Introduction
A critical area of obesity research is centered on
circulating proteins, the levels of which are altered by changes in
the body mass index. Understanding how these proteins affect
obesity-associated diseases may provide invaluable clues to
constrain the devastating consequences of the current obesity
epidemic (1). One such protein,
visfatin/eNampt/PBEF, has recently received much attention, but its
biological effects and mechanisms of action are not well
understood. Obesity is the risk factor of breast (2), endometrial (3) and hepatocellular cancer (4), as well as skin melanoma (5). It has been reported that visfatin
augments oxidative stress in differentiated myotubes (6) and endothelial cells (7). Nevertheless, the causal relationship
between these facts remains unclear. Visfatin is a novel fat
derived adipocytokine, secreted by visceral and subcutaneous fat
tissues (8,9), hepatocytes (10), monocytes and macrophages (11–13).
Visfatin has both intra- and extracellular forms in mammals
(14,15). An extracellular form of this protein
has been reported to act as a cytokine known as visfatin/PBEF
(16), an insulin-mimetic hormone
(17), or an extracellular NAD
biosynthetic enzyme, visfatin/eNampt (18). Only few studies have investigated
the effect of visfatin on cancer cells in vitro and in
vivo. Increased expression of visfatin was confirmed in ovarian
cancer (19); malignant astrocytoma
(20); the human prostate cancer
cell lines, LNCaP (androgen-sensitive) and PC3
(androgen-insensitive); as well as in human prostate cancer
(21).
Reactive oxygen metabolites (ROMs), such as
superoxide anions (O2*−) hydrogen peroxide
(H2O2), and hydroxyl radical
(OH*), malondialdehyde (MDA) and nitric oxide (NO) are
directly or indirectly involved in the multistage process of
carcinogenesis (22). They are
mainly involved in DNA damage leading to mutations in tumor
suppressor genes. They also act as initiators and/or promoters in
carcinogenesis (23). MDA, a
by-product of lipid peroxidation is likely involved in DNA adduct
formations and DNA damage which are believed to also be responsible
for carcinogenesis (24). To
counteract ROS-induced DNA damage, cells have developed defensive
mechanisms that prevent or repair such damage (25). Antioxidant enzyme systems belong to
those regulatory mechanisms whose role is to protect against
oxidative stress. Under oxidative stress, ROS are rapidly
eliminated by antioxidant enzymes, such as superoxide dismutase
(SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) (26).
No studies exploring the effects of adipokines (such
as visfatin) on oxidative stress, DNA damage in melanoma cells have
been conducted so far. This topic is of utmost interest owing to
the potential paracrine interactions between subcutaneous adipose
tissue and cancer cells. A better understanding of the role of
visfatin in melanoma redox states in vitro may provide sound
insight into the association between obesity-related fat adipokines
and the antioxidant defense system in vitro in cancer
progression.
Materials and methods
Cell culture and reagents
A human malignant melanoma Me45 cell line was
obtained from the Silesian University of Technology, as a kind gift
from Dr M. Wideł from the Marie Curie Memorial Cancer Centre and
Institute of Oncology, Gliwice, Poland. The cells were derived from
metastatic lesions (local lymph nodes) of a 35-year-old patient of
the Institute of Oncology (27).
Me45 cells were plated at the density of 1×106 cells per
25 cm2 flask and cultured in the Dulbecco’s modified
Eagle’s medium with L-glutamine (Sigma-Aldrich, St. Louis, MO, USA)
supplemented with 15% fetal bovine serum (Gibco, North Androver,
MA, USA), antibiotics: penicillin (10,000 IU/ml), streptomycin
(10,000 μg/ml), amphotericin B (2.5 μg/ml) (Sigma-Aldrich) under
the atmosphere of 95% air and 5% CO2 at 37°C. The Me45
cell line was free of mycoplasma, pathogenic viruses and bacteria.
Cultures were maintained for no longer than 4 weeks after recovery
from the frozen stock.
Visfatin (Alexis Biochemical, Plymouth, PA, USA) was
dissolved in phosphate buffered saline (PBS) without
Mg2+, Ca2+ (Sigma-Aldrich). The visfatin
solutions were prepared fresh, protected from light, and added to
the incubation medium obtaining the following final concentrations:
10, 50, 100 and 250 ng/ml. The purity of visfatin was 96–97%
(SDS-PAGE analysis) and contained <0.01 ng/μg LPS as determined
by the Limulus amebocyte lysate method.
Hydroxy-2-naphthalenylmethylphosphonic acid
tris-acetoxymethyl ester [HNMPA-(AM)3], and insulin were
also obtained from Sigma Chemicals Co.
Enzyme assay
After exposure of the cultured cells to visfatin for
24 h, antioxidative enzyme activities: MnSOD, Cu/ZnSOD, GSH-Px,
CAT, and the level of malondialdehyde (MDA) were measured in cell
supernatants. Cells were collected after 24 h of incubation with
different doses of visfatin (10–250 ng/ml) and were centrifuged
(2,000 rpm, 5 min) and supernatants were kept at −80°C until
analysis. All experiments were carried out in duplicate. Sample
size in all experiments was 12.
GSH-Px activity assay
The method of Paglia and Valentine, (28) was used with minor modifications.
Briefly, Me45 melanoma cells were pooled to a concentration of
5×106 cells/μl. Following centrifugation, the cell
pellet was mixed with cell lysis buffer and then sonicated for 10
sec. The protein concentration was measured using Bio-Rad protein
reagent (Bio-Rad Laboratories, Hercules, CA, USA). Equal volumes of
each sample containing 30 μg of protein were mixed with 2.68 ml of
0.05 M phosphate buffer (pH 7.0) containing 0.005 M
ethylenediaminetetraacetic acid (EDTA). The following solutions
were then added sequentially: 0.100 ml of 0.0084 M NADPH, 0.010 ml
of glutathione reductase (GR), 0.010 ml of 1.125 M sodium nitrate
(NaNO3), and 0.100 ml of 0.15 M reduced glutathione
(GSH). The enzymatic reaction was initiated by the addition of
0.100 ml of 0.0022 M H2O2. The conversion of
NADPH to oxidized NADP+ was followed by continuous
recording of the change in absorbency at 340 nm between 2 and 4 min
after the initiation of the reaction. The control measurements were
recorded in a simultaneous assay where the sample was replaced by
an equal volume of cell lysis buffer. GSH-Px enzyme activity (1 IU)
is defined as 1 mM NADPH converted to NADP+ per mg of
protein (IU/mg p).
SOD activity assay
The SOD activity assay was based on a minor
modification of the procedure described by Paoletti and Mocali
(29). The preparation of cell
lysates and the measurement of protein concentration were the same
as in the GSH-Px assay. Protein (30 μg) from each sample was mixed
with 0.8 ml of 1X triethanolamine-diethanolamine buffer (TDB); (pH
7.4), 40 μl of 7.5 mM NADPH (a reduced form of nicotinamide adenine
dinucleotide phosphate), and 25 μl of 100 mM EDTA-MnCl2.
The reaction was initiated by the addition of 0.1 ml of 10 mM
mercaptoethanol. The decrease in absorbance at 340 nm was recorded
over 20 min at room temperature. The control consisted of a
reaction mixture in which the sample was replaced by an equal
volume of cell lysis buffer. For the determination of MnSOD
activity, CuZnSOD activity was inhibited by incubating samples with
5 mM potassium cyanide (KCN) for 30 min with samples assayed for
activity within 2 h of adding cyanide to the sample mixture. Total
specific SOD and MnSOD (after CuZnSOD inhibition with KCN) activity
levels were measured, and then the CuZnSOD activity was calculated.
The enzymatic activity of both SOD isoenzymes was expressed in
nitric units (NU) per mg of protein (NU/mg p); 1 NU represents 50%
inhibition by SOD of the nitrosol ion formation under these
conditions.
CAT activity
Catalase activity was measured
spectrophotometrically as described by Aebi (30). Direct disappearance of 10 mM
hydrogen peroxide in 50 mM potassium phosphate buffer (pH 7.0), 1
mM EDTA was measured at 240 nm >30 sec on a Beckman DU-70
spectrophotometer. Enzyme activity was calculated based on the
molar extinction coefficient of hydrogen peroxide at 240 nm (ɛ =
39.4 M−1 cm−1) and reported as μmol hydrogen
peroxide decomposed per minute, recalculated to kIU per mg of
protein (kIU/mg p).
MDA assay
The measurement of MDA, a product of lipid
peroxidation, was determined by the thiobarbituric acid (TBA)
reaction as described by Placer et al(31). Aliquots of reaction buffer (1.5 ml)
that included 50 μg of protein from each sample and 1.4 ml of 0.2 M
Tris-0.16 M KCl (pH 7.4) were incubated at 37°C for 30 min. Next,
1.5 ml of TBA reagent were added, and the mixture was heated in a
boiling water bath for 10 min. After cooling with ice, 3.0 ml of
pyridine:n-butanol (3:1, v/v) and 1.0 ml of 1 M NaOH were
added and mixed by shaking, and the absorbance was read at 548 nm.
The blank control contained the same reaction mixture but was not
incubated. The level of MDA was expressed as μmol MDA per mg of
protein (μmol MDA/mg p).
Alkaline comet assay
All chemicals were purchased from Sigma. The comet
assay was carried out under alkaline conditions, as described by
Olive and Banáth (32). Fully
frosted slides were covered with 1% normal melting point agarose
(NMP agarose). Me45 cells (6×103), and the same number
of cells pretreated with visfatin at the concentration of 100 ng/ml
for 24 h were washed twice, trypsinized and resuspended in 2 ml
fresh medium. Half of the cells from each group were treated for 5
min with H2O2 (100 μM), and the genotoxic
factor was removed by centrifugation (2,000 rpm/3 min) with
supernatants. Subsequently, cells were resuspended in 2 ml medium
and incubated for 0 min (control for determination of the basal DNA
damage) or 5, 15, 30, 60, 120 and 180 min (for determination of the
residual DNA damage). The resuspended cells (50 μl) were mixed with
100 μl 0.5% low melting agarose at 37°C and transferred (100 μl)
onto microscope slides. The cells not treated with
H2O2 were collected at similar
time-points.
The slides were immersed for 1 h in the ice-cold,
freshly prepared lysis solution (2.5 M NaCl, 100 mM
Na2EDTA, 10 mM Tris-HCl, pH 10.0) with 1% Triton X-100
and 1% dimethylsulfoxide added fresh for cell destruction and DNA
unfolding. The slides were then randomly placed side by side in the
horizontal gel-electrophoresis tank, facing the anode. The unit was
filled with freshly prepared electrophoretic buffer (300 mM NaOH, 1
mM Na2EDTA, pH 13.0) and the slides were set in this
alkaline buffer for 15 min to allow DNA to unwind and express
alkali-labile sites. Denaturation and electrophoresis were
performed at 4°C under dim light. Electrophoresis was carried out
for 20 min at 25 V (300 mA). Following electrophoresis, the slides
were washed gently 2× at 5-min intervals with the neutralization
buffer (0.4 M Tris-HCl, pH 7.5) to remove excess alkali and
detergents. Slides were fixed in chilled 70% ethanol and stained
with ethidium bromide (20 μg/ml) and coverslipped. Slides were
stored at 4°C in humidified sealed containers. To prevent
additional DNA damage, handling samples and preparation of the
slides for the comet assay were conducted under yellow light or in
the dark. To avoid possible position effects during
electrophoresis, two parallel replicate slides per sample were
prepared. Each replicate was processed in a different
electrophoretic run.
The Comet images were visualized after ethidium
bromide staining (20 μg/ml) using a fluorescent microscope
(Axiophot M1, Zeiss, Jena, Germany) that was fitted with an
excitation filter of 515–535 nm and a barrier filter of 590 nm at
×40 magnification. The Comet images were captured using an online
charge-coupled device (CCD) camera and analyzed using the Comet
Score software (version 5.5) from Andor Technology (Belfast, UK).
The tail moments were quantified for a minimum of 100 random cells
per sample using the Comet Score software. The tail moment is
defined as the ratio of DNA in the comet tail to total DNA.
Assessment of cell proliferation
Me45 cell proliferation was assessed using
scintillation counting to measure the incorporation of
[3H]thymidine (2 μCi/ml) into trichloroacetic acid
(TCA)-insoluble material. Cells were plated at
2×104/well in 24-well plates and treated with 10–250
ng/ml visfatin or insulin (100 ng/ml) for 24 h. To study the
effects of insulin receptor (IR) inhibition, HNMPA-(AM)3
(50 μM) was added 3 h before visfatin or insulin, in the presence
of [3H]thymidine. After 24 h, the plates were washed
with PBS and 10% TCA was added to the wells. Incorporated
[3H]thymidine was released by washing with 0.2 N of
NaOH, and radioactivity was measured using β-scintillation counter
(Wallac 1410, Pharmacia, Freiburg, Germany). Results were shown as
counts per minute. To determine visfatin’s effect on the cell
number, melanoma cells were plated into 96-well plates at
2×103/well and treated with 10–250 ng/ml visfatin or 100
ng/ml insulin for 24 h. Cells were harvested by trypsinization, and
the viable ones were counted in an automated cell counter (model
no. TC10; Bio-Rad Laboratories) using 0.04% trypan blue.
Statistical analysis
The normality of distribution was evaluated by means
of Shapiro-Wilk’s test. All results are presented as the means ± SD
or a median (IQR). Results underwent statistical analysis, applying
the one-way ANOVA test with the Bonferroni’s post hoc test
or Kruskal-Wallis with Mann-Whitney U test, according to parameter
distribution. For repeated measures a Friedman test with Schaich
and Hamerle post hoc procedure was used in comet assay data.
P<0.05 was considered to indicate statistically significant
differences.
Results
Influence of visfatin on selected
antioxidative enzyme activities as well as malondialdehyde level in
human malignant melanoma Me45 cells
Visfatin treatment resulted in an increase in
antioxidative enzyme activities SOD: (MnSOD, CuZnSOD isoenzymes)
and CAT, GSH-Px compared to the control group.
SOD isoenzyme assay
SOD isoenzyme activities (MnSOD, CuZnSOD) after 24 h
of incubation with different concentrations of visfatin are shown
in Fig. 1. These experimental
groups also showed a statistically significant increase of the
activities of both isoenzymes, in relation to the control group.
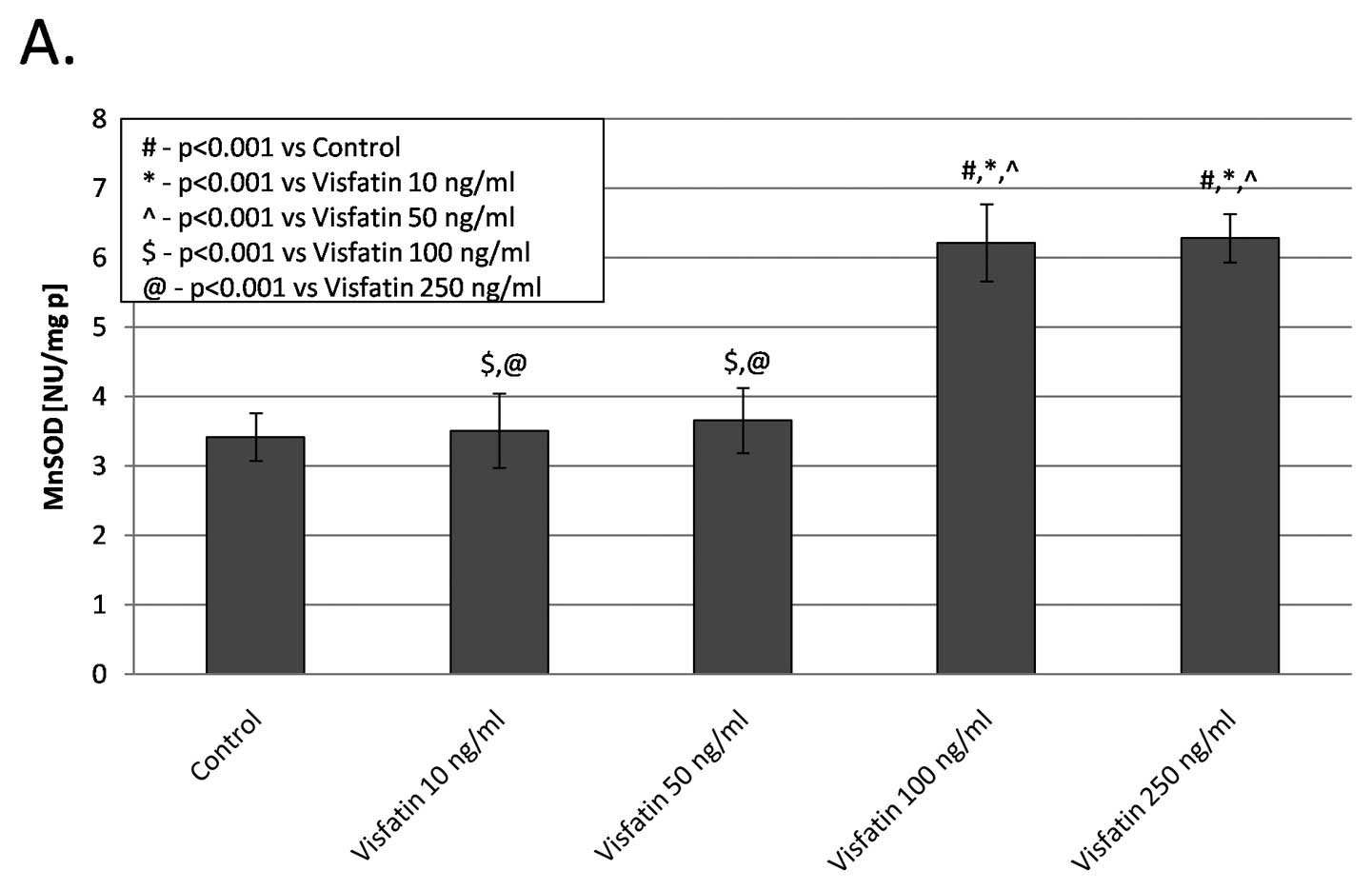
The highest MnSOD isoenzyme activity was observed in the group
treated with visfatin at a 100 and 250 ng/ml concentration compared
to the control group (6.22±0.55, 6.29±0.34 vs. 3.42±0.34 NU/mg p;
p<0.001); (Fig. 1A). The MnSOD
activity in the two other groups treated with visfatin remained
unaltered. All concentrations of visfatin led to a significant
2-fold increase of copper and zinc containing superoxide dismutase
isoenzyme (Cu/ZnSOD) activity when compared to the untreated group;
(3.42±0.36 3.43±0.52 3.52±0.75, 4.01±0.90 vs. 1.43±0.49 NU/mg p;
p<0.001); (Fig. 1B) The
dose-dependency in influencing CuZnSOD by various visfatin
concentrations was not observed.
CAT assay
In comparison to controls, a significant increase in
catalase (CAT) activity in all experimental groups was found at 24
h. CAT activity was ~2-and 2.5-fold higher in the group stimulated
with visfatin at 10 and 50 ng/ml than in the control group
(17.96±1.66, 19.12±1.50 vs. 7.55±1.37 kIU/mg p; p<0.001,
respectively). Moreover, a 4-fold increase in the CAT activity was
observed in the group treated with visfatin at a concentration of
100 and 250 ng/ml with respect to the control group; (32.1±2.83,
35.66±2.51 vs. 7.55±1.37 kIU/mg p; p<0.001; respectively)
(Fig. 2A).
 | Figure 2(A) Catalase (CAT); (B) Glutathione
peroxidase (GSH-Px) enzyme activities; and (C) malondialdehyde
(MDA) levels in the cell supernatants of the human malignant
melanoma Me45 cell line upon visfatin stimulation. C, control
(untreated) group. Data represent the means ± SD; n=12, and were
analyzed with the one-way ANOVA test and post hoc Bonferroni
correction [(enzymes activity; #p<0.001 vs. C,
*p<0.001 vs. 10 ng/ml visfatin,
^p<0.001 vs. 50 ng/ml visfatin,
$p<0.001 vs. 100 ng/ml visfatin,
@p<0.001 vs. 250 ng/ml visfatin); (MDA levels;
#p<0.001 vs. C, *p<0.001 vs. 10 ng/ml
visfatin, ^p<0.001 vs. 50 ng/ml,
$p<0.001 vs. 100 ng/ml, @p<0.05 vs. 250
ng/ml)]. |
GSH-Px assay
GSH-Px activity was ~2-fold higher in the groups
treated with visfatin at 50 and 100 ng/ml compared to the control
group (190.9±31.6, 197.1±20.3 vs. 93.51±6.1 IU/mg p;
@p<0.001, respectively). However a significant
decrease in GSH-Px activity was observed when cells were treated
with visfatin at a 250 ng/ml concentration (55.8±20.03 vs.
93.51±6.1 IU/mg p; p<0.001; respectively) (Fig. 2B).
MDA assay
We also studied the influence of visfatin on the MDA
level in the supernatants of Me45 cells. The MDA level was 2-fold
lower in the group stimulated with 100 ng visfatin compared to the
untreated group (0.79±0.10 vs. 1.49±0.17 μmol MDA/mg p; p<0.001,
respectively). On the contrary, the MDA concentration was increased
in Me45 cells stimulated by visfatin at a 250 ng/ml concentration
(1.69±0.5 vs. 1.49±0.17 μmol MDA/mg p; p<0.001, respectively)
(Fig. 2C).
Evaluation of DNA damage
We also examined the effect of visfatin at the
concentration of 100 ng/ml on DNA damage in Me45 cells. The DNA
damage after an incubation of these cells with the genotoxic factor
H2O2 was compared with cells pre-incubated
with 100 ng/ml of visfatin (24 h) and H2O2 at
different time-points (0–180 min). Fig.
3 shows the level of DNA damage at different time-points (0–180
min) after visfatin and H2O2 treatment, in
comparison to the untreated cells and the cells treated separately,
only with H2O2 or visfatin-control groups.
These results are presented as tail length (Fig. 3A) and median tail moment parameter
(Fig. 3B) with first and third
quartile. In every temporal point after H2O2
removal, the DNA damage (expressed as median tail moment or tail
length) was decreased in the group treated with visfatin and
H2O2, compared to the group treated solely
with H2O2. Me45 melanoma cells treated with
both visfatin and hydrogen peroxide exhibited significantly less
DNA damage than the cells treated solely with hydrogen peroxide
(p<0.05). One hundred and eighty minutes after
H2O2 treatment, the DNA damage level in cells
treated with both visfatin and H2O2 was lower
than in cells treated solely with H2O2
(p<0.05). Addition of visfatin to
H2O2-treated cells resulted in earlier
recovery in DNA damage resulting in similar levels of tail moment
and tail length 120 min after removal of
H2O2. The stationary DNA damage (without
adding H2O2) was also decreased in the
visfatin-treated group when compared to untreated controls (but
only in the tail moment parameter of the DNA damage); p<0.05
(Fig. 3).
Effects of visfatin on the proliferation
and viability of Me45 melanoma cells
The measurement of [3H]thymidine
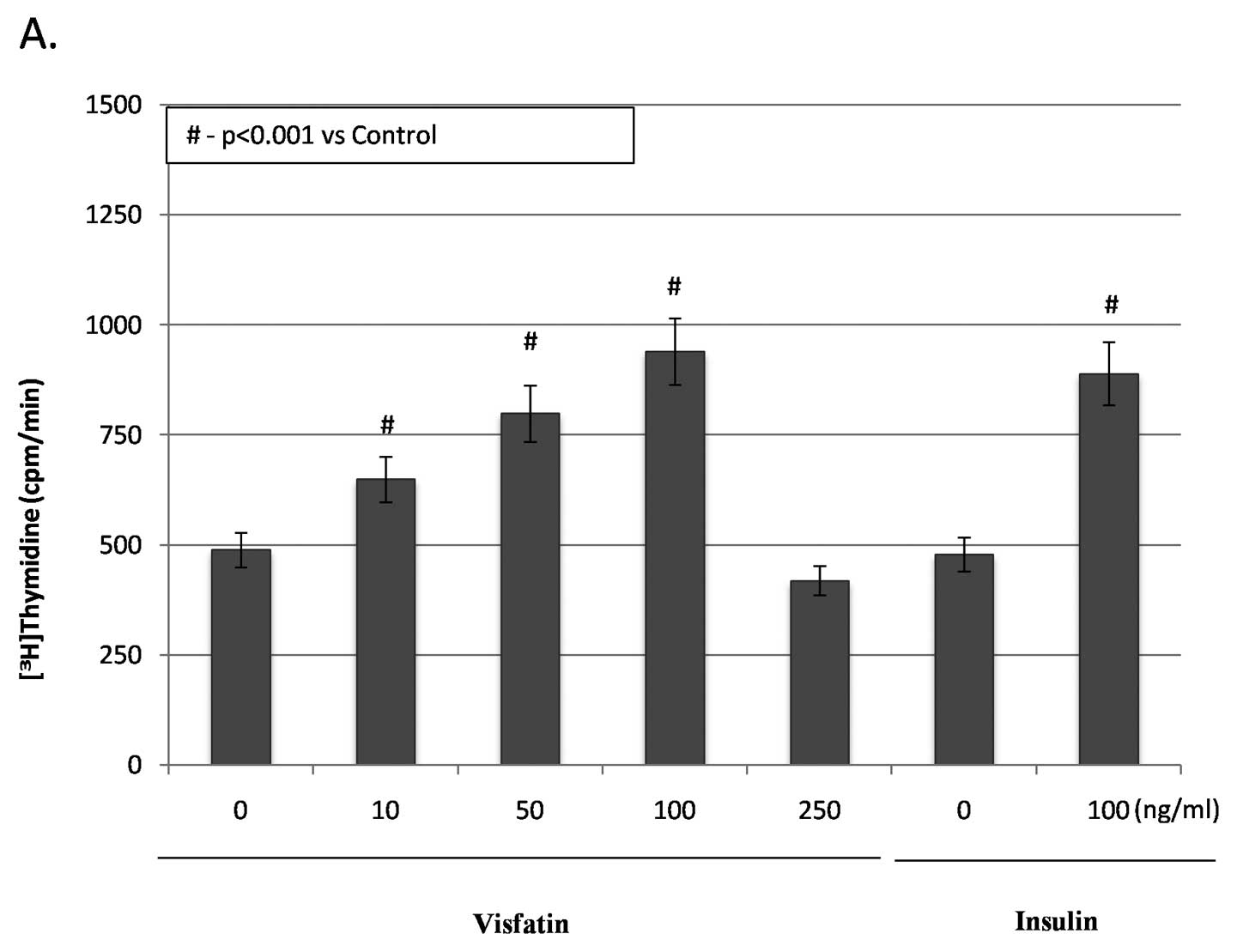
incorporation showed that visfatin at 10–100 ng/ml stimulated
proliferation of Me45 cells in a dose-dependent manner. Visfatin at
the concentration of 250 ng/ml led to a significant decrease in the
proliferation rate of these cells (15%) when compared to untreated
melanoma cells (p<0.05) (Fig.
4A). Compared to the control group, visfatin significantly
increased [3H]thymidine incorporation into melanoma
cells by 32.1% (10 ng/ml), 63% (50 ng/ml) and 91.2% (100 ng/ml)
(all p<0.05) (Fig. 4A).
Moreover, compared to the control group, visfatin at 10, 50 and 100
ng/ml increased the number of viable melanoma cells by 9, 15 and
36.3%, respectively (all p<0.05) (Fig. 4B). On the contrary, visfatin at 250
ng/ml decreased the number of viable melanoma cells by 15%
(Fig. 4B) in comparison to the
untreated group (p<0.05). Fig. 5
shows that insulin also promoted proliferation of Me45 cells.
Insulin at 100 ng/ml markedly increased [3H]thymidine
incorporation of Me45 cells by 85.4% with augment number of viable
cells (24% rise vs. untreated cells; p<0.05). No statistically
significant differences between study groups treated with various
concentrations of visfatin were observed at the level of α= 0.05
(excluding visfatin at the concentration of 250 ng/ml).
Insulin receptor signaling
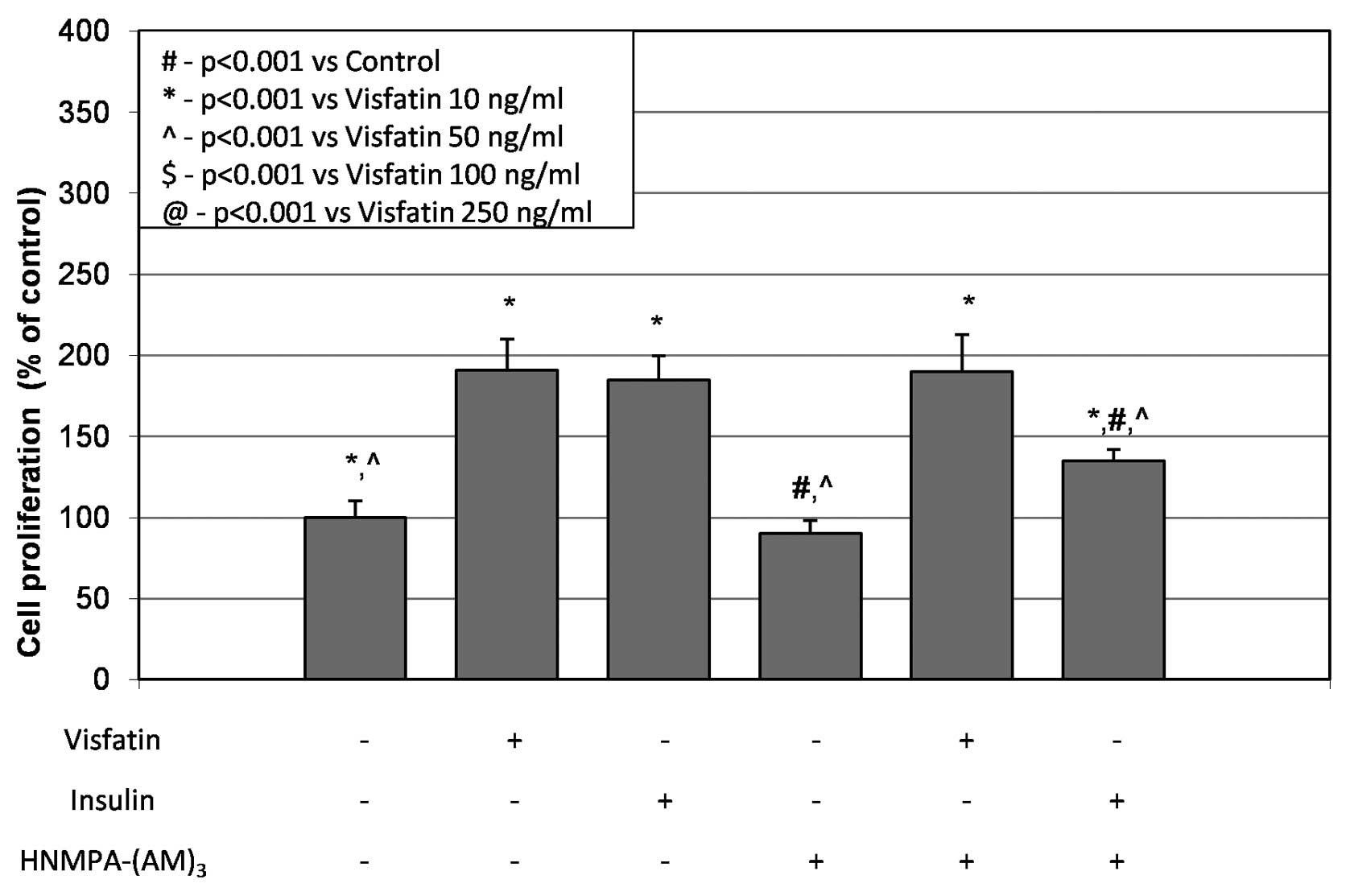
Melanoma cells pretreated with the IR tyrosine
kinase inhibitor HNMPA-(AM)3 did not show reduced
proliferation in visfatin-treated cells (Fig. 5). Additionally, the inhibitor
HNMPA-(AM)3 significantly inhibited insulin-stimulated
proliferation of melanoma Me45 cells by 27% (p<0.05) (Fig. 5). Furthermore, the immunoblot
analysis revealed that IR protein expression (95 kDa) could be
detected in Me45 melanoma lysates (not shown).
Discussion
There are no data describing the interactions in
subcutaneous microenvironments between adipocytes that secrete
various adipokines (such as visfatin) and neighboring melanocytes
as well as melanoma cells. This relationship might affect the
malignant transformation process of melanocytes. Therefore it seems
important to initiate studies that explore the potential paracrine
influence of adipokines on melanocytes and melanoma cells.
We studied the role of fat derived-visfatin on
antioxidative enzymatic activity and the level of lipid
peroxidation process in supernatants of Me45 melanoma cells. In the
present study, we observed increased activity of antioxidant
enzymes (SOD isoenzymes, catalase and glutathione peroxidase) in
cell supernatants upon stimulation with visfatin at the
concentration of 10–100 ng/ml. Increased mitochondrial MnSOD
isoenzyme activity after visfatin treatment suggests that visfatin
increases efficiency of the electron transport chain in the inner
mitochondrial membrane and stimulates complex I and IV of the
electron transport chain in the mitochondrion (33). Cells subjected to the action of the
visfatin at the concentration of 250 ng/ml for 24 h showed higher
mitochondrial oxidative stress compared to the other group. In our
previous study we also observed a marked increase in MnSOD activity
after exposure of AT478 tumor cells to extremely low frequency
electromagnetic field (ELF-EMF) and vitamin E. This action of
ELF-EMF and vitamin E is expected to increase efficiency of
oxidative phosphorylation and adenosine triphosphate production and
the high efficiency of ROS elimination by MnSOD itself (34). In this study we also reported
increase in CuZnSOD superoxide dismutase isoenzyme activity in Me45
cells subjected for 24 h to different concentrations of
visfatin.
SOD isoenzymes catalyze the conversion of
O2− to H2O2, which can then be
converted to water by catalase (CAT) or glutathione peroxidase
(GSH-Px) coupled with glutathione reductase (GR) (26). We observed a marked increase in CAT
activity in Me45 melanoma cells treated with all concentrations of
visfatin. Increased activity of GSH-Px or CAT in cancer cells can
make tumor cells less susceptible to anticancer drugs such as
doxorubicin (36,39). Samuels et al(35) demonstrated that the addition of
radical scavengers and compounds with peroxide activity can reduce
the cytotoxic effect of anticancer drugs in vitro. Increased
GSH-Px or CAT activity enables cells to survive with the high level
of ROS and maintain cellular viability (36).
In our study, application of visfatin (10–100 ng/ml)
also decreased the level of lipid peroxidation in Me45 cells with
augmented GSH-Px activity. On the contrary, MDA concentration was
increased in melanoma cells stimulated by visfatin in higher
concentrations and also decreased GSH-Px activity. ROS are known to
cause DNA damage and lipid peroxidation/oxidation in cellular
membranes (26). MDA is one of the
final products of polyunsaturated fatty acid peroxidation in cells.
An increase in free radicals causes overproduction of MDA (37). Glutathione peroxidase is an enzyme
that reduces hydrogen peroxide as well as organic superoxides. In
such reactions, an organic superoxide (ROOH) becomes reduced to an
appropriate alcohol (ROH). In the case of lipid superoxide this
means that it cannot become an initiator of lipid peroxidation and,
therefore, GSH-Px inhibits the lipid peroxidation process (31,37).
The results of our experiments with low concentrations of MDA in
the group stimulated with 10–100 ng/ml visfatin is concordant with
the high activity of the enzyme inhibiting lipid peroxidation
process-glutathione peroxidase (GSH-Px) in this study group when
compared to the control group. However, we speculate that the Me45
melanoma cells exposed to visfatin at the concentration of 250
ng/ml may show decreased resistance toward exogenic
H2O2 and higher oxidative stress in
comparison to the cells treated with lower concentrations of
visfatin.
We suppose that visfatin at the concentration of
10–100 ng/ml triggers a redox adaptation response, leading to an
upregulation of the antioxidant capacity (SOD isoenzymes, CAT and
GSH-Px enzymes).
Moreover, the increased oxidative-stress induces
oxidative DNA damage and lipid peroxidation process. We
hypothesized that visfatin mediated activation of antioxidant
enzymes with decreased levels of MDA may lead to the reduction of
DNA damage in Me45 melanoma cells. We pretreated these cells with
visfatin at the concentration of 100 ng/ml for 24 h. At every
time-point after H2O2 removal, the DNA damage
(expressed as median tail moment or tail length) was decreased in
the group treated with visfatin and H2O2
compared to the group treated solely with
H2O2. The results suggested a protective role
of visfatin in the Me45 melanoma cells exposed to the genotoxic
reagents. Pretreatment with visfatin before adding hydrogen
peroxide decreased DNA damage in the Me45 cells. Our observation of
increased antioxidant enzyme activity in melanoma cells after
visfatin stimulation shows that enzymatic defense mechanisms are
used for ROS scavenging and reduce the level of DNA damage in these
cells. Moreover, the stationary DNA damage (without adding
H2O2) was also decreased in the
visfatin-treated group when compared to the untreated group (but
only in tail moment parameter of DNA damage). Collectively, we
demonstrated for the first time the protective role of visfatin in
hydrogen peroxide-induced DNA damage in a human malignant melanoma
cell line. However, Wang et al(38) demonstrated that visfatin exerted
anti-apoptotic effects under a cell apoptotic state induced by
H2O2 in vascular smooth muscle cells
(38). Moreover, Adya et
al(7) reported that visfatin
exerted a protective role in H2O2-induced
endothelial cell apoptosis. Visfatin at 100 ng/ml in combination
with H2O2 (200 μM) markedly increased the
number of viable cells by 73% in comparison to the group treated
solely with hydrogen peroxide (only 19% of viable endothelial cells
in the culture) (7). This is
consistent with our report, which demonstrates that visfatin at 100
ng/ml also increased the level of viable melanoma cells in the
group (visfatin + H2O2) along with a
decreased level of DNA damage when compared to cells treated solely
with H2O2.
We also examined the effect of visfatin on Me45
melanoma cell proliferation. Previous studies reported that
visfatin stimulates proliferation of osteoblasts (39), vascular smooth muscle cells (VSMC)
(38) and prostate cancer cells
(PC-3 line) (21). The present
study demonstrated that visfatin (10–100 ng/ml), similar to
insulin, stimulates proliferation of Me45 cells in vitro in
a dose-dependent manner. Patel et al(21) also reported that visfatin (25–400
ng/ml) increases proliferation of the PC-3 prostate cancer cell
line in a dose-dependent manner after 24 h of incubation. The most
pronounced proliferation rate increase (approximately 2-fold) was
observed in the PC-3 cells subjected to visfatin at the
concentration of 400 ng/ml. In the present study, an almost 2-fold
increase of the proliferation rate was observed in Me45 cells
treated with 100 ng/ml visfatin with increased number of tumor
viable cells. The supraphysiological concentration of visfatin in
culture medium was similar to the concentrations used in other
in vitro studies (6,7,13,17,21,38,39).
These observation are concordant with a report which shows that
increased GSH-Px or CAT activity enables cells to survive with the
high level of ROS and maintain cellular viability (36). By contrast, in this case, high
concentration of visfatin (250 ng/ml) led to a significant decrease
of [3H]thymidine incorporation of Me45 cells with
respect to the untreated cells. The effect on proliferation was
demonstrated in a concentration-dependent manner and perhaps more
importantly at levels of visfatin similar to those that have been
demonstrated in obese patients (40). This provides novel evidence that
elevated levels of circulating visfatin may influence the growth of
melanoma cancer cells. According to other research studies,
visfatin can mimic the effects of insulin on the activation of
insulin signaling (17,39). In particular, two studies in
monocytes and in osteoblasts have indicated that IR may be involved
in visfatin function; both studies demonstrated that visfatin
effects could be blocked by IR tyrosine kinase inhibitor,
HNMPA-(AM)3(13,39). However, recent studies indicated
that visfatin cannot activate IR (41) and proposed that visfatin may act on
its own unidentified receptor (42). Therefore, to determine whether
visfatin-induced Me45 melanoma cell proliferation is mediated by
IR, we used HNMPA-(AM)3 - inhibitor of IR to elucidate
this matter, with insulin as a comparison. In our study melanoma
cultures pretreated with visfatin and the IR tyrosine kinase
inhibitor HNMPA-(AM)3 acted similarly to cells cultured
in the absence of the latter. These observations indicated that
visfatin-induced melanoma cell proliferation is not mediated by the
IR transduction signaling pathway. We also demonstrated that the
HNMPA-(AM)3 significantly inhibited insulin-stimulated
proliferation of melanoma Me45 cells.
Our study has a few limitations. Its in vitro
setting may not fully reflect more complex relationships in
organisms as a whole. Additionally, we observed an increased
proliferation rate occurring during cell culture. However the
magnitude of proliferation was relatively much lower than the
effect of visfatin on studied parameters, therefore we considered
the influence of this observation as negligible in the study
results.
In conclusion, this study provides the first
evidence that visfatin can reduce the level of DNA damage of Me45
melanoma cells, probably due to increased levels of antioxidative
enzyme activities, and can promote melanoma cell proliferation in a
dose-dependent manner.
Acknowledgements
This study was supported by grants from the Silesian
Medical University in Katowice, Poland. Grant nos. KNW-1-134/08,
KNW-1-078/09 and KNW-1-040/P/1/0.
References
|
1
|
Tilg H and Moschen AR: Role of adiponectin
and PBEF/visfatin as regulators of inflammation: involvement in
obesity-associated diseases. Clin Sci. 114:275–288. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cappellani A, Di Vita M, Zanghi A,
Cavallaro A, Piccolo G, Veroux M, Berretta M, Malaguarnera M,
Canzonieri V and Lo Menzo E: Diet, obesity and breast cancer: an
update. Front Biosci (Schol Ed). 4:90–108. 2012. View Article : Google Scholar
|
|
3
|
Thomas CC, Wingo PA, Dolan MS, Lee NC and
Richardson LC: Endometrial cancer risk among younger, overweight
women. Obstet Gynecol. 114:22–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larsson SC and Wolk A: Overweight, obesity
and risk of liver cancer: a meta-analysis of cohort studies. Br J
Cancer. 97:1005–1008. 2007.PubMed/NCBI
|
|
5
|
Dennis LK, Lowe JB, Lynch CF and Alavanja
MC: Cutaneous melanoma and obesity in the Agricultural Health
Study. Ann Epidemiol. 18:214–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oita RC, Ferdinando D, Wilson S, Bunce C
and Mazzatti DJ: Visfatin induces oxidative stress in
differentiated C2C12 myotubes in an Akt- and MAPK-independent,
NFκB-dependent manner. Pflugers Arch. 459:619–630. 2010.PubMed/NCBI
|
|
7
|
Adya R, Tan BK, Punn A, Chen J and Randeva
HS: Visfatin induces human endothelial VEGF and MMP-2/9 production
via MAPK and PI3K/Akt signalling pathways: novel insights into
visfatin-induced angiogenesis. Cardiovasc Res. 78:356–365. 2008.
View Article : Google Scholar
|
|
8
|
Berndt J, Klöting N, Kralisch S, Kovacs P,
Fasshauer M, Schön MR, Stumvoll M and Blüher M: Plasma visfatin
concentrations and fat depot-specific mRNA expression in humans.
Diabetes. 54:2911–2916. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanaka M, Nozaki M, Fukuhara A, Segawa K,
Aoki N, Matsuda M, Komuro R and Shimomura I: Visfatin is released
from 3T3-L1 adipocytes via a non-classical pathway. Biochem Biophys
Res Commun. 359:194–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garten A, Petzold S, Barnikol-Oettler A,
Körner A, Thasler WE, Kratzsch J, Kiess W and Gebhardt R:
Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is
constitutively released from human hepatocytes. Biochem Biophys Res
Commun. 391:376–381. 2010. View Article : Google Scholar
|
|
11
|
Friebe D, Neef M, Kratzsch J, Erbs S,
Dittrich K, Garten A, Petzold-Quinque S, Blüher S, Reinehr T,
Stumvoll M, Blüher M, Kiess W and Körner A: Leucocytes are a major
source of circulating nicotinamide phosphoribosyltransferase
(NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and
inflammation in humans. Diabetologia. 12:1200–1211. 2011.
|
|
12
|
Laudes M, Oberhauser F, Schulte DM, Freude
S, Bilkovski R, Mauer J, Rappl G, Abken H, Hahn M, Schulz O and
Krone W: Visfatin/PBEF/Nampt and resistin expressions in
circulating blood monocytes are differentially related to obesity
and type 2 diabetes in humans. Horm Metab Res. 42:268–273. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dahl TB, Yndestad A, Skjelland M, Øie E,
Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, Bendz
B, Tonstad S, Gullestad L, Frøland SS, Krohg-Sørensen K, Russell D,
Aukrust P and Halvorsen B: Increased expression of visfatin in
macrophages of human unstable carotid and coronary atherosclerosis:
possible role in inflammation and plaque destabilization.
Circulation. 115:972–980. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sommer G, Garten A, Petzold S,
Beck-Sickinger AG, Blüher M, Stumvoll M and Fasshauer M:
Visfatin/PBEF/Nampt: structure, regulation and potential function
of a novel adipokine. Clin Sci. 115:13–23. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dahl TB, Holm S, Aukrust P and Halvorsen
B: Visfatin/NAMPT: a multifaceted molecule with diverse roles in
physiology and pathophysiology. Annu Rev Nutr. 32:229–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moschen AR, Kaser A, Enrich B, Mosheimer
B, Theurl M, Niederegger H and Tilg H: Visfatin, an adipocytokine
with proinflammatory and immunomodulating properties. J Immunol.
178:1748–1758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukuhara A, Matsuda M, Nishizawa M, Segawa
K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T,
Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S,
Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R,
Matsuzawa Y and Shimomura I: Visfatin: a protein secreted by
visceral fat that mimics the effects of insulin. Science.
307:426–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Imai S: Nicotinamide
phosphoribosyltransferase (Nampt): a link between NAD biology,
metabolism, and diseases. Curr Pharm Des. 15:20–28. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shackelford RE, Bui MM, Coppola D and
Hakam A: Over-expression of nicotinamide phosphoribosyltransferase
in ovarian cancers. Int J Clin Exp Pathol. 3:522–527.
2010.PubMed/NCBI
|
|
20
|
Reddy PS, Umesh S, Thota B, Tandon A,
Pandey P, Hegde AS, Balasubramaniam A, Chandramouli BA, Santosh V,
Rao MR, Kondaiah P and Somasundaram K: PBEF1/NAmPRTase/Visfatin: a
potential malignant astrocytoma/glioblastoma serum marker with
prognostic value. Cancer Biol Ther. 7:663–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel ST, Mistry T, Brown JE, Digby JE,
Adya R, Desai KM and Randeva HS: A novel role for the adipokine
visfatin/pre-B cell colony-enhancing factor 1 in prostate
carcinogenesis. Peptides. 31:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park SH, Ozden O, Jiang H, Cha YI,
Pennington JD, Aykin-Burns N, Spitz DR, Gius D and Kim HS: Sirt3,
mitochondrial ROS, ageing, and carcinogenesis. Int J Mol Sci.
12:6226–6239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kong W, Kuester RK, Gallegos A and Sipes
IG: Induction of DNA damage in human urothelial cells by the
brominated flame retardant 2,2-bis(bromomethyl)-1,3-propanediol:
role of oxidative stress. Toxicology. 290:271–277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Angeli JP, Garcia CC, Sena F, Freitas FP,
Miyamoto S, Medeiros MH and Di Mascio P: Lipid
hydroperoxide-induced and hemoglobin-enhanced oxidative damage to
colon cancer cells. Free Radic Biol Med. 51:503–515. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hegde ML, Hazra TK and Mitra S: Early
steps in the DNA base excision/single-strand interruption repair
pathway in mammalian cells. Cell Res. 18:27–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: a radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kramer-Marek G, Serpa C, Szurko A, Widel
M, Sochanik A, Snietura M, Kus P, Nunes RM, Arnaut LG and Ratuszna
A: Spectroscopic properties and photodynamic effects of new
lipophilic porphyrin derivatives: efficacy, localisation and cell
death pathways. J Photochem Photobiol B. 84:1–14. 2006. View Article : Google Scholar
|
|
28
|
Paglia DE and Valentine WN: Studies on the
quantitative and qualitative characterization of erythrocyte
glutathione peroxidase. J Lab Clin Med. 70:158–169. 1967.PubMed/NCBI
|
|
29
|
Paoletti F and Mocali A: Determination of
superoxide dismutase activity by purely chemical system based on
NAD(P)H oxidation. Methods Enzymol. 186:209–220. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aebi H: Catalase in vitro. Methods
Enzymol. 105:121–126. 1984. View Article : Google Scholar
|
|
31
|
Placer ZA, Cushman LL and Johnson BC:
Estimation of product of lipid peroxidation (malonyl dialdehyde) in
biochemical systems. Anal Biochem. 16:359–364. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Olive PL and Banáth JP: Detection of DNA
double-strand breaks through the cell cycle after exposure to
X-rays, bleomycin, etoposide and 125IdUrd. Int J Radiat
Biol. 64:349–356. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Polaniak R, Bułdak RJ, Karoń M, Birkner K,
Kukla M, Zwirska-Korczala K and Birkner E: Influence of an
extremely low frequency magnetic field (ELF-EMF) on antioxidative
vitamin E properties in AT478 murine squamous cell carcinoma
culture in vitro. Int J Toxicol. 29:221–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Samuels BL, Murray JL, Cohen MB, Safa AR,
Sinha BK, Townsend AJ, Beckett MA and Weichselbaum RR: Increased
glutathione peroxidase activity in a human sarcoma cell line with
inherent doxorubicin resistance. Cancer Res. 51:521–527.
1991.PubMed/NCBI
|
|
36
|
Lenehan PF, Gutiérrez PL, Wagner JL, Milak
N, Fisher GR and Ross DD: Resistance to oxidants associated with
elevated catalase activity in HL-60 leukemia cells that overexpress
multidrug-resistance protein does not contribute to the resistance
to daunorubicin manifested by these cells. Cancer Chemother
Pharmacol. 35:377–386. 1995. View Article : Google Scholar
|
|
37
|
Gaweł S, Wardas M, Niedworok E and Wardas
P: Malondialdehyde (MDA) as a lipid peroxidation marker. Wiad Lek.
57:453–455. 2004.
|
|
38
|
Wang P, Xu TY, Guan YF, Su DF, Fan GR and
Miao CY: Perivascular adipose tissue-derived visfatin is a vascular
smooth muscle cell growth factor: role of nicotinamide
mononucleotide. Cardiovasc Res. 81:370–380. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xie H, Tang SY, Luo XH, Huang J, Cui RR,
Yuan LQ, Zhou HD, Wu XP and Liao EY: Insulin-like effects of
visfatin on human osteoblasts. Calcif Tissue Int. 80:201–210. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Haider DG, Schindler K, Schaller G, Prager
G, Wolzt M and Ludvik B: Increased plasma visfatin concentrations
in morbidly obese subjects are reduced after gastric banding. J
Clin Endocrinol Metab. 91:1578–1581. 2006. View Article : Google Scholar
|
|
41
|
Revollo JR, Körner A, Mills KF, Satoh A,
Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR,
Milbrandt J, Kiess W and Imai S: Nampt/PBEF/Visfatin regulates
insulin secretion in beta cells as a systemic NAD biosynthetic
enzyme. Cell Metab. 6:363–375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH
and Koo TH: Visfatin enhances ICAM-1 and VCAM-1 expression through
ROS-dependent NF-kappaB activation in endothelial cells. Biochim
Biophys Acta. 1783:886–895. 2008. View Article : Google Scholar : PubMed/NCBI
|