Introduction
Cell cycle progression is a highly regulated process
involving the sequential activation of a series of cell cycle
control proteins, the cyclins, and their catalytic subunits, the
cyclin-dependent kinases (Cdks). Different cyclin/Cdk complexes are
required at different phases of the cell cycle. In mammalian cells,
cyclin A is produced in the late G1 phase, and its expression
accumulates during the S and G2 phases (1,2). The
onset of the M phase is regulated by the maturation-promoting
factor (MPF), which consists of at least two subunits, a regulatory
subunit, cyclin B, and its catalytic subunit, Cdc2 (also known as
Cdk1). The kinase activity of cyclin B/Cdc2 is activated
specifically at the G2/M transition and thereafter inactivated at
the onset of anaphase. At the end of mitosis, the activity of the
Cdc2 kinase is abolished suddenly by proteolysis of cyclin B
(3,4). The progression from the G2 phase into
mitosis is negatively regulated by Cdc2 phosphorylation on
threonine-14 (Thr14) and tyrosine-15 (Tyr15)
residues (4). Wee1 is one of the
inhibitory protein kinases and is capable of phosphorylating Cdc2
on Tyr15, but not Thr14 (5).
The cyclin/Cdk complexes may be further regulated by
certain types of proteins, the Cdk inhibitors, which bind to the
complexes and inhibit their activity (2,6). The
first mammalian Cdk inhibitor identified,
p21WAF1/CIP1, is an important mediator of cell
cycle arrest imposed by the tumor suppressor, p53, in response to
DNA damage (7,8). Accumulating data have implicated
growth arrest accompanied by the upregulation of p21 not only in
inhibiting proliferation, but also in promoting differentiation
(2,7,9). p21
is also known to be an inhibitor of DNA replication by inhibiting
the proliferating cell nuclear antigen, which is required for cell
cycle progression (2,9,10).
Cell death occurs either by necrosis or apoptosis.
Necrosis is usually considered to result from physical injury and
not genetically controlled, whereas apoptosis, programmed cell
death, is a deliberate and genetically controlled cellular response
to specific development or environmental stimuli, such as DNA
damage, viral infection, cellular damage, and loss of cell-cell or
cell-substrate contact (11–13).
For cells that have sustained irreversible levels of DNA damage,
apoptosis is an important means for elimination of these cells,
whose presence may ultimately be damaging to the organism.
Apoptosis occurs either in a cell cycle-dependent or -independent
manner. Deregulation of cell cycle progression and inappropriate
Cdk activity may be signals that trigger apoptotic cell death
(14,15). The molecular mechanism implicated in
apoptosis has been partially elucidated, and the induction of
apoptosis is partly mediated intracellularly by a number of genes,
such as p53, Bcl-2 and Bax. The p53 protein functions in part by
responding to DNA damage and inducing apoptosis, which is likely to
be a crucial aspect of the function of p53 as a tumor suppressor.
Wild-type p53 protein arrests DNA-damaged cells in the late G1
phase by inducing p21 activity, and non-repaired cells may be
eliminated by apoptosis, by inducing Bax activity and repressing
Bcl-2 activity (16,17). However, the mechanism of apoptosis
occurring in the G2/M phase is not well understood.
Paclitaxel (taxol), a diterpene with a natural
chemotherapeutic activity first described in 1971, has been
isolated from the bark of the Pacific Western yew tree, Taxus
brevifolia (18). This
antineoplastic drug is one of the most effective antitumor agents
currently used for therapy against ovarian and breast cancer, as
well as non-small cell lung cancer, melanoma and lymphoma (19,21).
Unlike other antimicrotubulin agents, taxol is characterized by a
strong affinity for tubulin protein. Thus, taxol achieves its
antitumor effect by promoting tubulin dimerization and inhibiting
the depolymerization of microtubules, resulting in the formation of
abnormally stable and non-functional microtubules (19,22,23).
As a result of this unique mechanism of action, the continuous
exposure to taxol prevents the completion of mitosis, resulting in
mitotic metaphase arrest and cellular toxicity (24,25).
The cell death induced by taxol appears to result from apoptosis,
which is the mechanism of most antineoplastic agents (25,26).
However, despite these accumulating data, the precise mechanism of
taxol-induced apoptosis remains poorly understood.
In the present study, we investigated the mechanism
of taxol-induced growth arrest and apoptosis in the MCF-7 and
MDA-MB-231 human breast carcinoma cells, which have different
estrogen receptor (ER) and tumor suppresser p53 statuses. We
demonstrate that taxol treatment induces a similar inhibition of
cell growth, due to cell cycle arrest in the G2/M phase and
apoptosis in both cell lines, which are associated with a decrease
in Wee1 expression and the induction of p21 activity. Our data
suggest that taxol induces apoptosis in cells arrested in the G2/M
phase, which may be explained at least in part by the induction of
Cdk inhibitor p21 activity in an ER- and p53-independent manner in
human breast carcinoma cells.
Materials and methods
Cell culture and taxol treatments
The MCF-7 and MDA-MB-231 human breast carcinoma cell
lines were purchased from the American Type Culture Collection
(Rockville, MD). They were cultured at 37°C in a humidified
atmosphere containing 5% CO2. Taxol was purchased from
Sigma Chemical Co. (St. Louis, MO). A stock solution in dimethyl
sulfoxide (DMSO) was prepared and stored at 4°C. The final
concentration of DMSO did not exceed 0.05% in the culture medium,
which did not affect growth.
Growth study and morphology
For growth inhibition analysis, the cells were
plated at 0.5×104 cells per 100-mm plate and incubated
for 24 h. The cells were cultured in the presence or absence of
taxol. After every 12 h of culture, the cells were trypsinized and
washed with phosphate-buffered saline (PBS), and the viable cells
were scored using a Neubauer hemocytometer with trypan blue
exclusion. For the morphological study, cells were grown on
coverslips, treated with taxol for 48 h, and Wright-stained (Fisher
Scientific, Pittsburgh, PA), as recommended by the
manufacturer.
DAPI staining
The cells were washed with PBS and fixed with 3.7%
paraformaldehyde in PBS for 10 min at room temperature. The fixed
cells were washed with PBS and stained with
4,6-diamidino-2-phenylindole (DAPI, Sigma) solution for 10 min at
room temperature. The cells were washed two more times with PBS and
analyzed via a fluorescence microscope at ×400 magnification.
Assessment of DNA degradation
The cells were incubated with taxol for 24 and 48 h
and then trypsinized. Cells were washed with PBS and resuspended in
lysis buffer [1 mM EDTA, 10 mM Tris (pH 8.0), 1% SDS, 1 μg/ml
proteinase K]. After 1 h of incubation at 37°C, RNase A was added,
and incubation was continued for another hour. Crude DNA
preparations were extracted twice with phenol:chloroform:isoamyl
alcohol (25:24:1). Cell lysate samples were subsequently run at 120
V on a 0.8% agarose gel containing ethidium bromide (EtBr, Sigma).
The gel was examined on an ultraviolet light source and
photographed (27).
DNA flow cytometric analysis
The cells were incubated for various periods of time
in the presence of taxol and detached with trypsin. After
centrifugation, cell pellets were resuspended gently in cold PBS
and fixed with ice-cold 70% ethanol for 30 min. The cells were
resuspended in citrate buffer (250 mM sucrose, 40 mM trisodium
citrate, 5% DMSO, pH 7.6), and frozen at −80°C. Before staining,
the cells were thawed rapidly and treated with RNase A at room
temperature for 30 min. The nuclei were stained with propidium
iodide (PI, Sigma). The fluorescence intensity of the PI-stained
DNA in each cell nucleus was examined with a FACScan flow cytometer
(Becton-Dickinson, San Jose, CA).
Immunoprecipitation and western blot
analysis
Total cell lysates were lysed in extraction buffer.
The supernatant was collected, and protein concentration was
determined with a Bio-Rad protein assay kit (Bio-Rad, Hercules,
CA). Samples were stored at −80°C or immediately used for
immunoblotting and immunoprecipitations. For immunoprecipitation,
cell extracts were incubated with immunoprecipitating antibodies in
extraction buffer for 1 h at 4°C. The immunocomplexes were
precipitated with protein A-Sepharose beads (Sigma) for 1 h and
washed five times with extraction buffer prior to boiling in an SDS
sample buffer. The immunoprecipitated proteins or aliquots
containing 40 μg protein were separated on SDS-polyacrylamide gels
and transferred onto nitrocellulose membranes (Schleicher &
Schuell, Keene, NH). Western blot analysis was performed as
previously described (28).
Monoclonal antibodies to Cdc2, cyclin A, cyclin B1 and pRB, and
polyclonal antibodies to Cdk2 and Wee1 were purchased from Santa
Cruz Biotechnology Inc. (Santa Cruz, CA). The monoclonal
anti-poly(ADP-) polymerase (PARP) and anti-p53 antibodies were
purchased from Calbiochem (Cambridge, MA). Monoclonal anti-p21
antibody was obtained from Transduction Laboratories (Lexington,
KY). Peroxidase-labeled donkey anti-rabbit immunoglobulin and
peroxidase-labeled sheep anti-mouse immunoglobulin were purchased
from Amersham Corp. (Arlington Heights, IL).
p21 promoter-luciferase constructs and
transfection assay
The cells were transiently transfected with p21
promoter-luciferase reporter constructs using Lipofectamine (Gibco
BRL, Gaithersburg, MD), as recommended by the manufacturer.
Following transfection, the cells were incubated for 12 h, the
medium was exchanged, and the cells were incubated for various
periods of time in the presence of 20 nM taxol. The cells were then
lysed, and luciferase activity in the lysates was assayed using a
Dynatech ML1000 luminometer (Dynatech Laboratories, Chantilly, VA).
Luciferase activity was normalized to β-galactosidase activity,
which was assayed using the β-galactosidase Enzyme Assay System
(Promega, Madison, WI). All the luciferase assays were carried out
at least in triplicate, and the experiments were repeated at least
three times.
Statistical analysis
Data are presented as the means ± SD of at least
three separate experiments. Comparisons between groups were
analyzed using Student’s t-test. P-values <0.05 were considered
to indicate statistically significant differences.
Results
Growth inhibition by taxol
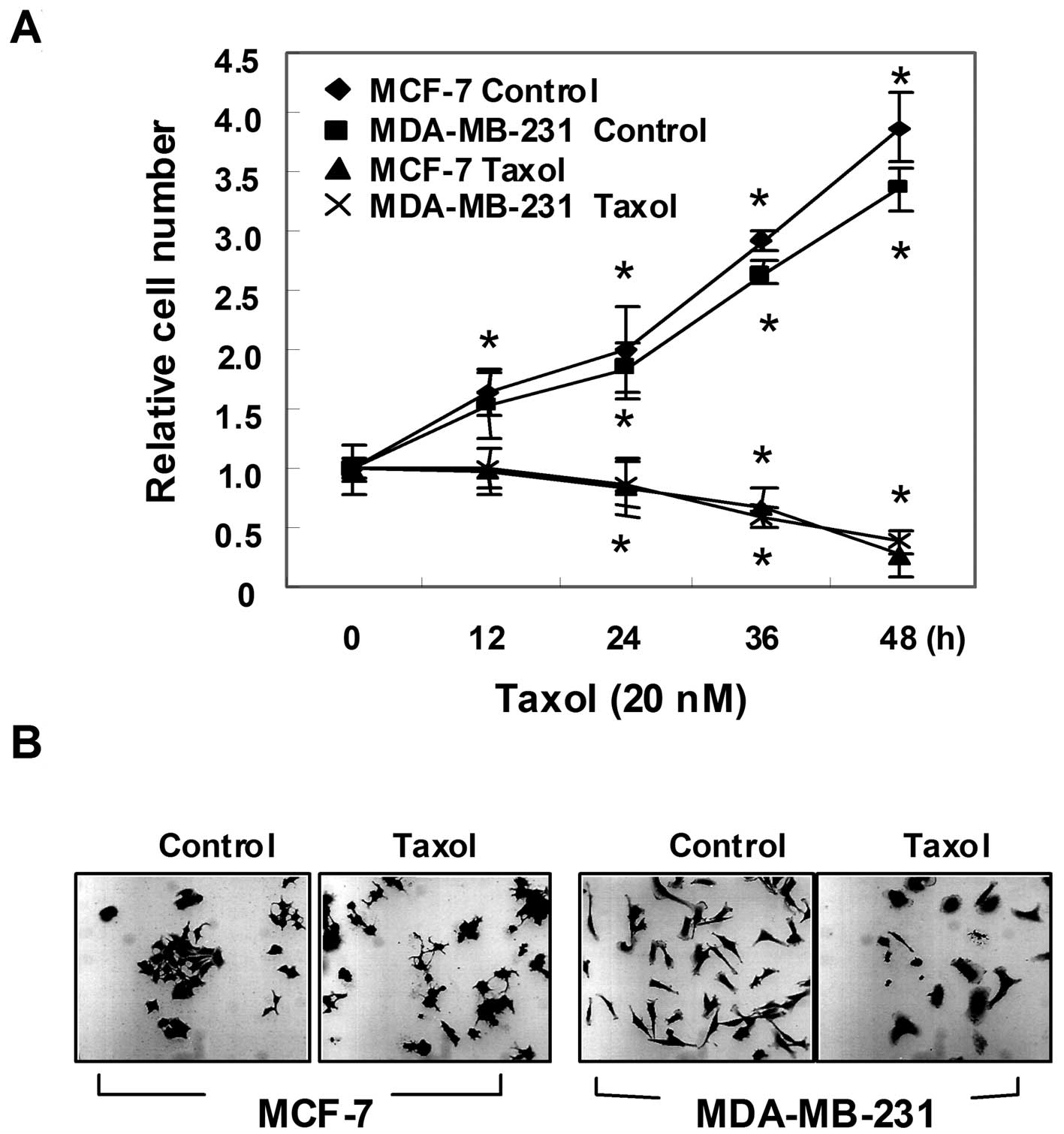
To evaluate the effect of taxol on the proliferation
of human breast carcinoma cell lines, we initially determined the
effect of taxol on the growth of MCF-7 and MDA-MB-231 cells. The
cells were cultured in the absence or presence of 20 nM taxol for
various time-periods, and viable cells were scored by a
hemocytometer (Fig. 1A). The
results showed that the untreated control cells displayed
exponential growth during the 48-h incubation, whereas taxol
treatment resulted in a dramatic decrease in the number of viable
cells. This growth inhibition was accompanied by membrane shrinking
and cells rounding up (Fig. 1B).
These distinct morphological changes were even more pronounced with
higher concentrations and prolonged exposure to taxol (data not
shown).
Apoptosis induction by taxol
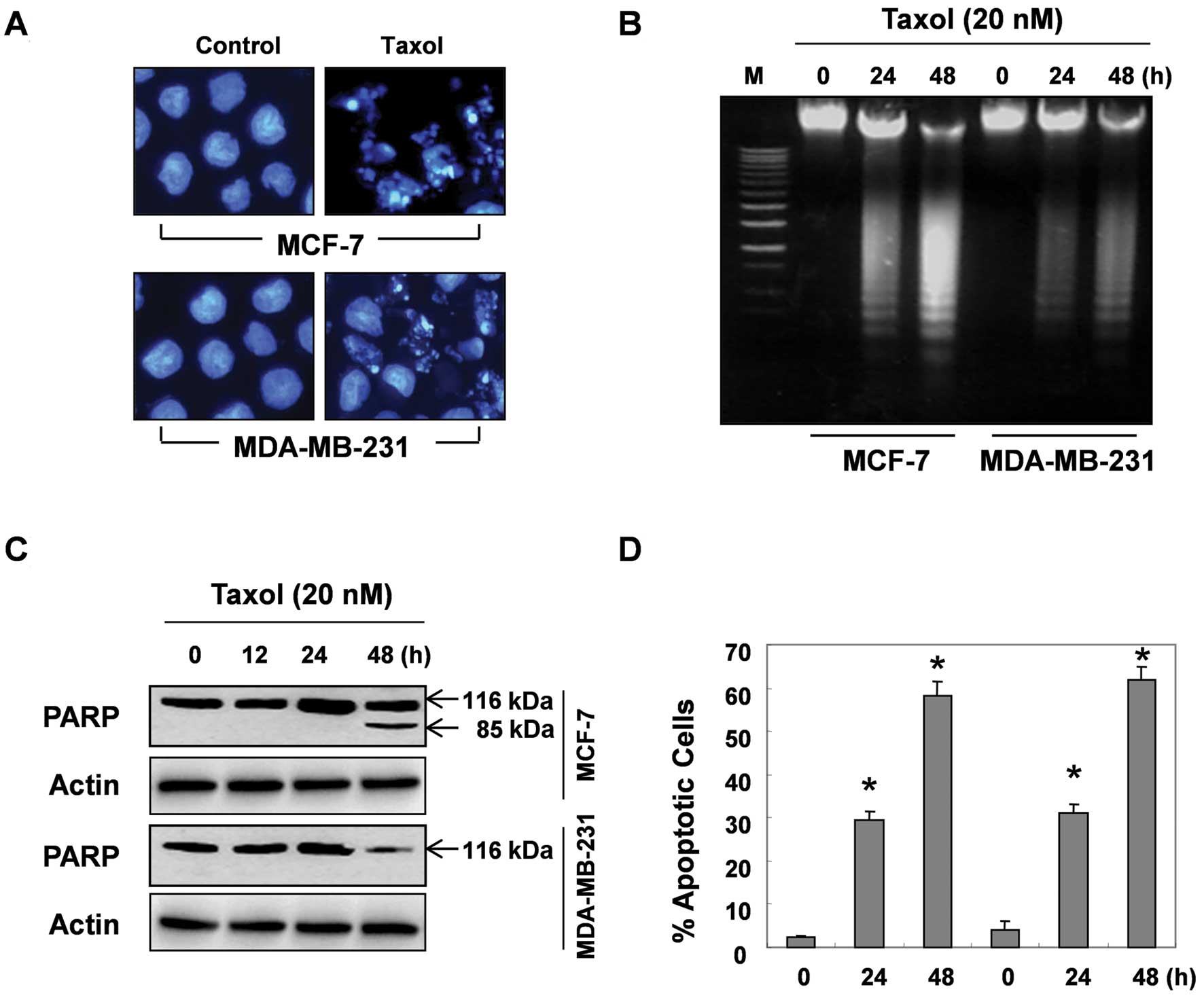
We then investigated whether taxol can induce
apoptosis in human breast cancer cells, using DAPI staining, DNA
fragmentation, PARP cleavage and flow cytometry. Fig. 2A shows the morphological changes in
the MCF-7 and MDA-MB-231 cells incubated with or without taxol. The
control cells displayed an intact nuclear structure, while the
cells treated with taxol displayed chromosomal condensation and the
formation of apoptotic bodies, indicating that the prolonged
exposure to taxol induced programmed cell death. As reported,
during the apoptotic process, endonucleases are activated, and they
degrade chromosomal DNA to small nucleosomal fragments; the
biochemical hallmark for apoptosis is the cleavage of DNA at an
internucleosomal ladder. Thus, we then determined whether taxol can
induce this type of chromatin destruction, by using agarose gel
electrophoresis. After a 24-h and 48-h treatment of the cells with
taxol, typical DNA laddering was clearly visible in the
EtBr-stained gels in a time-dependent manner, which indicated the
occurrence of apoptosis (Fig. 2B).
Western blot analysis of PARP cleavage also showed that the PARP
protein was degraded, with the concomitant disappearance of
full-size (116 kd) molecules and accumulation of 85-kd fragments in
the MCF-7 cells after 48 h of taxol treatment (Fig. 2C). The 85-kd PARP fragments in the
MDA-MB-231 cells treated with taxol were not detected. However, the
steady-state levels of PARP protein were markedly decreased between
24 and 48 h, suggesting that the PARP protein was degraded by taxol
(Fig. 2C). The levels of apoptosis
were also quantified by means of flow cytometry following PI
staining. As shown in Fig. 2D, the
percentage of apoptotic cells increased in a time-dependent manner
in both cell lines following taxol treatment.
Cell cycle arrest by taxol
We subseqently examined the effect of taxol on cell
cycle progression. At the indicated time-points, cell aliquots were
collected and analyzed for DNA content by a fluorescence-activated
cell sorter. As demonstrated in Table
I, the percentage of G2/M cells in the untreated controls was
approximately 23% for MCF-7 cell line and approximately 27% for the
MDA-MB-231 cell line. However, >60% of MCF-7 cells and 50% of
MDA-MB-231 cells were in the G2/M phase following 24 h of taxol
treatment, which suggested the existence of a block at the G2/M
phase of the cell cycle. Taken together, these results indicate
that the growth inhibition in response to taxol in both cell lines
is due to growth arrest and cell death.
 | Table ITaxol inhibits cell cycle progession
at the G2/M phase in MCF-7 and MDA-MB-231 cells.a |
Table I
Taxol inhibits cell cycle progession
at the G2/M phase in MCF-7 and MDA-MB-231 cells.a
| | No. of cells (%) in
each phase of the cell cycle |
|---|
| |
|
|---|
| Cell line | Time (h) | G1 | S | G2/M |
|---|
| MCF-7 | 0 | 57.55 | 19.19 | 23.26 |
| 12 | 25.69 | 18.12 | 56.19 |
| 24 | 20.20 | 15.00 | 64.80 |
| 48 | 34.89 | 27.85 | 37.25 |
| MDA-MB-231 | 0 | 49.68 | 23.09 | 27.23 |
| 12 | 27.19 | 29.80 | 43.01 |
| 24 | 23.77 | 25.47 | 50.76 |
| 48 | 30.91 | 36.00 | 33.09 |
Effect of taxol on G2/M cell cycle
regulatory proteins
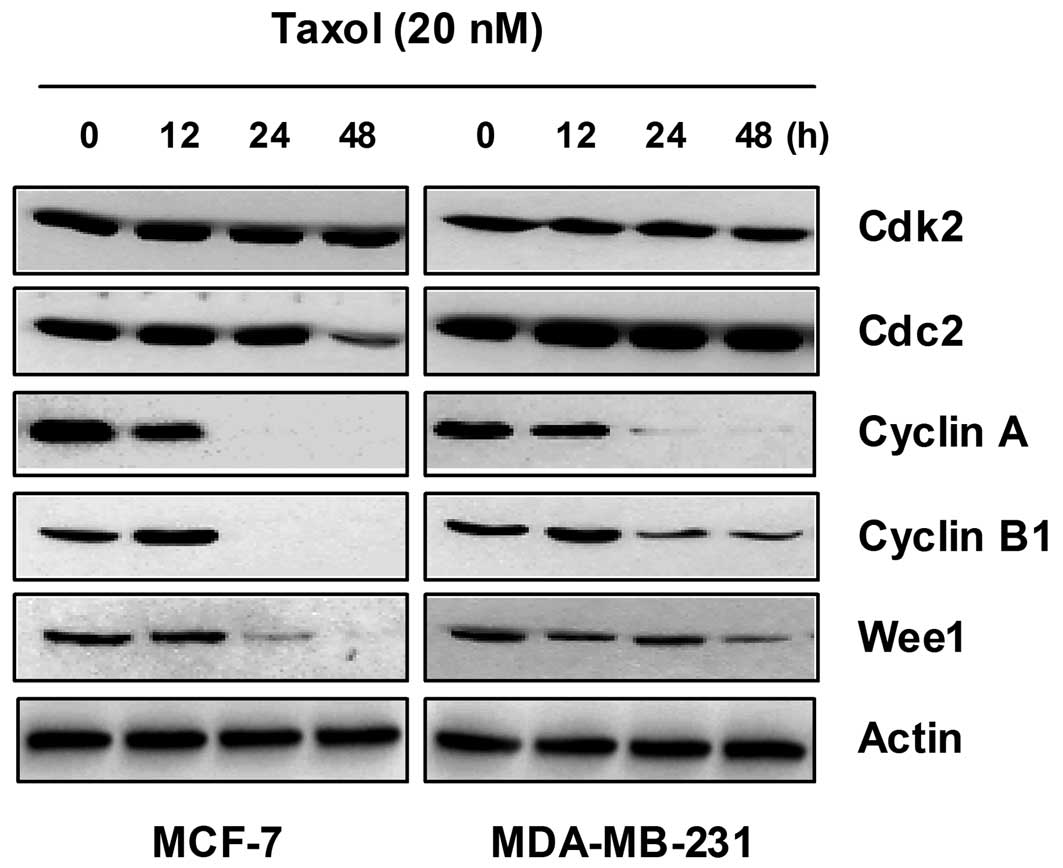
Since taxol blocked cell cycle progression in the
G2/M phase, we determined the expression of cell cycle regulatory
components at the G2/M boundary, such as cyclin A, cyclin B1, Cdk2,
Cdc2 and Wee1. Western blot analysis of the samples obtained from
taxol treatment for various time-periods showed no significant
change in the intracellular protein levels of Cdk2 and Cdc2
(Fig. 3A), whereas taxol treatment
resulted in a marked decrease in cyclin A expression between 12 and
24 h (Fig. 3). The expression
levels of cyclin B1 first increased at 12 h and then decreased at
24 h following treatment with 20 nM taxol (Fig. 3). Under the same conditions, there
was a dramatic reduction in the expression of Wee1 kinase protein
(Fig. 3). These results indicate
that the G2/M arrest induced by taxol is associated with a marked
alteration in the expression of G2/M cell cycle regulatory
proteins, such as cyclin A, cyclin B1 and Wee1.
Induction of Cdk inhibitor p21 by
taxol
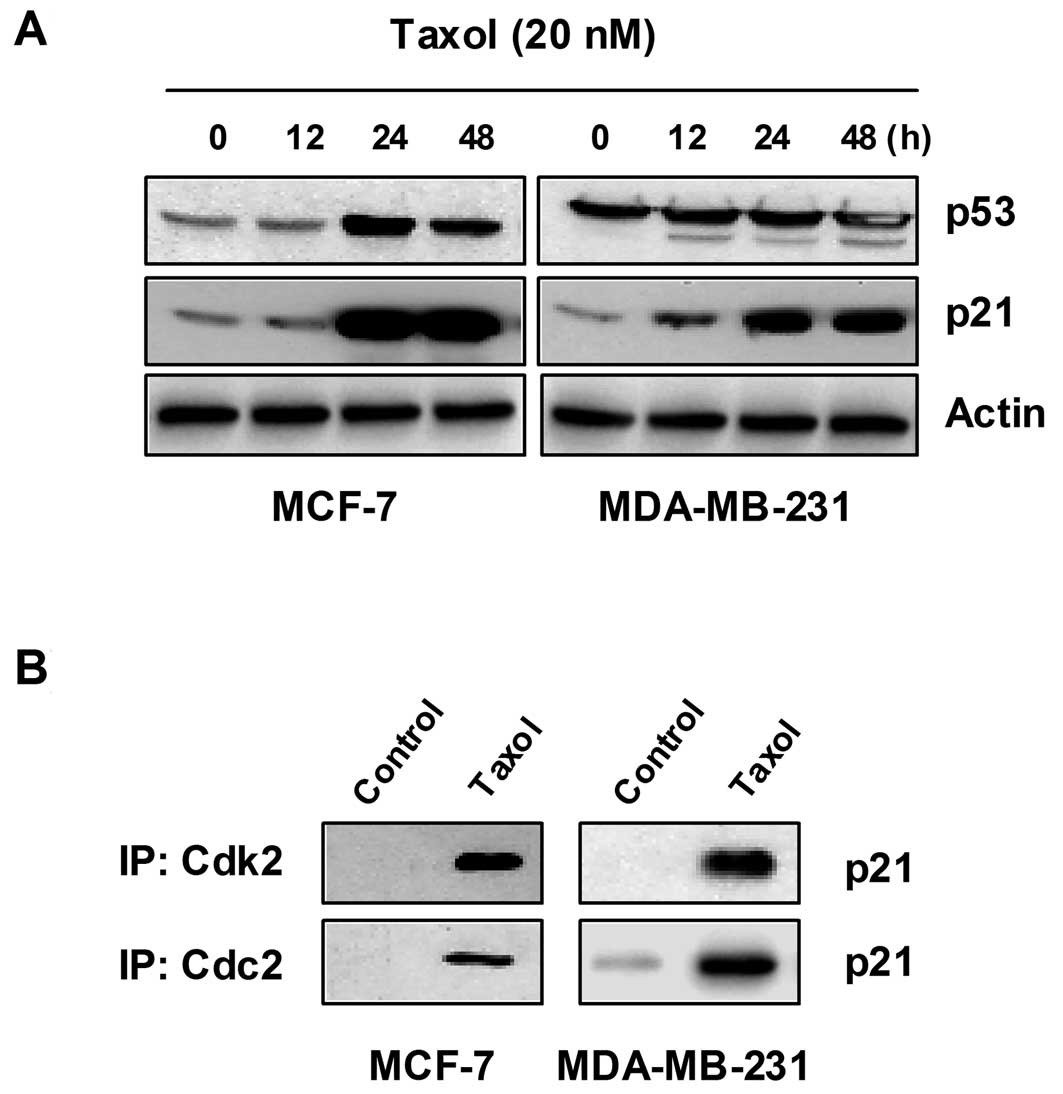
We then investigated whether Cdk inhibitors are
involved in taxol-induced growth arrest and apoptosis. As shown in
Fig. 4A, the incubation of cells
with taxol caused a striking time-dependent induction of p21
protein activity in the MCF-7 and MDA-MB-231 cells. However, other
Cdk inhibitors, such as p27 and p16, were not detectable in either
cell line, without or with taxol treatment (data not shown). MCF-7
cells harbor wild-type p53, whereas MDA-MB-231 cells express
abundant mutant p53. In this experiment, we used anti-p53 antibody,
which reacts with both human wild-type and mutant p53. In the MCF-7
cells, treatment with taxol induced a consistent increase in the
level of p53 with respect to the untreated control cells; however,
the expression of p53 in the MDA-MB-231 cells was not affected by
taxol (Fig. 4A). As the p53 gene is
mutated in MDA-MB-231 cells, it is highly likely that the induction
of p21 activity by taxol is mediated in a p53-independent
manner.
Since it was well known that the Cdk inhibitor, p21,
inhibits the activity of Cdks by direct association with various
cyclin/Cdk complexes, the complex formation of cyclins/Cdks/p21 is
increased in cells arrested by DNA damaging agents. As shown in
Fig. 4B, the association of p21
with Cdks was almost undetectable by co-immunoprecipitation
analysis of the untreated log phase cells. However, the treatment
of cells with taxol resulted in a significant increase in the
binding of Cdk2 and Cdc2 with p21 in both cell lines.
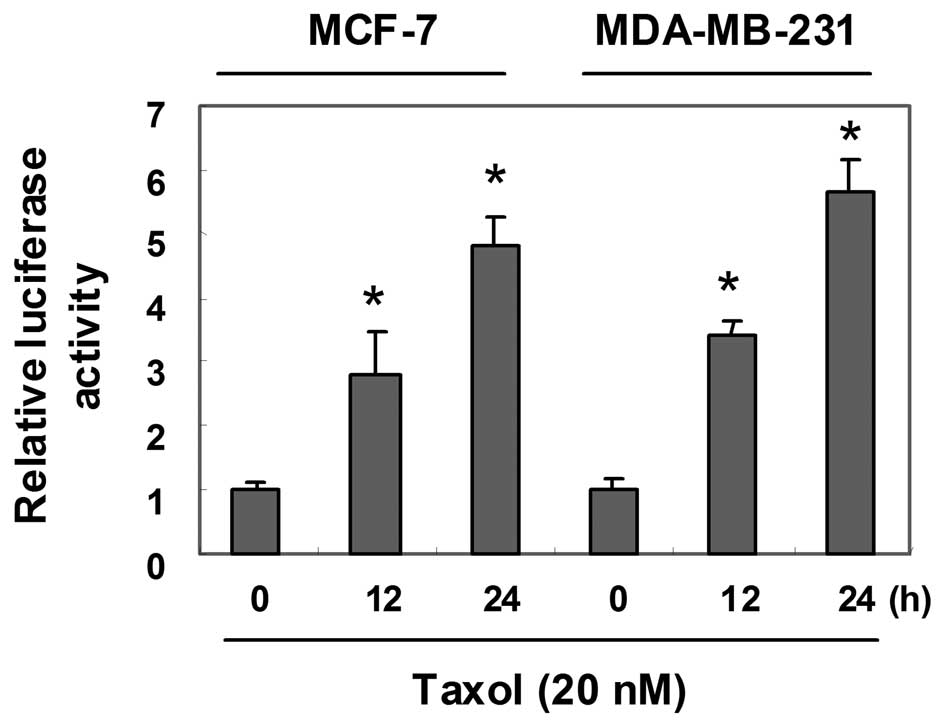
Taxol stimulates p21 promoter
activity
Since p21 expression was markedly induced by taxol
in the MCF-7 and MDA-MB-231 cells, we subsequently investigated
whether the upregulation of p21 expression by taxol involves the
transcriptional regulation of the p21 gene promoter. The cells were
transiently transfected with wild-type p21 promoter-luciferase
fusion plasmids, and luciferase activity was measured in the
untreated control cells and in the cells treated with taxol
(Fig. 5). Following exposure to
taxol, the activity of the p21 promoter was time-dependently
activated in both cell lines.
Discussion
Taxol is one of the most effective antitumor agents
currently used in the treatment of drug refractory tumors (19–21).
This drug is a potent inhibitor of microtubule depolymerization,
which results in the formation of abnormally stable and
non-functional microtubules, mitotic arrest at the metaphase of the
cell cycle and apoptotic cell death (22,23).
However, the precise molecular mechanism of taxol-induced apoptosis
remains to be fully elucidated. The purpose of the present study
was to investigate the mechanisms involved in the inhibition of
proliferation and the induction of apoptosis by taxol in the
ER-positive MCF-7 and ER-negative MDA-MB-231 human breast carcinoma
cell lines. Taxol similarly inhibited the growth of the ER-positive
and -negative cells (Fig. 1). Flow
cytometry confirmed that this growth inhibition was associated with
G2/M cell cycle arrest up to 24 h after taxol exposure in both cell
lines (Table I).
Apoptosis is an active cellular death process
induced by normal physiological or pathological factors for the
elimination of unwanted or damaged cells. It has been proposed that
DNA fragmentation results from the loss of the compartmentalization
of DNase I, which would reach the nucleus due to the breakdown of
the endoplasmic reticulum and the nuclear membrane (29). Fragmentation of DNA at the
internucleosomal linker regions has been observed in cells
undergoing apoptosis induced by a variety of agents. This cleavage
produces ladders of DNA fragments that are the size of integer
multiples of a nucleosome length (180–200 bp) (30,31).
Since their characteristic patterns are shown by agarose gel
electrophoresis, these nucleosomal DNA ladders are widely used as
biochemical markers of apoptosis. The cleavage of PARP has also
been used as a marker of chemotherapy-induced apoptosis (32). In addition, flow cytometry analysis
has been conducted to detect and quantify cells undergoing
apoptosis (12). An increase in the
number of apoptotic cells was recognized following exposure to
taxol by flow cytometry analysis, chromatin condensation, cleavage
of PARP and DNA ladder formation (Fig.
2). At 48 h, flow cytometry demonstrated the highest peak of
sub-G1 fraction, implying that the apoptotic cells outnumbered the
cells arrested in G2/M. Thus, taxol arrested the examined cells in
G2/M, and then induced apoptosis.
In eukaryotic cells, two cyclins, cyclin A and
cyclin B, play a critical role in regulating cell cycle progression
from the G2 through the M phase, including exit from the M phase.
Cyclin A is associated with and primarily activates Cdk2 in the
regulation of S and G2/M phase transition (1,2). It
has been shown that entry into and exit from the M phase of the
cell cycle are controlled by fluctuations in the activity of MPF,
of which Cdc2 is the catalytic subunit and cyclin B is a regulatory
element. Cyclin B accumulates during the S and G2 phases, and its
induction and activation of MPF are necessary to initiate the
transition from the G2 to the M phase. The cyclin B/Cdc2 complex
allows for the phosphorylation of Cdc2 at several sites,
maintaining it in an inactive state and ensuring that mitosis does
not occur prematurely (3,4). This phosphorylation is regulated by
protein kinases, such as Wee1 (5).
Based on these data, we investigated the effects of taxol on the
expression of Cdk2, Cdc2, cyclin A, cyclin B and Wee1 proteins
using western blot analysis. Taxol did not affect the levels of
Cdk2 and Cdc2 expression; however, the expression levels of cyclin
A and cyclin B1 decreased after 24 h (Fig. 3), suggesting that the synthesis of
cyclin A and cyclin B1 was suppressed following the onset of
taxol-induced apoptosis. Wee1 kinase protein expression also
decreased in a time-dependent manner in both cell lines (Fig. 3). These results demonstrate that the
G2/M arrest of the cell cycle and apoptosis are associated with the
inhibition of cyclin A and cyclin B1 expression, as well as a
decrease in Wee1 expression.
Wild-type p53 protein arrests DNA damaged cells in
the late G1 phase by inducing the Cdk inhibitor, p21 (7,8), and
non-repaired cells may be eliminated by apoptosis by inducing Bax
and repressing Bcl-2 activity (33). It has been suggested that the ratio
between the levels of pro-apoptotic Bax protein and the
anti-apoptotic factor, Bcl-2, determines whether a cell responds to
an apoptotic signal (34). However,
Tang et al (35) found that
Bcl-2 did not affect taxol-induced microtubular binding and G2/M
arrest in human pre-B leukemia cells, although it did delay the
induction of apoptosis. Thus, it is possible that apoptosis
provoked by DNA damage is mediated, in other cases, by mechanisms
independent of p53 (36). On the
contrary, an increased level of p21 in cyclin-containing complexes
is associated with decreasing cyclin-dependent activity in damaged
cells destined for apoptosis.
In addition to being induced by p53, p21 is also be
induced by other factors in p53-independent pathways (2,9,10).
However, the mechanism of p21-involved apoptosis occurring in the
G2/M phase is not yet well understood. We therefore investigated
whether taxol can induce the levels of p53 and p21. As shown in
Fig. 5A, the levels of both p21 and
p53 protein expression significantly increased after 24 h in the
MCF-7 cells, which harbor wild-type p53. However, MDA-MB-231 cells
harbor mutant p53 and express abundant mutant p53 protein (37). The treatment of MDA-MB-231 cells
with taxol did not change the level of mutant p53, whereas p21
activity was markedly induced by taxol (Fig. 4A). The p21 proteins levels that
increased due to 36-h treatment with taxol were significantly
associated with Cdk2 and Cdc2 (Fig.
4B). Since p21 expression was markedly induced by taxol in
MCF-7 and MDA-MB-231 cells, we subsequently investigated whether
the upregulation of p21 expression by taxol involves the
transcriptional regulation of the p21 gene promoter. Both cell
lines were transiently transfected with wild-type p21
promoter-luciferase fusion plasmids, and luciferase activity was
measured in both the untreated control cells and the cells treated
with taxol (Fig. 5). Following 24 h
of exposure to taxol, promoter activity increased by approximately
5.5-fold compared to the control in both cell lines. Since p21 is
usually associated with G1 arrest, the G2/M arrest and apoptosis
noted in this study may be specifically related to a unique taxol
pathway.
In conclusion, the findings from our present study
demonstrate that taxol has a signficant and similar inhibitory
effect on cell proliferation in both ER-positive and -negative
human breast carcinoma cell lines. This inhibitory effect on cell
proliferation is the result of G2/M arrest and is accompanied by
the downregulation in the expression of G2/M regulatory proteins,
such as cyclin A, cyclin B1 and Wee1. The anti-proliferative effect
of taxol also resulted in the onset of apoptosis, which followed
the induction of p21 activity and G2/M arrest. It is possible that
the induction of p21 activity occurs through a p53-independent
pathway, and it may be one of the molecular mechanisms through
which taxol induces apoptosis. Thus, taxol-induced cell death may
provide a model of the apoptosis that occurs in the G2/M phase in
an ER-independent manner, which is associated with upregulation of
p21 in a p53-independent manner.
Acknowledgements
This study was supported by National Research
Foundation of Korea grant funded by the Korea government (2012
0000897).
References
|
1
|
Burhans WC and Heintz NH: The cell cycle
is a redox cycle: linking phase-specific targets to cell fate. Free
Radic Biol Med. 47:1282–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shapiro GI: Cyclin-dependent kinase
pathways as targets for cancer treatment. J Clin Oncol.
24:1770–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ruetz S, Fabbro D, Zimmermann J, Meyer T
and Gray N: Chemical and biological profile of dual Cdk1 and Cdk2
inhibitors. Curr Med Chem Anticancer Agents. 3:1–14. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stark GR and Taylor WR: Control of the
G2/M transition. Mol Biotechnol. 32:227–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han SJ and Conti M: New pathways from PKA
to the Cdc2/cyclin B complex in oocytes: Wee1B as a potential PKA
substrate. Cell Cycle. 5:227–231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Cárcer G, Pérez de Castro I and
Malumbres M: Targeting cell cycle kinases for cancer therapy. Curr
Med Chem. 14:969–985. 2007.PubMed/NCBI
|
|
7
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown L, Boswell S, Raj L and Lee SW:
Transcriptional targets of p53 that regulate cellular
proliferation. Crit Rev Eukaryot Gene Expr. 17:73–85. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boulaire J, Fotedar A and Fotedar R: The
functions of the cdk-cyclin kinase inhibitor p21WAF1. Pathol Biol.
48:190–202. 2000.PubMed/NCBI
|
|
10
|
Cazzalini O, Scovassi AI, Savio M, Stivala
LA and Prosperi E: Multiple roles of the cell cycle inhibitor
p21(CDKN1A) in the DNA damage response. Mutat Res. 704:12–20. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lou Z and Chen J: Mammalian DNA damage
response pathway. Adv Exp Med Biol. 570:425–455. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Krysko DV, Vanden Berghe T, D’Herde K and
Vandenabeele P: Apoptosis and necrosis: detection, discrimination
and phagocytosis. Methods. 44:205–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sinclair A, Yarranton S and Schelcher C:
DNA-damage response pathways triggered by viral replication. Expert
Rev Mol Med. 8:1–11. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwartz GK and Shah MA: Targeting the
cell cycle: a new approach to cancer therapy. J Clin Oncol.
23:9408–9421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raucher D, Moktan S, Massodi I and Bidwell
GL III: Therapeutic peptides for cancer therapy. Part II - cell
cycle inhibitory peptides and apoptosis-inducing peptides. Expert
Opin Drug Deliv. 6:1049–1064. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hainaut P: The tumor suppressor protein
p53: a receptor to genotoxic stress that controls cell growth and
survival. Curr Opin Oncol. 7:76–82. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maddika S, Ande SR, Panigrahi S,
Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E
and Los M: Cell survival, cell death and cell cycle pathways are
interconnected: implications for cancer therapy. Drug Resist Updat.
10:13–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wani MC, Taylor HL, Wall ME, Coggon P and
McPhail AT: Plant antitumor agents. VI The isolation and structure
of taxol, a novel antileukemic and antitumor agent from Taxus
brevifolia. J Am Chem Soc. 93:2325–2327. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steed H and Sawyer MB: Pharmacology,
pharmacokinetics and pharmacogenomics of paclitaxel.
Pharmacogenomics. 8:803–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jassem J, Carroll C, Ward SE, Simpson E
and Hind D: The clinical efficacy of cytotoxic agents in locally
advanced or metastatic breast cancer patients pretreated with an
anthracycline and a taxane: a systematic review. Eur J Cancer.
45:2749–2758. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Montana M, Ducros C, Verhaeghe P, Terme T,
Vanelle P and Rathelot P: Albumin-bound Paclitaxel: the benefic of
this new formulation in the treatment of various cancers. J
Chemother. 23:59–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Trudeau ME: Docetaxel: a review of its
pharmacology and clinical activity. Can J Oncol. 6:443–457.
1996.PubMed/NCBI
|
|
23
|
Pienta KJ: Preclinical mechanisms of
action of docetaxel and docetaxel combinations in prostate cancer.
Semin Oncol. 28:S3–S7. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Horwitz SB: Mechanism of action of taxol.
Trends Pharmacol Sci. 13:134–136. 1992. View Article : Google Scholar
|
|
25
|
Donaldson KL, Goolsby GL, Kiener PA and
Wahl AF: Activation of p34cdc2 coincident with
taxol-induced apoptosis. Cell Growth Differ. 5:1041–1050. 1994.
|
|
26
|
Liu Y, Bhalla K, Hill C and Priest DG:
Evidence for involvement of tyrosine phosphorylation in
taxol-induced apoptosis in a human ovarian tumor cell line. Biochem
Pharmacol. 48:1265–1272. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang BR, Kim H, Nam SH, Yun EY, Kim SR,
Ahn MY, Chang JS and Hwang JS: CopA3 peptide from Copris
tripartitus induces apoptosis in human leukemia cells via a
caspase-independent pathway. BMB Rep. 45:85–90. 2012.
|
|
28
|
Jang JY, Kim MK, Jeon YK, Joung YK, Park
KD and Kim CW: Adenovirus adenine nucleotide translocator-2 shRNA
effectively induces apoptosis and enhances chemosensitivity by the
down-regulation of ABCG2 in breast cancer stem-like cells. Exp Mol
Med. 44:251–259. 2012. View Article : Google Scholar
|
|
29
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: Apoptosis: mechanisms and relevance in cancer. Ann
Hematol. 84:627–639. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Widłak P: The DFF40/CAD endonuclease and
its role in apoptosis. Acta Biochim Pol. 47:1037–1044. 2000.
|
|
31
|
Higuchi Y: Chromosomal DNA fragmentation
in apoptosis and necrosis induced by oxidative stress. Biochem
Pharmacol. 66:1527–1535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miyashita T, Krajewski S, Krajewska M,
Wang HG, Lin HK, Liebermann DA, Hoffman B and Reed JC: Tumor
suppressor p53 is a regulator of bcl-2 and bax gene expression
in vitro and in vivo. Oncogene. 9:799–805.
1994.PubMed/NCBI
|
|
34
|
Rodriguez I, Ody C, Araki K, Garcia I and
Vassalli P: An early and massive wave of germinal cell apoptosis is
required for the development of functional spermatogenesis. EMBO J.
16:2262–2270. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang C, Willingham MC, Reed JC, Miyashita
T, Ray S, Ponnathpur V, Huang Y, Mahoney ME, Bullock G and Bhalla
K: Highlevels of p26BCL-2 oncoprotein retard
taxol-induced apoptosis in human pre-B leukemia cells. Leukemia.
8:1960–1169. 1994.
|
|
36
|
Strasser A, Harris AW, Jacks T and Cory S:
DNA damage can induce apoptosis in proliferating lymphoid cells via
p53-independent mechanisms inhibitable by Bcl-2. Cell. 79:329–339.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Negrini M, Sabbioni S, Haldar S, Possati
L, Castagnoli A, Corallini A, Barbanti-Brodani G and Croce CM:
Tumor and growth suppression of breast cancer cells by chromosome
17-associated functions. Cancer Res. 54:1818–1824. 1994.PubMed/NCBI
|