell line A2780AKT+ and
A2780cisAKT+ expressed phosphorylated AKT-1
(phospho-AKT) at high levels, while cells transfected with the
AKT-1 antisense plasmid (A2780AKT−,
A2780cisAKT−) displayed only moderate expression of
phospho-AKT (Fig. 1). Expression of
phospho-AKT was higher in A2780cis than in A2780 cells, as
described previously (9).
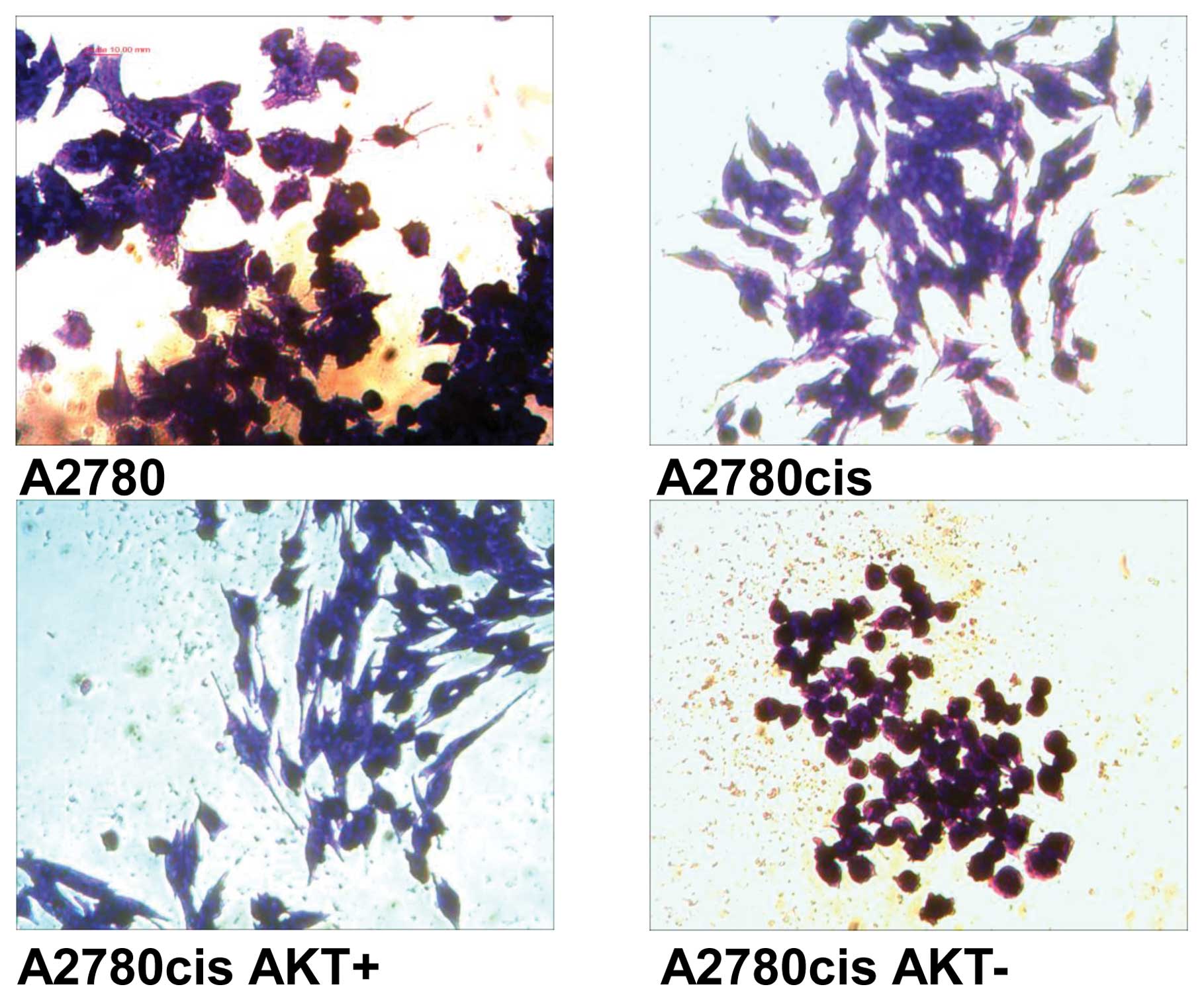
Morphological changes are visible especially in cell
lines derived from A2780cis cells with Akt-overexpression and
downregulation (Fig. 2).
AKT-overexpression results in cells (A2780cisAKT+) with
more spindle-shaped appearance in comparison to A2780cis cells.
Whereas, AKT-downregulation results in cells
(A2780cisAKT−) with spheroidal form. In spite of their
rounded appearance, cells were viable as determined by MTT and
clonogenicity assays.
Cytotoxic effects of cisplatin in ovarian
cancer cells with phospho-AKT-overexpression and
downregulation
IC50s subsequent to treatment with
cisplatin were determined for wild-type A2780 (WT), A2780cis and
transfected cells (Table I). As
expected, IC50 in WT A2780 cells is significantly lower
than in A2780cis cells. Stable transfection with AKT-1
significantly (p<0.05) increases IC50 in A2780 after
cisplatin addition, but does only slightly increase IC50
in A2780cis cells, which are already platinum-resistant. On the
other hand, stable transfection with antisense AKT-1 slightly but
significantly decreases IC50 in A2780 cells, but
significantly (p<0.05) decreases IC50 in
platinum-resistant A2780cis cells. With A2780cis,
A2780cisAKT+ and A2780cisAKT− cells, similar
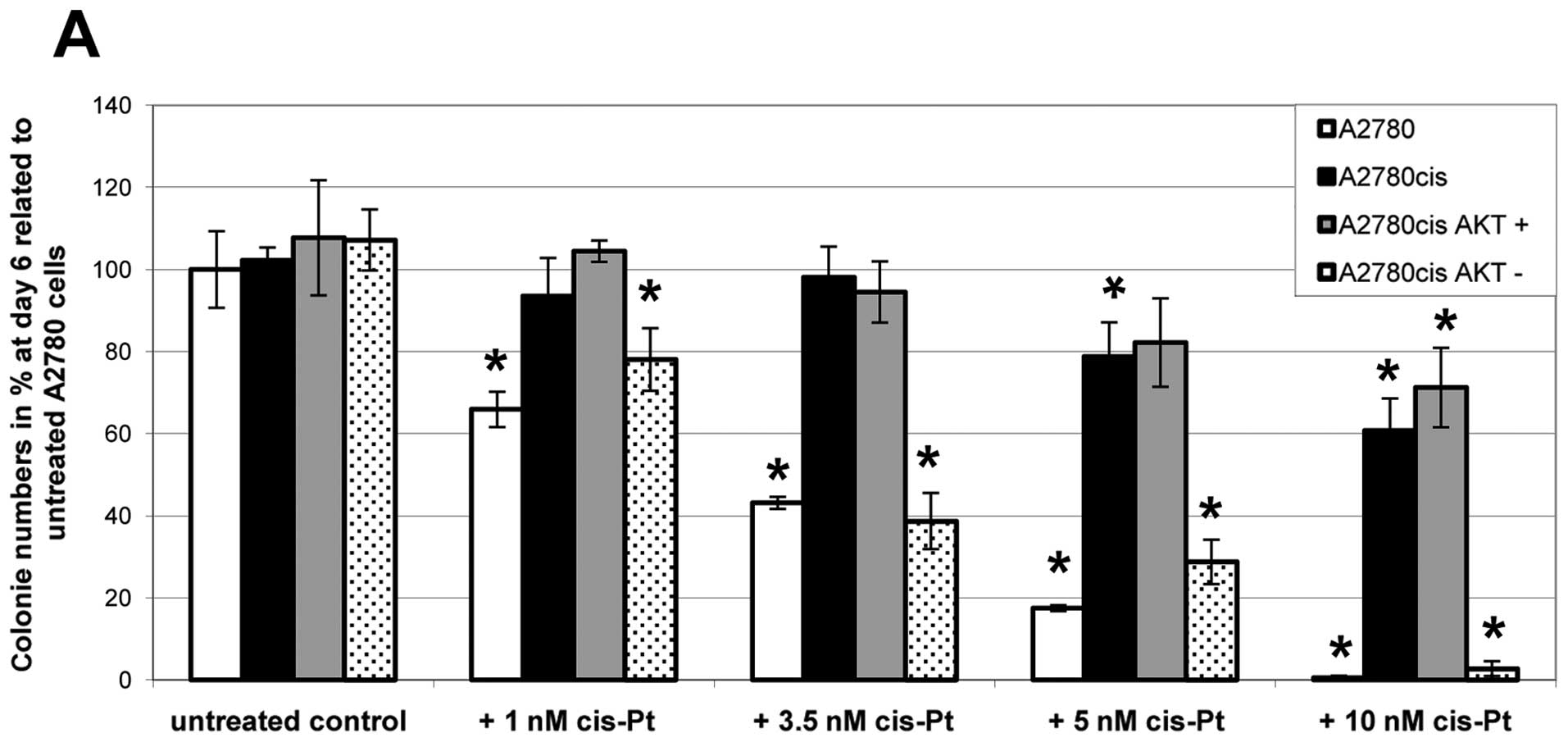
results were obtained by clonogenicity assays (Fig. 3), confirming our hypothesis that
AKT-downregulation can break platinum-resistance in ovarian cancer
cells. In untreated controls all cell lines exhibit the same
growing capacity. After addition of increasing amounts of
cisplatin, however, colony formation differs significantly between
the cell lines; nevertheless all cell lines show a dose-dependent
decrease in growing capacity. Parental A2780 and
A2780cisAKT− cells cannot grow in the presence of 10 nM
cisplatin. In contrast, A2780cis and A2780cisAKT+ cells
are still growing in the presence of 10 nM cisplatin, forming
between 60 and 70% of colonies compared to untreated controls.
 | Table IIC50 of ovarian cancer cell
with and without overexpression and downregulation of Akt
subsequent to treatment with cisplatin.a |
Table I
IC50 of ovarian cancer cell
with and without overexpression and downregulation of Akt
subsequent to treatment with cisplatin.a
| 24 h |
|---|
| A2780 | 9.4±0.6 |
| A2780cis | >40.0 |
| A2780
AKT+ | 31.0±0.3a |
| A2780 AKT
− | 7.6±0.2a |
| A2780cis
AKT+ | >40.0 |
| A2780cis
AKT− | 18.9±0.3a |
Irradiation with 2.5 Gy results in reduced colony
formation in all cell lines. Once again the growing capacity of
A2780cisAKT− cells is comparable to parental A2780
cells. Combination of irradiation with 3.5 nM cisplatin inhibits
the growth of WT A2780 and A2780cisAKT− cells
completely. This combination treatment decreases colony formation
only ~40% in the A2780cis and A2780cisAKT+ cells.
Effects of cisplatin on cell cycle
distribution in ovarian cancer cells with
phospho-AKT-overexpression and downregulation
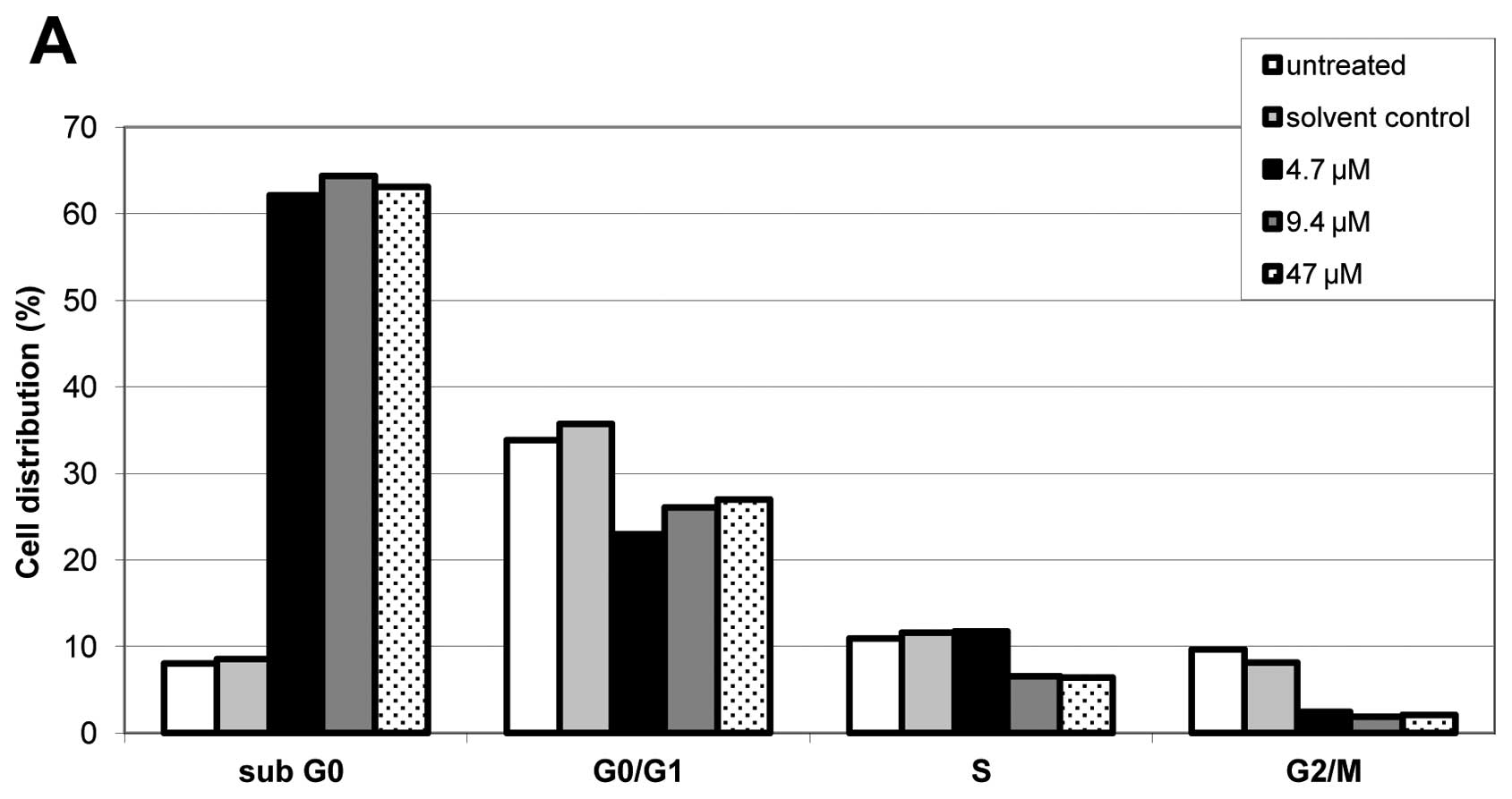
Effects of different doses of cisplatin in A2780cis,
A2780cisAKT+ and A2780cisAKT− on cell cycle
distribution were determined by FACS analysis. Cisplatin induced
apoptosis in all phases of the cell cycle, however, to a greater
extent in the S and the G2/M phase. This effect occurred
regardless of AKT overexpression or downregulation in A2780cis
cells (Fig. 4). FACS analysis also
shows that induction of apoptosis (reflected by a dose-dependent
increase of the sub-G0 fraction) occurs at substantially
lower doses of cisplatin in A2780cisAKT−, than in
A2780cisAKT+ and A2780cis cells, which is in good
accordance with the results of our cytoxicity and clonogenic
assays.
Discussion
The effect of the phosphatidylinositol-3-kinase
(PI-3K)/AKT cascade on proapoptotic protein BAD, a known substrate
of AKT, has been studied in both cisplatin-resistant Caov-3 and
-sensitive A2780 human ovarian cancer cells (18). Treatment of Caov-3 and A2780 cells
with cisplatin stimulated the activation of AKT, and the PI-3K
inhibitor wortmannin blocked the cisplatin-induced AKT-activation.
Cisplatin treatment also stimulated phosphorylation of BAD at the
Ser-112 and Ser-136 sites in Caov-3 and A2780 cells. Whereas
phosphorylation of BAD at Ser-136 was blocked by treatment with
wortmannin, its phosphorylation at Ser-112 was blocked by a MAP/ERK
kinase inhibitor, PD98059. Exogenous transient expression of a
dominant-negative AKT in both Caov-3 and A2780 cells decreased cell
viability after treatment with cisplatin. In contrast, no
sensitization to cisplatin was observed in cells expressing
wild-type AKT. These findings suggested that cisplatin-induced DNA
damage causes the phosphorylation of BAD via an extracellular
signal-regulated protein kinase (ERK) cascade and via a PI-3K/AKT
cascade. Inhibition of either of these cascades sensitizes ovarian
cancer cells to cisplatin, thus providing the first evidence, that
the AKT-pathway is involved in cisplatin-resistance in ovarian
cancers (18). Additional results
suggest that AKT confers platinum-resistance, in part, by
modulating the direction of p53 on the caspase-dependent
mitochondrial death pathway (19).
Thus, in ovarian cancers p53 is a determinant of platinum
sensitivity and AKT contributes to chemoresistance, in part, by
attenuating p53-mediated PUMA upregulation and phosphorylation of
p53 (20). Recent results suggest
that in platinum sensitive ovarian cancer cells, cisplatin-induced
apoptosis can also proceed, in part, via a caspase-independent
mechanism involving apoptosis inducing factor (AIF), and that AKT
activation additionally confers resistance to cisplatin-induced
apoptosis by blocking this pathway (21).
The anti-tumor effect of tangeretin, a citrus
flavonoid known to inhibit cancer cell proliferation, was
investigated in combination with cisplatin in in vitro
models of A2780/CP70 and 2008/C13 cisplatin-resistant human ovarian
cancers (22). Pretreatment of
cells with tangeretin before cisplatin treatment synergistically
inhibited cancer cell proliferation. Interestingly, phospho-AKT and
its downstream substrates, e.g., NF-κB, phospho-GSK-3β, and
phospho-BAD, were downregulated upon tangeretin-cisplatin
treatment. The tangeretin-cisplatin-induced apoptosis in A2780/CP70
cells was increased by PI3K inhibition and siRNA-mediated AKT
silencing, but reduced by overexpression of constitutively
activated AKT. Although the overall results can only be interpreted
with caution, as natural compounds such as tangeretin may display
different effects apart from AKT-inhbition, tangeretin exposure
preconditions cisplatin-resistant human ovarian cancer cells for
cisplatin-induced cell death. This effect may occur through
downregulation of the PI3K/AKT signaling pathway.
In addition to these in vitro studies, in a
series of 98 patients with amplification of PI3K, an upstream
component of the AKT-pathway was associated with resistance to
platinum-based chemotherapy (23).
Accordingly, Woenckhaus et al found that PIK3CA
amplification was a strong predictor for early tumor-associated
death in ovarian cancer patients (24).
Recent work by our group evaluated the anti-tumor
efficacy of the AKT inhibitor perifosine in platinum-sensitive and
-resistant human ovarian cancer cells (9). We used the platinum-sensitive A2780
cell line and the A2780cis cell line, which displays secondary
resistance to cisplatin and was generated from the parental A2780
cells by exposure to increasing doses of cisplatin (15). Thus, in two cell lines with the same
genetic background we could show that A2780cis cells express
substantially higher levels of phospho-AKT and are more sensitive
to treatment with AKT-inhibitor perifosin. Furthermore,
coincubation with perifosine sensitized A2780cis cells to treatment
with cisplatin. AKT-inhibitor perifosine has been tested in phase
II studies in patients with breast, prostate, pancreatic, head and
neck cancer, malignant melanoma, multiple myeloma, colorectal
cancer and soft tissue sarcoma (25–31). A
recent phase I study with perifosine combined with radiotherapy
performed in patients with advanced solid tumors has shown
preliminary evidence of anti-cancer activity, including complete
responses (32). Thus, perifosine
is an attractive compound for further clinical studies in tumor
entities, such as platinum-resistant ovarian cancers.
The clinical observation, that the AKT-inhibitor
perifosine displays synergy with radiotherapy is in accordance with
our results obtained by clonogenicity assays, as the effect of
radiation was most pronounced in tumor cells with low or
downregulated AKT (e.g., A2780cisAKT− cells) (Fig. 3B).
In order to further elucidate the effect of
AKT-expression on platinum-resistance, we established four stable
ovarian cancer lines by transfection of platinum-resistant A2780cis
and parental A2780 cells with either an Akt-1 or an Akt-1-antisense
expression vector. Furthermore, in order to avoid clonal
variations, cells with Akt-1-overexpression (A2780AKT+
and A2780cisAKT+) and downregulation
(A2780AKT− and A2780cisAKT−) were selected
from pooled populations of transfected cells. Thus, we could show
in two cell lines with the same genetic background that
AKT-overexpression rendered platinum-sensitive A2780 cells
platinum-resistant. Still more importantly, platinum-resistance of
A2780cis cells could be reversed by downregulation of AKT. However,
AKT-overexpression in platinum-resistant A2780cis cells did not
increase platinum-resistance any further. These effects were shown
by MTT-assay as well as by clonogenicity assay and FACS analysis.
Additionally, we could show by FACS that cisplatin induces cell
cycle arrest predominantly in the S and the G2/M phase but also in
the G1 phase regardless of the AKT-expression status, as previously
described (33). However, required
doses of cisplatin were substantially higher in cell lines with
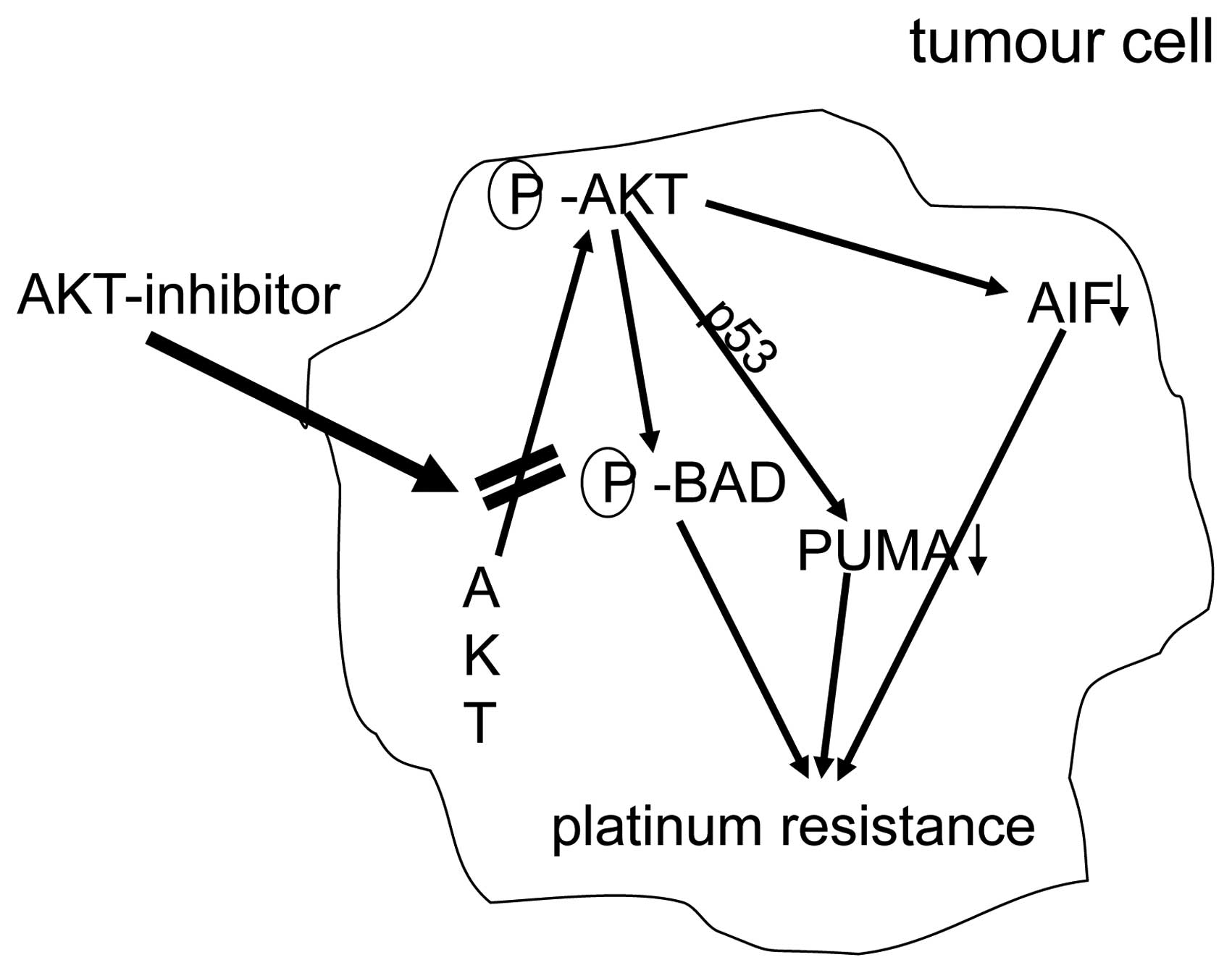
AKT-overexpression. Fig. 5
summarizes mechanisms of platinum-resistance due to overxpression
of AKT.
In conclusion, the present study provides strong
evidence that in ovarian cancers resistance to cisplatin is
mediated by AKT-overexpression and can be overcome by
AKT-downregulation. In context with the already existing evidence,
our data could be a rationale for phase II/III studies in patients
with advanced platinum-resistant ovarian cancers with
AKT-inhbitors, such as perifosine and cisplatin.
Acknowledgements
We appreciate the permission to use the INTAS
ChemoStar Imager (Department of Microbiology, University of
Würzburg). Therefore we thank especially Professor T. Rudel and Dr
B. Bergmann. This work was supported by IZKF Würzburg.
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Pignata S, Cannella L, Leopardo D, Pisano
C, Bruni GS and Facchini G: Chemotherapy in epithelial ovarian
cancer. Cancer Lett. 303:73–83. 2011. View Article : Google Scholar
|
|
3
|
Gonzalez-Martin AJ: Medical treatment of
epithelial ovarian cancer. Expert Rev Anticancer Ther. 4:1125–1143.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsuo K, Lin YG, Roman LD and Sood AK:
Overcoming platinum resistance in ovarian carcinoma. Expert Opin
Investig Drugs. 19:1339–1354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Altomare DA, Wang HQ, Skele KL, et al: AKT
and mTOR phosphorylation is frequently detected in ovarian cancer
and can be targeted to disrupt ovarian tumor cell growth. Oncogene.
23:5853–5857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng JQ, Lindsley CW, Cheng GZ, Yang H
and Nicosia SV: The Akt/PKB pathway: molecular target for cancer
drug discovery. Oncogene. 24:7482–7492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steelman LS, Chappell WH, Abrams SL, et
al: Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in
controlling growth and sensitivity to therapy-implications for
cancer and aging. Aging. 3:192–222. 2011.PubMed/NCBI
|
|
8
|
Levine DA, Bogomolniy F, Yee CJ, et al:
Frequent mutation of the PIK3CA gene in ovarian and breast cancers.
Clin Cancer Res. 11:2875–2878. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engel JB, Schonhals T, Hausler S, et al:
Induction of programmed cell death by inhibition of AKT with the
alkylphosphocholine perifosine in in vitro models of platinum
sensitive and resistant ovarian cancers. Arch Gynecol Obstet.
283:603–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santiskulvong C, Konecny GE, Fekete M, et
al: Dual targeting of phosphoinositide 3-kinase and mammalian
target of rapamycin using NVP-BEZ235 as a novel therapeutic
approach in human ovarian carcinoma. Clin Cancer Res. 17:2373–2384.
2011. View Article : Google Scholar
|
|
11
|
Westfall SD and Skinner MK: Inhibition of
phosphatidylinositol 3-kinase sensitizes ovarian cancer cells to
carboplatin and allows adjunct chemotherapy treatment. Mol Cancer
Ther. 4:1764–1771. 2005. View Article : Google Scholar
|
|
12
|
Benedetti V, Perego P, Luca Beretta G, et
al: Modulation of survival pathways in ovarian carcinoma cell lines
resistant to platinum compounds. Mol Cancer Ther. 7:679–687. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sato S, Fujita N and Tsuruo T: Modulation
of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA.
97:10832–10837. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Katayama K, Fujita N and Tsuruo T:
Akt/protein kinase B-dependent phosphorylation and inactivation of
WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell
Biol. 25:5725–5737. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Behrens BC, Hamilton TC, Masuda H, et al:
Characterization of a cis-diamminedichloroplatinum(II)-resistant
human ovarian cancer cell line and its use in evaluation of
platinum analogues. Cancer Res. 47:414–418. 1987.PubMed/NCBI
|
|
16
|
Engels K, Knauer SK, Loibl S, et al: NO
signaling confers cytoprotectivity through the survivin network in
ovarian carcinomas. Cancer Res. 68:5159–5166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Campling PJ, Galbraith PR and Cole SP: Use
of the MTT assay for rapid determinarion of chemosensitivity of
human leukemic blast cells. Leuk Res. 12:823–831. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayakawa J, Ohmichi M, Kurachi H, et al:
Inhibition of BAD phosphorylation either at serine 112 via
extracellular signal-regulated protein kinase cascade or at serine
136 via Akt cascade sensitizes human ovarian cancer cells to
cisplatin. Cancer Res. 60:5988–5994. 2000.
|
|
19
|
Yang X, Fraser M, Moll UM, Basak A and
Tsang BK: Akt-mediated cisplatin resistance in ovarian cancer:
modulation of p53 action on caspase-dependent mitochondrial death
pathway. Cancer Res. 66:3126–3136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fraser M, Bai T and Tsang BK: Akt promotes
cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function. Int J
Cancer. 122:534–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang X, Fraser M, Abedini MR, Bai T and
Tsang BK: Regulation of apoptosis-inducing factor-mediated,
cisplatin-induced apoptosis by Akt. Br J Cancer. 98:803–808. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arafa el SA, Zhu Q, Barakat BM, et al:
Tangeretin sensitizes cisplatin-resistant human ovarian cancer
cells through downregulation of phosphoinositide 3-kinase/Akt
signaling pathway. Cancer Res. 69:8910–8917. 2009.
|
|
23
|
Kolasa IK, Rembiszewska A, Felisiak A, et
al: PIK3CA amplification associates with resistance to chemotherapy
in ovarian cancer patients. Cancer Biol Ther. 8:21–26. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Woenckhaus J, Steger K, Sturm K, Munstedt
K, Franke FE and Fenic I: Prognostic value of PIK3CA and
phosphorylated AKT expression in ovarian cancer. Virchows Arch.
450:387–395. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marsh Rde W, Rocha Lima CM, Levy DE,
Mitchell EP, Rowland KM Jr and Benson AB III: A phase II trial of
perifosine in locally advanced, unresectable, or metastatic
pancreatic adenocarcinoma. Am J Clin Oncol. 30:26–31.
2007.PubMed/NCBI
|
|
26
|
Leighl NB, Dent S, Clemons M, et al: A
phase 2 study of perifosine in advanced or metastatic breast
cancer. Breast Cancer Res Treat. 108:87–92. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Snyder EL, Bailey D, Shipitsin M, Polyak K
and Loda M: Identification of CD44v6(+)/CD24− breast
carcinoma cells in primary human tumors by quantum dot-conjugated
antibodies. Lab Invest. 89:857–866. 2009.
|
|
28
|
Argiris A, Cohen E, Karrison T, et al: A
phase II trial of perifosine, an oral alkylphospholipid, in
recurrent or metastatic head and neck cancer. Cancer Biol Ther.
5:766–770. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Knowling M, Blackstein M, Tozer R, et al:
A phase II study of perifosine (D-21226) in patients with
previously untreated metastatic or locally advanced soft tissue
sarcoma: a National Cancer Institute of Canada Clinical Trials
Group trial. Invest New Drugs. 24:435–439. 2006. View Article : Google Scholar
|
|
30
|
Posadas EM, Gulley J, Arlen PM, et al: A
phase II study of perifosine in androgen independent prostate
cancer. Cancer Biol Ther. 4:1133–1137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ernst DS, Eisenhauer E, Wainman N, et al:
Phase II study of perifosine in previously untreated patients with
metastatic melanoma. Invest New Drugs. 23:569–575. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vink SR, Schellens JH, Beijnen JH, et al:
Phase I and pharmacokinetic study of combined treatment with
perifosine and radiation in patients with advanced solid tumours.
Radiother Oncol. 80:207–213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He G, Kuang J, Khokhar AR and Siddik ZH:
The impact of S- and G2-checkpoint response on the fidelity of
G1-arrest by cisplatin and its comparison to a non-cross-resistant
platinum(IV) analog. Gynecol Oncol. 122:402–409. 2011. View Article : Google Scholar : PubMed/NCBI
|