Introduction
Gefitinib and erlotinib, epidermal growth factor
receptor tyrosine kinase inhibitors (EGFR-TKIs), have been widely
used to treat NSCLC in the clinic. However, their efficacy has been
limited by both natural and acquired resistance. Autophagy is known
as a type II programmed cell death. It has been found that cell
death can occur concomitantly with features of autophagy, and
excessive stimulation of autophagy through over-expression of
beclin1 suppresses tumorigenesis (1,2).
Autophagy is a multi-step process consisting of
initiation, autophagosome formation (nucleation, elongation, and
completion), maturation, and degradation (3). Autophagy initiation is complete with
the accumulation of the ULK1/2- ATG13-FIP200 complex, which results
in development of the isolation membrane, also known as a
phagophore. The generation of the complex is regulated by mammalian
target of rapamyacin (mTOR), which lies downstream of the class I
phosphatidylinositol 3-kinase (PI3K)/Akt pathway. mTOR senses
mitogenic stimuli, nutrient conditions, and ATP. The development of
the autophagosome is dependent on the class III PI3K complex, which
consists of the proteins Vps-34, beclin1, and p150, which all
localize to the phagophore and recruit further autophagy-related
genes (ATGs) to allow for elongation and completion of the
autophagosome. Once the autophagosome is developed, its maturation
is complete upon fusion with a lysosome to form an
autophagolysosome (4,5).
Constitutive activation of the PI3K/Akt pathway
occurs in 90% of NSCLC cell lines, thus, promoting cell survival
and resistance to chemotherapy or γ-irradation (6). As a result, inhibition of PI3K/Akt
signaling is not only important for induction of autophagic cell
death but also essential for finding new treatment for NSCLC.
In our preliminary screening, OP-B was found to be
effective in reducing the viability of a panel of human NSCLC
cells. Further investigation of its anticancer mechanisms in
NCI-H157 and H460 cells showed that OP-B primarily induces
autophagy but not apoptosis. Examination of the PI3K/Akt/mTOR
signaling pathway showed that OP-B selectively inhibits
phosphorylation of Akt both at Ser473 and Thr308 in both of the two
cell lines, suggesting that OP-B may be a potential inhibitor of
the PI3K/Akt pathway for the treatment of NSCLC.
Materials and methods
Materials and reagents
Ophiopogonin B was purchased from Nanjing Ze Lang
medical technology company. The compound was initially dissolved in
dimethyl sulfoxide (DMSO) (Sigma, USA) as a stock solution before
use. For treatment of cells, it was diluted in culture medium to
the appropriate concentrations, and the final concentration of DMSO
was less than 0.01%. The chemicals used were rapamycin, LY294002
(Cell Signaling Technology), staurosporine, insulin, PI, Alamar
blue, and Hoechst 33258 (Sigma). We also used the Alexa Fluor 488
Annexin-V/ Dead cell apoptosis kit (Invitrogen, USA).
Cell culture
Human non-small cell lung cancer cells lines A549,
NCI-H460, NCI-H157, H1299, H1792-2, H1944, NCI-226, H358, H292-G,
Hop62, and H522 were obtained from Professor Haian Fu (Emory
University School of Medicine, Atlanta, GA, USA). Cells were grown
in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin-streptomycin mixed antibiotics,
and cultured under 5% CO2 at 37°C.
In vitro viability assay
Cells were seeded into 384-well plates using a
Liquid dispenser in a bio-safety cabinet. Using the liquid handling
system, cells were treated with drug the next day for 72 h. The
final concentrations used in the assay were 50, 25, 12.5, 6.25,
3.125, 1.56, 0.78 and 0.39 μmol/l in triplicate. A volume of 5
μl/well Alamar blue was transferred into the assay plates for a
final concentration of 10%. The plates were exposed to an
excitation wavelength of 530 nm, and the emission at 590 nm was
recorded to determine whether any of the test compounds fluoresce
at the emission wavelength and thus interfere with the assay.
Plates were returned to the incubator and the fluorescence was read
at 4 h. Data were calculated as the percentage of cell viability by
the following formula: the percentage of cell viability = (At/As)
×100%. At and As indicated the absorbance of the test substances
and solvent control, respectively. The mean value and standard
error for each treatment were determined and the % cell viability
relative to control (0.01% DMSO) was calculated. The
IC50 is defined as the concentration of drug that kills
50% of the total cell population as compared to control cells at
the end of the incubation period.
Cell cycle analysis and apoptosis
detection
Cells were treated with or without OP-B for 24 h,
and then harvested by centrifugation, washed with ice-cold
phosphate-buffered saline (PBS), and fixed in ice-cold 70% ethanol
overnight. The cells were then treated with 40 μg/ml RNase at 37°C
and then stained with 40 μg/ml PI for 30 min. The percentage of
cells in each phase (SubG1, G0/G1, S, and G2/M) was calculated
(Becton Dickinson).
For Hoechst 33258 nuclear staining, exponentially
growing cells were seeded at a density of 105 cells per
well onto heat-sterilized coverslips in 6-well plates. After
attachment, cells were treated with or without 10 μmol/l OP-B or 1
μmol/l Staurosporine for 24 h. Subsequently, the cells were fixed
(methanol: glacial acetic acid = 3:1) for 10 min, and then dyed
(Hoechst 33258, 10 μg/ml) for another 10 min at 37°C. After washing
three times with PBS, the cells were observed under a fluorescence
microscope (Olympus, Japan).
Apoptosis and dead cells induced by OP-B were
assayed using the high content screening (HCS) Kinetic Scan Reader
(ThermoFisher Scientific, USA). The principle of the assay is that
cells are labeled with a cocktail of fluorescent dyes (including
Hoechst 33258 and Alexa Fluor 488 Annexin-V) that indicate the
cellular properties of interest, including nuclear structure, cell
membrane permeability, and early and late apoptosis. All procedures
were performed according to the manufacturer’s instructions. The
cells were plated at a density of 8×103 cells/well in
each well of a 96-well plate. After culturing for 24 h, cells were
incubated with 10 μmol/l OP-B for another 24 h. Thirty minutes
before the completion of incubation, a cocktail of fluorescent dyes
was added to each well. The cells were then fixed with prewarmed
Fixation Solution and washed twice with PBS. Plates were then
sealed and ran on an HCS Reader to acquire images. Images were
analyzed with HCS software, and the fluorescence intensity of
Hoechst 33258 and Annexin-V/PI were calculated.
Transmission electron microscopy
(TEM)
After being exposed to 10 μmol/l OP-B for 48 h, the
cells were trypsinized, washed with PBS and fixed in 2.5%
glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) overnight at 4°C.
The next day, cells were washed three times with 0.1 M phosphate
buffer. Thereafter, the cells were fixed in 1% aqueous osmium,
dehydrated with increasing concentrations of ethanol (30, 50, 70,
80, 90 and 100%), and embedded in araldite. The ultrathin sections
were prepared with a microtome (Leica, Germany) and mounted on
copper grids. The samples were stained with 2% aqueous uranyl
acetate and lead citrate and observed in a transmission electron
microscope (Jeol, Japan).
Western blot analysis
After treating with different concentrations of
OP-B, the cells were lysed in RIPA buffer containing 50 mM Tris/HCl
(pH 8.0), 150 mM NaCl, 1% Nonidet-P40, 1% sodium deoxycholate, 0.1%
SDS, 0.1 mM DTT, 0.05 mM PMSF, 0.002 mg/ml aprotinin, 0.002 mg/ml
leupeptin, and 1 mM NaVO3. The protein concentrations of
the supernatants were determined by the BCA protein assay. Equal
amounts of protein were loaded and separated by 10 or 12% SDS-PAGE
and then transferred onto polyvinylidene difluoride membranes. The
membranes were incubated overnight with appropriate primary
antibodies against p-PDK1(Ser241), p-Akt (Ser473), Akt,
p-p70S6kinase (Thr389), p70S6kinase, p-4E-BP1 (Thr37/46), 4E-BP1,
LC3A/B, caspase-3, Bcl-2, cleaved-caspase-3, or β-actin overnight
at 4°C, and then with HRP-conjugated secondary antibodies
(anti-rabbit or mouse immunoglobulin G) for an additional 1 h at
room temperature. Immunoreactivity was detected by enhanced
chemiluminescence (ECL, Bio-Rad). β-actin was used as a loading
control. Immunoblot experiments were performed at least three
times. Quantitative analysis was performed by Image Lab™
software.
Statistical analysis
Unless otherwise stated, data were expressed as the
mean ± SD, and analyzed by Student’s t test. P-value <0.05 was
considered statistically significant.
Results
Effect of OP-B on proliferation of NSCLC
cells
NSCLC, including squamous carcinoma, adenocarcinoma,
and large cell carcinoma, represents ~80–87% of all lung cancer
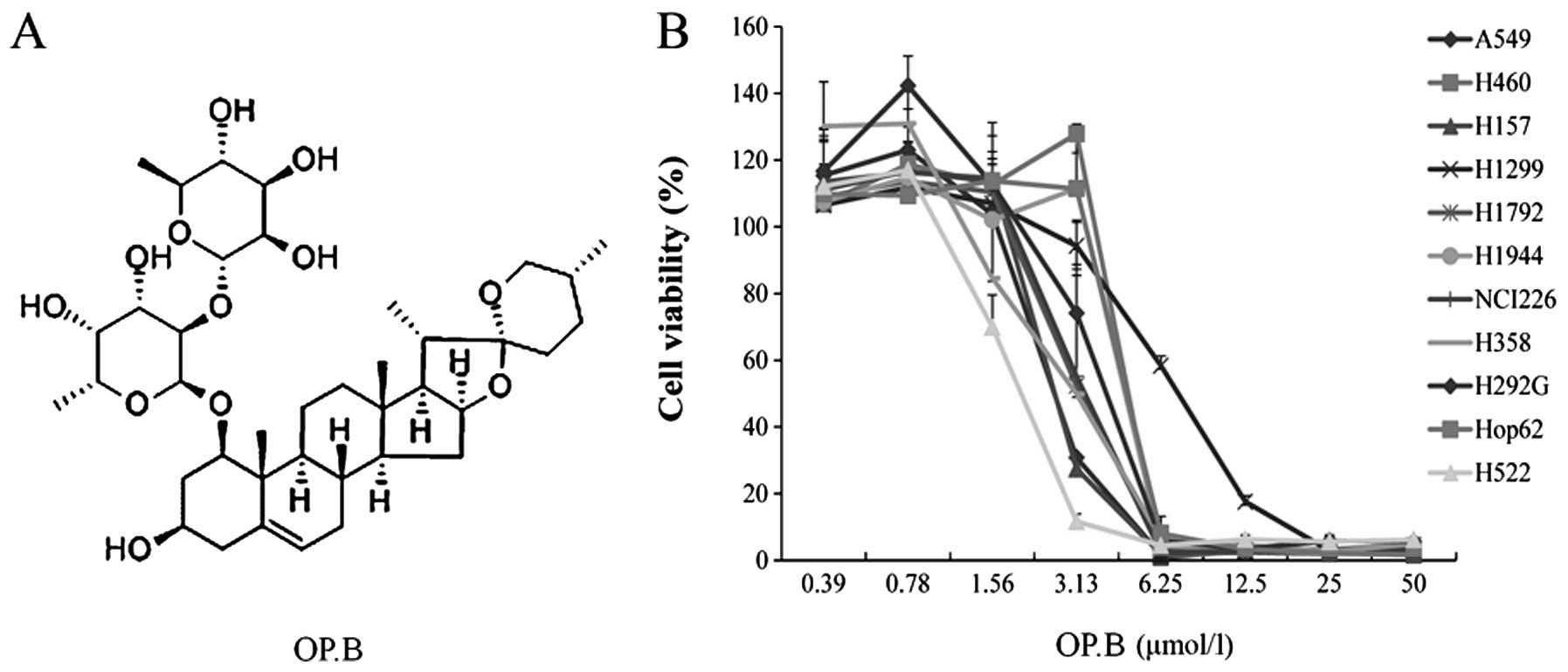
cases (7). To determine whether
OP-B (structure shown in Fig. 1A)
has any therapeutic effect on NSCLC cells, we performed a cell
viability assay using eleven human NSCLC cell lines. After 72 h of
treatment, OP-B significantly decreased cell viability of all cell
lines tested in a dose-dependent manner, and the IC50
was <4 μmol/l (~3.87 μmol/l) (Fig.
1B).
Effect of OP-B on cell morphology, cell
cycle, and apoptosis in NCI-H157 and H460 cells
NCI-H157 and H460 cells represent the main subtypes
of NSCLC and are derived from squamous cell carcinoma and large
cell carcinoma, respectively. In our experiment, we found that
these two cell lines were more sensitive to OP-B (IC50
of H157 and H460 was 2.86 and 4.61 μmol/l, respectively) than all
other NSCLC cell lines tested. Therefore, we chose these lines to
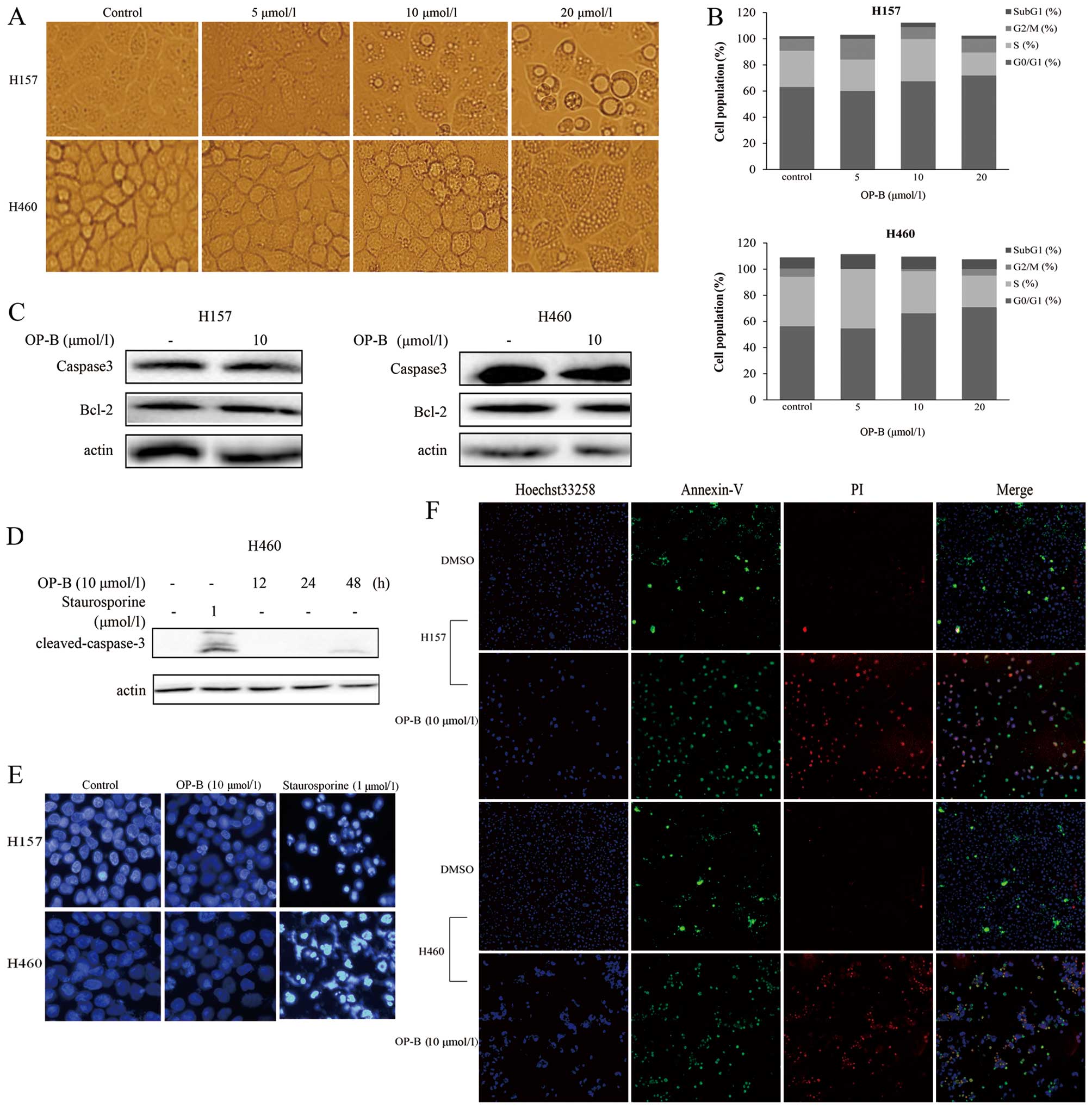
further investigate the pharmacological effect of OP-B. Following
10 μmol/l OP-B treatment for 24 h, many vacuoles appeared in both
cell lines but especially in H157 cells (Fig. 2A). When the concentration of OP-B
was increased to 20 μmol/l, the vacuoles became even larger and
occupied almost all of the space outside the nucleus in NCI-H157
cells.
Flow cytometric analysis of cells stained with
propidium iodide showed a mild increase in the cell population in
G0–G1 phase after the cells were treated with different
concentrations of OP-B for 24 h in medium containing 10% FBS
(Fig. 2B). To further investigate
whether the cell cycle arrest was associated with apoptosis, we
measured levels of caspase-3 and Bcl-2. Treatment with
staurosporine served as a positive control for cleaved caspase-3
staining. The results showed that there was no change compared to
the negative control (Fig. 2C), and
cleaved caspase-3 was only slightly increased in NCI-H460 cells
following 48 h of treatment with OP-B (Fig. 2D); however, no change was detected
in NCI-H157 cells (data not shown). Nuclear staining with Hoechst
33258 also showed no characteristics of apoptosis, such as cell
shrinkage, nuclear condensation, and fragmentation (Fig. 2E). Furthermore, the cells labeled
with a cocktail of fluorescent dyes (including Hoechst 33258 and
Alexa Fluor 488 Annexin-V/Dead cell apoptosis kit) and scanned with
high content screening (HCS) Kinetic Scan Reader (ThermoFisher
Scientific) (Fig. 2F) also
suggested that OP-B did not induce apoptosis or necrosis in either
of the two cell lines.
OP-B induces autophagy in NCI-H157 and
H460 cells
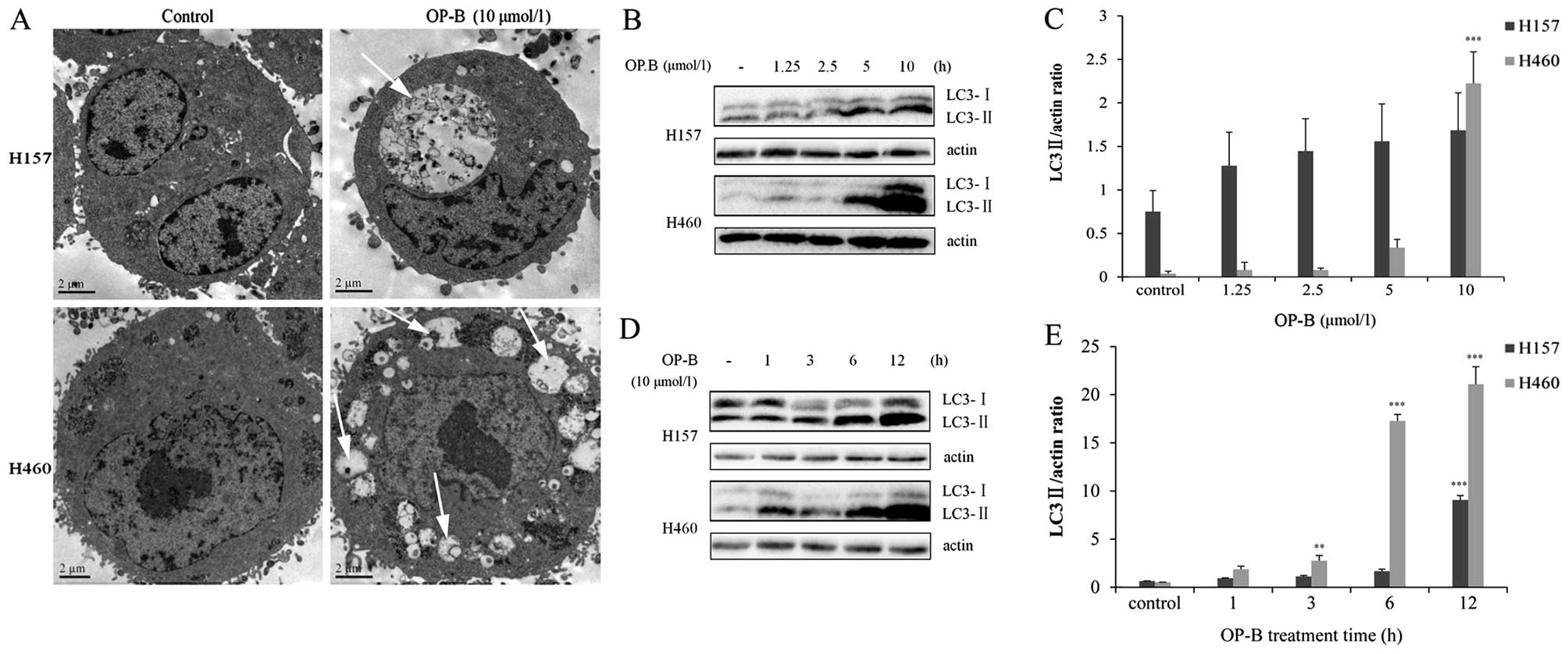
Results from transmission electron microscopy (TEM)
showed that the cytoplasmic vaculoes had double-layered membranes
and that many of them contained cytoplasmic organelles or myelin
figures. Furthermore, the vacuoles increased in size and number and
fused into larger vacuoles, while the nucleus remained intact
(Fig. 3A). Detection of LC3 by
immunoblotting showed that OP-B treatment increased the conversion
of LC3- I to LC3-II in a dose- and time-dependent manner (Fig. 3B-E). Thus, we speculated that
treatment with OP-B induced autophagy of NCI-H157 and H460
cells.
Effects of OP-B on PI3K/Akt/mTOR/p70S6K
signaling pathway and induction of autophagy in NCI-H157 and H460
cells
The PI3K/Akt/mTOR/p70S6K signaling pathway, which is
often associated with tumorigenesis and activated in numerous
tumors, is well-known to regulate autophagy (8–10).
Thus, the pathway was examined in relation to OP-B-induced
autophagy in NCI-H157 and H460 cells.
As shown in Fig. 4A and
B, when cells were treated with at least 10 μmol/l OP-B for at
least 1h, p-Akt (Ser473) was significantly inhibited in both
NCI-H157 and H460 cells, but p-p70S6K (Thr389) was inhibited only
in H460 cells under the same conditions. Under all treatments, p-
4EBP1 (Thr37/46) was not affected.
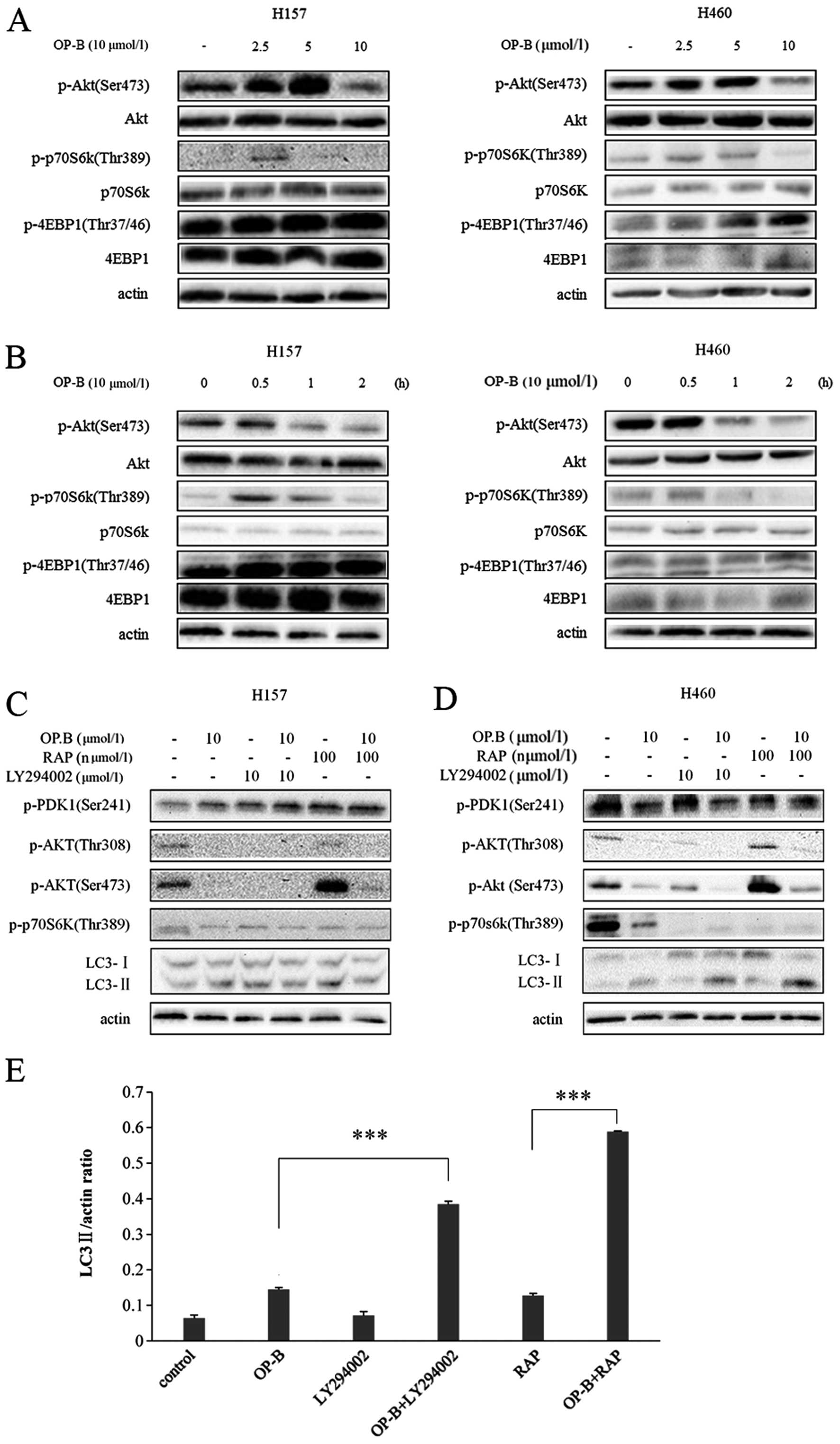 | Figure 4Effect of OP-B on PI3K/Akt/mTOR/
p70s6k signaling pathway in NCI-H157 and H460 cells. (A and B) The
NCI-H157 and H460 cells treated with 0, 2.5, 5, 10 μmol/l OP-B for
1.5 h or 10 μmol/l of OP-B for 0, 0.5, 1 or 2 h were analyzed by
immunoblotting with antibodies against p-Akt (Ser473), p-p70S6K
(Thr389), p-4EBP1 (Thr37/46), Akt, p70S6K, 4EBP1, and actin. (C and
D) The NCI-H157 and H460 treated with 10 μmol/l OP-B, 10 μmol/l
LY294002 or 10 μmol/l Rapmycin for 4 h were analyzed by
immunoblotting with antibodies against p-PDK1 (Ser241), p-Akt
(Thr308), p-Akt (Ser473), p-p70S6K (Thr389), LC3, and actin. (E)
Densitometry analysis of LC3-II levels relative to actin in H460
cells was performed using three independent experiments. Error
bars, SD; **p<0.01; ***p<0.001. |
LY294002 is a well-characterized inhibitor of PI3K,
and rapamycin is an inhibitor of mTORC1. Next, we tested the effect
of OP-B on the PI3K/Akt/mTOR/p70S6K pathway in both H157 and H460
cells. In H157 cells, similarly to LY294002, OP-B inhibited
phosphorylation of Akt both at Ser473 and Thr308, and it weakened
the feedback activation of rapamycin on p-Akt at Ser473. In
contrast, p-PDK1 (Ser241), p-p70S6K (Thr389), and LC3, were not
affected by any of the above treatments (Fig. 4C).
Similarly, in H460 cells, OP-B showed enhanced
inhibition of p-Akt (Ser473 and Thr308) after co-treatment with
LY294002. Additionally, it weakened the feedback activation of
rapamycin on the two sites. Unlike in H157 cells, the
phosphorylation of p70S6K (Thr389) was inhibited by treatment with
OP-B, LY294002, or rapamycin. Conversion of LC3 I to LC3-II was
induced by OP-B, inhibited by LY294002, and enhanced by
co-treatment with OP-B and LY294002. In fact, co-treatment with
OP-B and rapamycin had an even more significant effect than single
treatment with OP-B or rapamycin alone (Fig. 4D and E).
The above results show that in NCI-H157 and H460
cell lines, OP-B has similar pharmacological effects on the
inhibition of p-Akt (Ser473and Thr308). However, there are varying
degrees of inhibition on the PI3K/Akt/mTOR/p70S6K signaling
pathway. Taken together, NCI-H460 was more sensitive to OP-B not
only based on inhibition of the pathway but also based on induction
of autophagy.
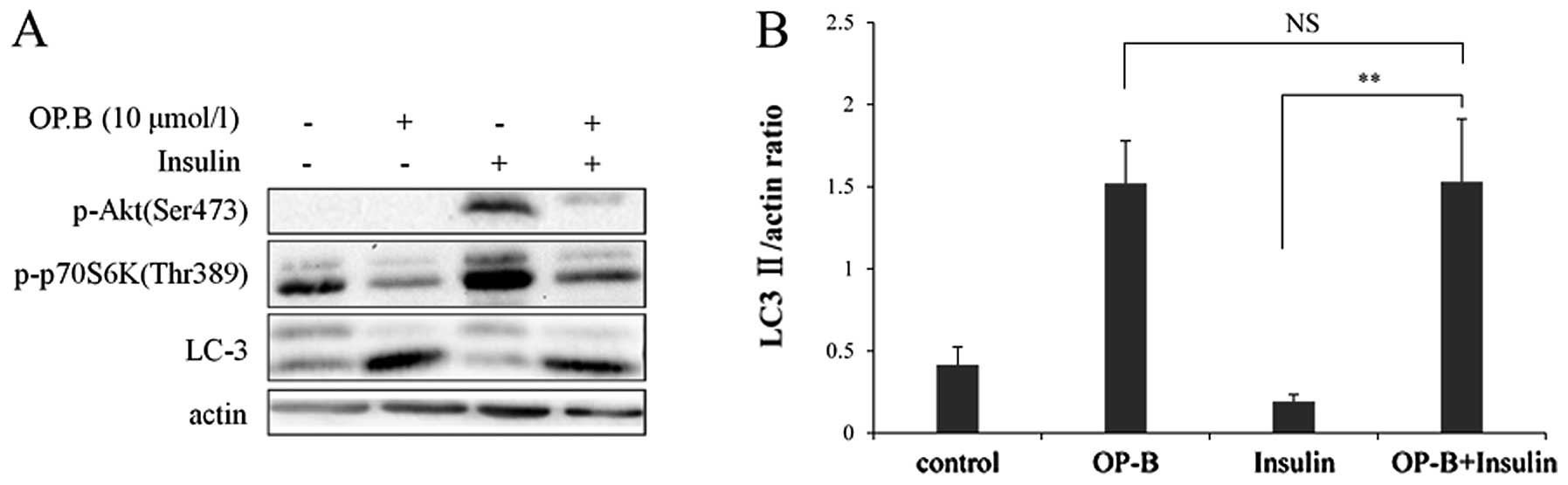
Correlation between inhibition of the
PI3K/Akt/mTOR/p70S6K pathway and induction of autophagy from OP-B
in H460 cells
Insulin upregulates PI3K and its downstream targets,
including Akt and mTOR, and also suppresses autophagy (11–13).
As shown in Fig. 5B, 30 min of
insulin treatment significantly phosphorylates Akt at Ser473 and
p70S6K atThr389. In contrast, when cells were pretreated with OP-B
and then stimulated with insulin, the phosphorylation was
significantly inhibited. Otherwise, no significant differences in
the LC3-II/ actin ratio between OP-B treatment and OP-B treatment
with insulin were observed (Fig.
5).
Discussion
OP-B is a natural active compound extracted from the
Chinese herbal medicine ophiopogon. In this study, we found that
OP-B successfully inhibited cell proliferation in a panel of NSCLC
cell lines. The IC50 in all lines tested was <4
μmol/l. In order to further investigate the pharmacological effect
of OP-B on NSCLC, we chose NCI-H157 and NCI- H460 cells as our cell
line models since they represent the most commonly used NSCLC cells
and because they were all sensitive to OP-B in our preliminary
studies. Of note, we found that after 24 h of treatment with at
least 10 μmol/l OP-B in medium containing 10% FBS, a large number
of vacuoles accumulated in the cytoplasm of cells. From the volume
of the vacuoles, we judged that NCI-H157 seemed to be more
sensitive to OP-B than NCI-H460 (Fig.
2A).
Cell cycle analysis by flow cytometry showed that
OP-B induced a modest increase in G0/G1 phase in both cell lines
(Fig. 2B). However, expression of
caspase-3 and Bcl-2, detected by western blot (Fig. 2C), nuclear morphology (stained by
Hoechst 33258), and fluorescence intensity (labeled by AnnexinV/PI)
detected by high content screening (HCS) Kinetic Scan Reader
(Fig. 2F) all showed that OP-B did
not induce apoptosis or necrosis in either cell line.
In order to determine if the vacuoles were
associated with autophagy, we measured several markers.
Importantly, TEM is able to distinguish autophagic cytoplasmic
vacuoles from cellular vesicles, such as endosomes, lysosomes, and
apoptotic blebs (14). We first
observed cell morphology. The vacuoles had double membranes, the
internal contents were degraded by lysosomal hydrolases, and only
some myelin figures remained (Fig.
3A). These results are consistent with earlier publications
(15). The presence of LC3 in
autophagosomes, and the conversion of LC3 I to LC3-II are known
indicators of autophagy (16).
Detection of LC3 using western blot showed that in both cell lines,
the OP-B increased the conversion of LC3 I to II in a dose- and
time-dependent manner (Fig. 3B and
D). Unexpectedly, the conversion rate of LC3 I to II was more
significant in NCI-H460 than in H157 (Fig. 3C and E). To further investigate the
reason behind this, we assessed the PI3K/Akt/mTOR signaling
pathway, which is the main pathway involved in the regulation of
autophagy. Within 4 h of OP-B treatment in NCI-H157 cells, p-Akt
was inhibited and autophagy was not induced. However, in H460
cells, p-PDK1, p-Akt, and p-p70S6K were all inhibited by OP-B and
autophagy was induced (Fig. 4C and
D). In addition, activation of the pathway using Insulin showed
that the autophagy induced by OP-B correlated with an active
signaling pathway (Fig. 5).
Since activation of PI3K/Akt occurs in 90% of NSCLC
cell lines, it has become an important target for the development
of anticancer drugs. It is well known that LY294002 and rapamycin
are inhibitors of PI3K and mTORC1, respectively. However, these
compounds have some drawbacks. For example, LY294002 does not
distinguish between class I and class III PI3K, and its inhibition
of class III PI3K also inhibits autophagy (17–19).
Rapamycin inhibits mTORC1, but has a negative feedback on Akt
(20). Herein, we found that at
least in NCI-H460 cells, OP-B was an ideal inhibitor of the
PI3K/Akt/mTOR/p70S6K pathway. It inhibited all components of the
pathway and even had a synergistic effect with LY294002 on Akt. It
also decreased the activation of rapamycin on Akt and had a
synergistic effect on induction of autophagy.
Taken together, OP-B displayed significant
cytotoxicity on a panel of NSCLC cell lines at a relatively low
concentration. In NCI H157 and H460 cells, it inhibited p-Akt both
at Ser308 and Thr473 and significantly induced autophagy. In
NCI-H460 cells, it inhibited the PI3K/Akt/mTOR/p70S6K pathway more
thoroughly than in H157 cells. Thus, we speculate that OP-B may be
an alternative agent in the classification and treatment of
NSCLC.
Acknowledgements
We thank members of the Fu laboratory, Emory
University and Jiangsu key laboratory for TCM formulae research,
Nanjing University of Chinese Medicine for assistance and
enlightening discussions. This study was supported in part by
National Science and Technology Pillar Program in the 11th
Five-year Plan of China 2006BAI11B08-01 (to H.F. and X.Z), the
Priority Academic Program Development (PAPD) of Jiangsu Higher
Education Institutions (to X.Z), the Research and Innovation
Program of Postgraduates in Jiangsu Province (to M.C.).
References
|
1
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liang XH, Jackson S, Seaman M, et al:
Induction of autophagy and inhibition of tumorigenesis by beclin 1.
Nature. 402:672–676. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
4
|
Baehrecke EH: Autophagy: dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levy JM and Thorburn A: Targeting
autophagy during cancer therapy to improve clinical outcomes.
Pharmacol Ther. 131:130–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
West KA, Brognard J, Clark AS, et al:
Rapid Akt activation by nicotine and a tobacco carcinogen modulates
the phenotype of normal human airway epithelial cells. J Clin
Invest. 111:81–90. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khan N, Afaq F, Khusro FH, Mustafa Adhami
V, Suh Y and Mukhtar H: Dual inhibition of phosphatidylinositol
3-kinase/Akt and mammalian target of rapamycin signaling in human
nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int
J Cancer. 130:1695–1705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saiki S, Sasazawa Y, Imamichi Y, et al:
Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Codogno P and Meijer AJ: Autophagy and
signaling: their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inoki K, Li Y, Xu T and Guan KL: Rheb
GTPase is a direct target of TSC2 GAP activity and regulates mTOR
signaling. Genes Dev. 17:1829–1834. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inoki K, Li Y, Zhu T, Wu J and Guan KL:
TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signalling. Nat Cell Biol. 4:648–657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garami A, Zwartkruis FJ, Nobukuni T, et
al: Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP
signaling, is inhibited by TSC1 and 2. Mol Cell. 11:1457–1466.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clarke PG: Developmental cell death:
morphological diversity and multiple mechanisms. Anat Embryol.
181:195–213. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fujiwara K, Iwado E, Mills GB, Sawaya R,
Kondo S and Kondo Y: Akt inhibitor shows anticancer and
radiosensitizing effects in malignant glioma cells by inducing
autophagy. Int J Oncol. 31:753–760. 2007.PubMed/NCBI
|
|
16
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blommaart EF, Krause U, Schellens JP,
Vreeling-Sindelarova H and Meijer AJ: The phosphatidylinositol
3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in
isolated rat hepatocytes. Eur J Biochem. 243:240–246. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
|
|
19
|
Arico S, Petiot A, Bauvy C, et al: The
tumor suppressor PTEN positively regulates macroautophagy by
inhibiting the phosphatidylinositol 3-kinase/protein kinase B
pathway. J Biol Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O’Reilly KE, Rojo F, She QB, et al: mTOR
inhibition induces upstream receptor tyrosine kinase signaling and
activates Akt. Cancer Res. 66:1500–1508. 2006.PubMed/NCBI
|