Introduction
Cyclooxygenases (COXs) are catalytic enzymes that
are necessary for the conversion of arachidonic acid into
prostaglandin (PG) G2 and subsequently to
PGH2, which is a precursor for the synthesis of
prostanoids, including PGs, prostacyclins and thromboxanes. There
are three COX isozymes: COX-1, COX-2 and COX-3. COX-1, which is
constitutively expressed in various cell types, plays an important
role in homeostatic PG synthesis. For example, COX-1-derived
prostanoids contribute to platelet aggregation and cytoprotective
effects in the stomach. COX-3 is a COX-1 splice variant and is
expressed primarily in the brain and spinal cord; however, the
detailed function of COX-3 remains unknown (1). The COX-2 isozyme is induced during
inflammation, and the development of several types of cancer, such
as colon, breast and prostate cancer, is closely associated with
chronic inflammation (2–5). Notably, the chronic use of aspirin has
been shown to reduce the incidence and progression of colorectal
cancer, including familial adenomatous polyposis (6). Furthermore, breast cancer patients
using aspirin exhibit a reduced risk of distant recurrence and
mortality related to breast cancer (7). However, traditional non-steroidal
anti-inflammatory drugs (NSAIDs) such as aspirin may produce
adverse gastrointestinal effects due to the inhibition of COX-1.
Newer selective COX-2 inhibitors, such as celecoxib, which is used
to ameliorate adverse gastrointestinal tract-related effects, may
be useful as chemopreventive agents and anticancer drugs against
various cancers due to increased COX-2-specific targeting.
Therefore, COX-2 may provide a potential therapeutic target for the
chemoprevention of cancer.
Canine mammary tumors have long been considered a
suitable animal model for human breast cancer. Recent research has
focused on the similarities and differences in the molecular
alterations that occur in canine vs. human mammary tumors. Canine
mammary tumors are the most common tumors in female dogs, and ~50%
are diagnosed as malignant tumors. Furthermore, one study reported
that the mammary tumors in 58% of dogs were relapsed tumors that
appeared after initial removal by a regional mastectomy (8). Clinically, mammary tumors are a
prominent canine disease, and the establishment of a new treatment
for canine mammary tumors is urgently required. Similar to the
findings for human breast cancer, the elevation of COX-2 expression
in canine mammary tumors, compared with benign tumors (adenoma),
tends to be associated with increasing malignancy (adenocarcinoma)
(9). Furthermore, COX-2 expression
has been shown to be absent or weak in normal mammary gland tissue
(9,10). Therefore, these observations suggest
the potential utility of COX-2 as a therapeutic target, which may
also reduce the negative impact on normal cells.
COX-2 has attracted attention as a potential tool
for chemoprevention and chemotherapy for canine mammary tumors
(11). However, the antitumor
effects of selective COX-2 inhibitors against canine mammary tumor
cells are essentially unknown. Therefore, the objective of the
present study was to determine the antitumor effect of selective
COX-2 inhibitors in canine mammary tumor cells. Furthermore, using
canine mammary tumor cells, we compared the antitumor-effect
intensity of three selective COX-2 inhibitors: celecoxib, etodolac
and meloxicam.
Materials and methods
COX inhibitors
To evaluate the antitumor effect of selective COX-2
inhibitors against canine mammary tumor cell lines, we used
COX-2-selective NSAIDs (celecoxib, etodolac and meloxicam).
Celecoxib and etodolac were purchased from Sigma Aldrich (Tokyo,
Japan). Meloxicam was purchased from Wako Pure Chemicals Ltd.
(Osaka, Japan). The stock solutions of the COX-2-selective
inhibitors were dissolved in DMSO. The final concentration of DMSO
in the culture medium was adjusted to 0.1% in all the experiments.
The control cells were treated with 0.1% DMSO. The term ‘parent
cells’ was used to refer to non-treated cells.
Cell culture
Canine mammary tumor (CF33) cells were purchased
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The cells were cultured as previously described (12,13).
AZACB cells were purchased from Primary Cell Co., Ltd. (Hokkaido,
Japan). The AZACB cells were cultured in a manner similar to that
used for the CF33 cells.
Cell proliferation analysis
To evaluate the effect of COX-2 selective inhibitors
on cell proliferation, the untreated and treated cells were
subjected to a WST-8 assay using Cell Counting Kit-8 (Dojindo
Laboratories, Kumamoto, Japan). The CF33 cells were seeded at a
density of 1×103 cells/well into 96-well plates (BD
Falcon, Tokyo, Japan). At each time-point (days 0–4), 10 μl of
CCK-8 reagent was added. After a 2-h incubation, the absorbance was
measured at 450 nm using a Benchmark Plus microplate reader
(Bio-Rad Laboratories, Tokyo, Japan). In these experiments, 5
replicate wells were used for each time-point.
Cell cycle and apoptosis analyses
CF33 cells were seeded at a density of
2.5×105 cells/100-mm dish (BD Falcon). After the cells
were treated, they were harvested and washed with PBS, resuspended
in 70% ethanol in PBS, and incubated at −30°C overnight. Prior to
the analysis, the cells were mixed and incubated in the dark for 15
min in PI/RNase staining buffer (BD Pharmingen, San Jose, CA, USA).
The suspension was then filtered through a 5-ml polystyrene
round-bottom tube with a cell-strainer cap (Becton-Dickinson,
Franklin Lakes, NJ, USA) and analyzed using FACSCanto (Becton
Dickinson) and FlowJo 7 (Tree Star, Ashland, OR, USA).
Trypan blue exclusion assay
Cells were seeded at a density of 1×105
cells in 60-mm dishes at 24 h before DMSO or COX-2 inhibitor
treatment. The cells were incubated in the presence of meloxicam
(100 μM), etodolac (100 μM) and celecoxib (100 μM) for 24 h. The
cells were washed with PBS, trypsinized and resuspended in PBS. The
suspended cells were incubated with 0.4% (w/v) trypan blue solution
(Wako Pure Chemical Industries) for 1 min at room temperature, and
the number of stained cells was counted.
Real-time RT-PCR
Total RNA was extracted from cells using the TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions and our previously described methods
(12–14). cDNA was synthesized using a
PrimeScript™ RT reagent kit (Perfect Real-Time) (Takara Bio, Shiga,
Japan) according to the manufacturer’s protocols. Real-time PCR was
performed using SYBR Premix Ex Taq™II (Tli RNaseH Plus) (Takara
Bio) and the ABI Prism 7500 Real-Time PCR system (Applied
Biosystems, Foster City, CA, USA) under the following conditions:
95°C for 30 sec and 40 cycles each of 95°C for 5 sec and 60°C for
34 sec. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression
was used as an internal control. The primer sequences are shown in
Table I. The primers for BAX and
Bcl-2 were purchased from Takara Bio, and the primer for GAPDH was
obtained from Operon Biotechnologies (Tokyo, Japan). All the
samples were amplified in triplicate in each experiment. The
relative levels of mRNA were calculated using the ΔΔCt method.
 | Table IPrimer sequences for real-time
RT-PCR. |
Table I
Primer sequences for real-time
RT-PCR.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|
| GAPDH |
ATTCTATCCACGGCAAATCC |
GGACTCCACAACATACTCAG |
| BAX |
CGCATCGGAGATGAACTGGA |
ACCAGTTTGCTGGCAAAGTAGAAG |
| Bcl-2 |
TGAACCGGCATCTGCACAC |
GAGCAGCGCCTTCAGAGACA |
Western blotting
Whole cell lysates were harvested using RIPA buffer
containing protease inhibitors. The total cell lysate extraction
was performed as previously described (14). The protein concentration of the cell
lysates was measured using a Pierce® BCA Protein Assay
kit (Pierce, Rockford, IL, USA). Then, the cell lysates (15–20 μg)
were boiled for 5 min in Laemmli sample buffer before being
subjected to electrophoresis on 12% gel (BAX) and 10% (COX-2)
SDS-PAGE gels and were subsequently transferred to PVDF membranes
(Bio- Rad). The primary antibody against BAX was purchased from
Cell Signaling Technology (Tokyo, Japan), the anti-COX-2 antibody
was obtained from Abcam (Tokyo, Japan), and the anti-actin antibody
was purchased from Sigma Aldrich. The immune complex was detected
using WesternBright Sirius Western Blotting Detection kit
(Advansta, Menlo Park, CA, USA) according to the manufacturer’s
instructions.
Quantification of the caspase-3 and
caspase-7 activities
To determine the activity of caspase-3 and
caspase-7, we analyzed the treated and untreated cells using
Caspase-Glo®3/7 assays (Promega, Tokyo, Japan) according
to the manufacturer’s instructions. The fluorescence was measured
using an ARVO™1420 Multilabel Counter (Perkin-Elmer, Tokyo,
Japan).
Identifying the stages of apoptosis
The stages of apoptosis were analyzed using the
ApoAlert® Annexin V-FITC Apoptosis kit (Clontech
Laboratories, Inc., Palo Alto, CA, USA) according to the
manufacturer’s instructions. The cells were seeded at a density of
2.5×105 cells/well in 100-mm dishes (BD Falcon). Both
adherent and non-adherent cells were harvested (trypsin, 0.25%) and
centrifuged. After washing and then resuspending the cell pellets
in binding buffer, the cells were incubated with Annexin V-FITC and
PI for 15 min in the dark at room temperature. The samples were
analyzed using FACSCanto and FlowJo 7.
Statistical analysis
To determine significant differences between the
selective COX-2 inhibitor-treated cells and the control cells,
statistical analysis was performed using paired two-tailed
Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
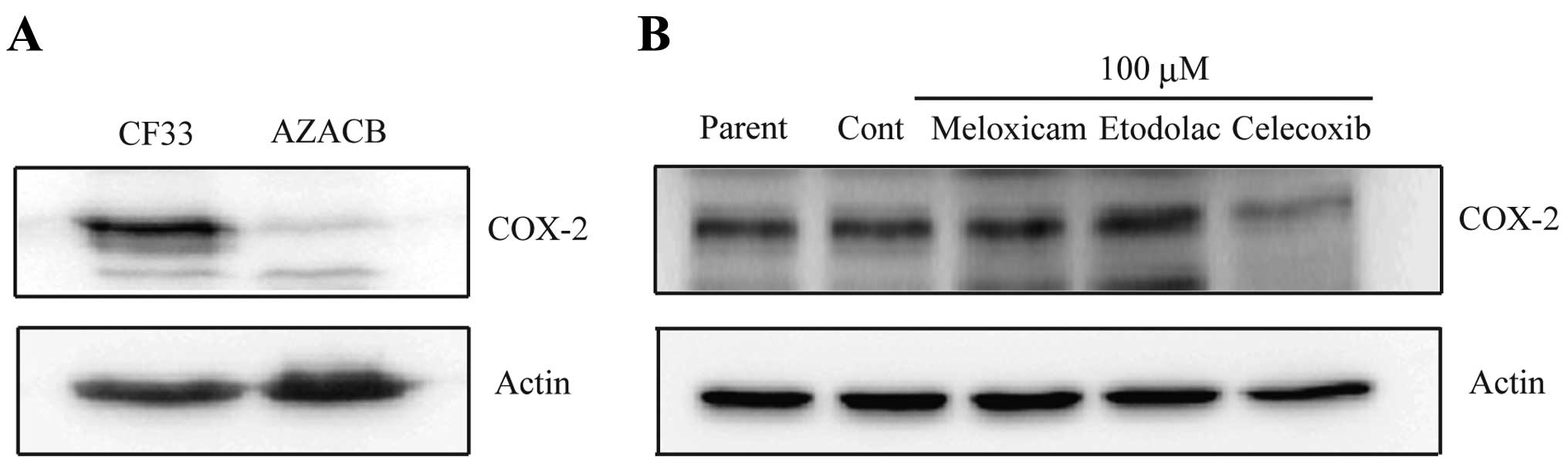
Celecoxib reduces COX-2 expression in
CF33 cells
Immunohistochemical analysis has shown a tendency of
an increase in COX-2 expression in canine mammary tumors compared
to benign tumors (9). Therefore, we
examined and compared COX-2 protein expression in CF33 and AZACB
cells. The CF33 cells exhibited higher COX-2 protein levels than
the AZACB cells (Fig. 1A). This
result may indicate that COX-2 contributes to maintaining the
malignant phenotype of CF33 cells. To determine the antitumor
effect of selective COX-2 inhibitors in canine mammary tumor cells,
we used CF33 cell lines that highly expressed COX-2. To clarify
whether selective COX-2 inhibitors affect COX-2 expression
patterns, we compared COX-2 protein levels in selective COX-2
inhibitor-treated CF33 cells. COX-2 protein expression levels were
markedly downregulated after 24 h of celecoxib treatment (100 μM)
(Fig. 1B). However, meloxicam or
etodolac treatment did not alter COX-2 expression (Fig. 1B). This finding may suggest that
celecoxib effectively inhibits the function of COX-2 in canine
mammary tumor cells. Similar results have also been observed in
human cancer cell lines (15).
Celecoxib-induced COX-2 downregulation is thought to occur at the
transcriptional level due to the inhibition of NF-κB activity
(16).
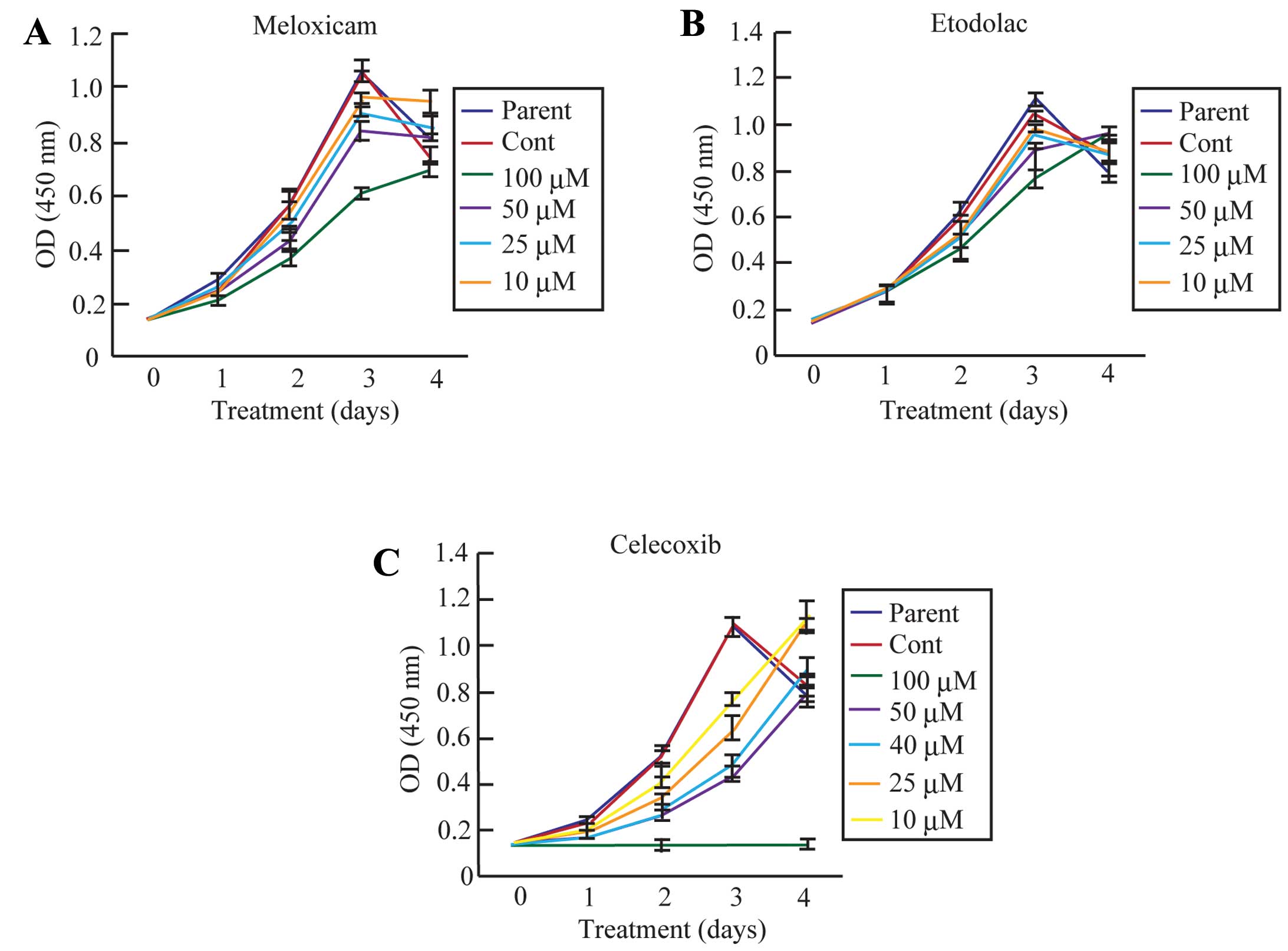
Selective COX-2 inhibitors, especially
celecoxib, markedly inhibit CF33 cell proliferation via a decrease
in the percentage of S-phase cells and induction of G0/G1
arrest
A number of studies have provided conclusive
evidence that selective COX-2 inhibitors possess potential
chemopreventative and chemotherapeutic activity in human breast
cancer patients. To analyze the effect of selective COX-2
inhibitors on CF33 cell proliferation, we utilized a WST-8 assay at
0, 1, 2, 3 and 4 days following the addition of each selective
COX-2 inhibitor (meloxicam, etodolac and celecoxib) (Fig. 2). The number of parent and control
cells increased steadily and nearly equally for 4 days after
plating. However, the rate of CF33 cell proliferation was
suppressed in a dose-dependent manner following meloxicam, etodolac
and celecoxib treatment (Fig. 2A).
Notably, CF33 cell proliferation was completely blocked from days
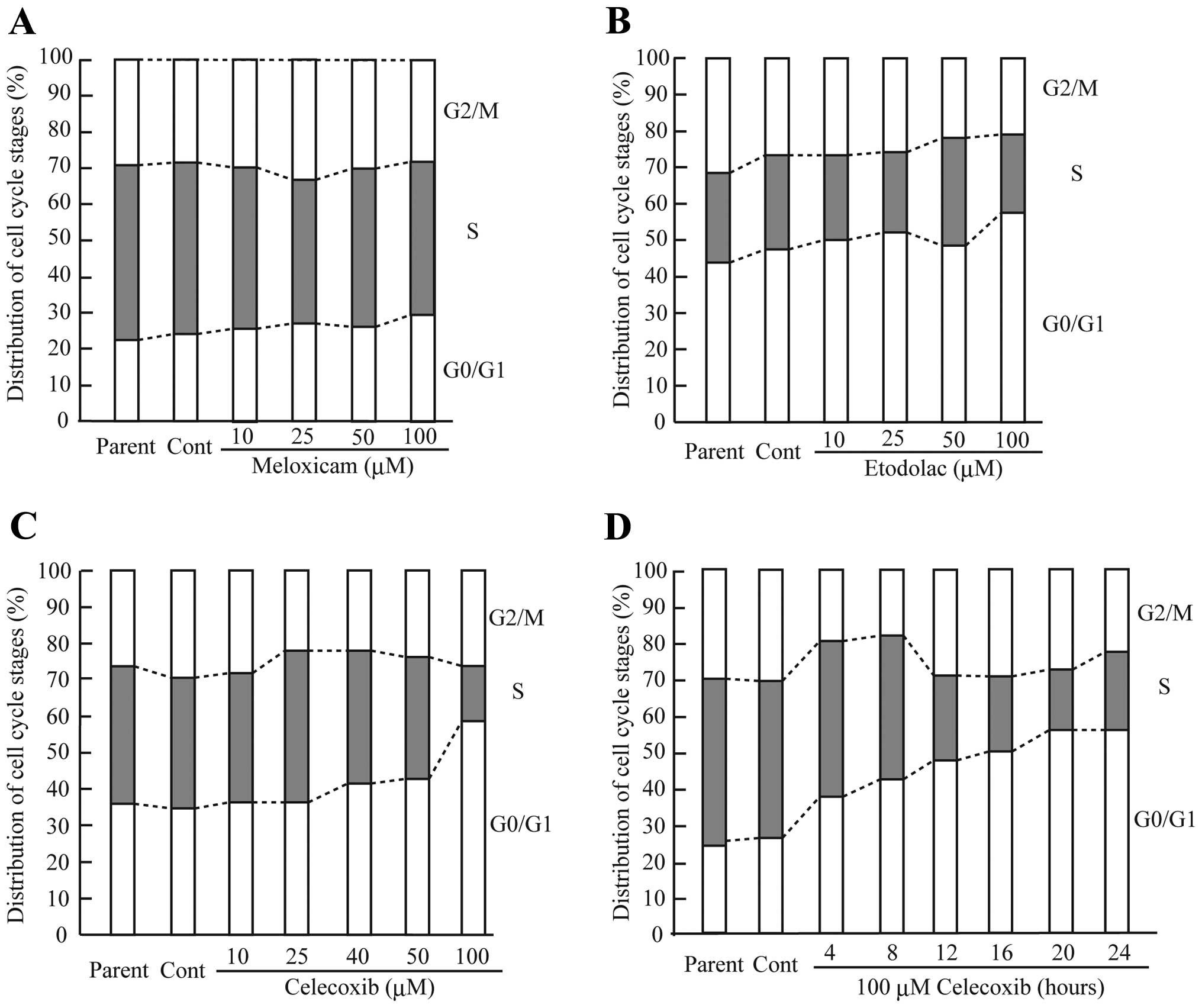
1–4 after the celecoxib (100 μM) treatment (Fig. 2C). The cells treated with celecoxib
(100 μM) exhibited a marked decrease in the percentage of S-phase
cells and an increase in G0/G1 arrest (Fig. 3C). Moreover, the celecoxib (100
μM)-treated cells exhibited a marked alteration in the percentage
of S-phase and G0/G1-phase cells.
Meloxicam (100 μM) and etodolac (100 μM) treatment
also strongly inhibited CF33 cell proliferation (Fig. 2A and B). However, G0/G1 arrest was
slightly induced in the CF33 cells treated with a higher dose of
meloxicam and etodolac (Fig. 3A and
B). These data suggest that celecoxib strongly induced the
inhibition of cell proliferation in canine mammary tumor cells.
Furthermore, these results indicate that selective COX-2 inhibitors
are capable of inhibiting the cell proliferation of canine mammary
tumor cells, similar to the inhibition observed in human breast
cancer cells from previous studies.
Celecoxib is a more powerful apoptosis
inducer than etodolac or meloxicam in canine mammary tumor
cells
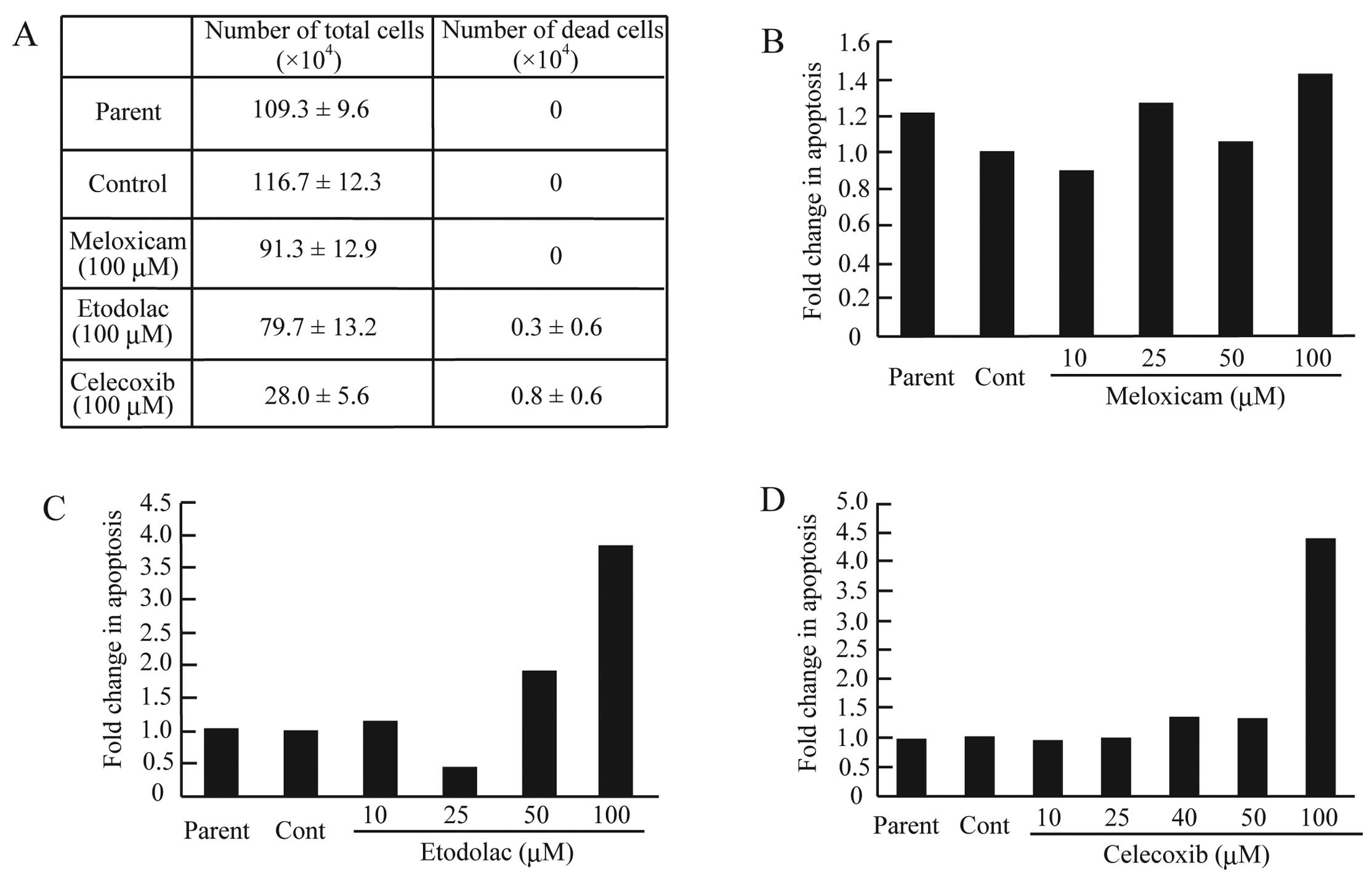
Selective COX-2 inhibitors markedly suppressed the
proliferation of CF33 cells in our experiments. To determine
whether selective COX-2 inhibitors induced apoptosis in the CF33
cells, the cells were exposed to meloxicam, etodolac or celecoxib
for 24 h and were then analyzed using a trypan blue exclusion
assay. In the meloxicam-treated cells, dead or apoptotic cells were
not observed (Fig. 4A and B).
However, treatment with etodolac (100 μM) or celecoxib (100 μM)
increased the number of dead cells [(0.3±0.6) × 104 and
(0.8±0.6) × 104, respectively] (Fig. 4A). To determine whether the
etodolac- or celecoxib-induced CF33 cell death was a result of
apoptosis, the cells were stained with PI and analyzed using FACS.
Apoptosis was induced more frequently in the CF33 cells treated
with a high dose of etodolac or celecoxib (Fig. 4C and D). Specifically, the treatment
with 100 μM of etodolac or celecoxib elevated the proportion of
apoptotic cells by ~3.7- and 4.6-fold, respectively (Fig. 4C and D). The imbalance between
pro-apoptotic molecules (BAX and BAK) and anti-apoptotic molecules
(Bcl-2 and Bcl-XL) induces apoptosis mediated by the
stimulation of mitochondrial outer membrane permeabilization (MOMP)
(17). To further clarify the
effect of selective COX-2 inhibitors on apoptosis, we measured
BAX and Bcl-2 mRNA and BAX protein expression levels
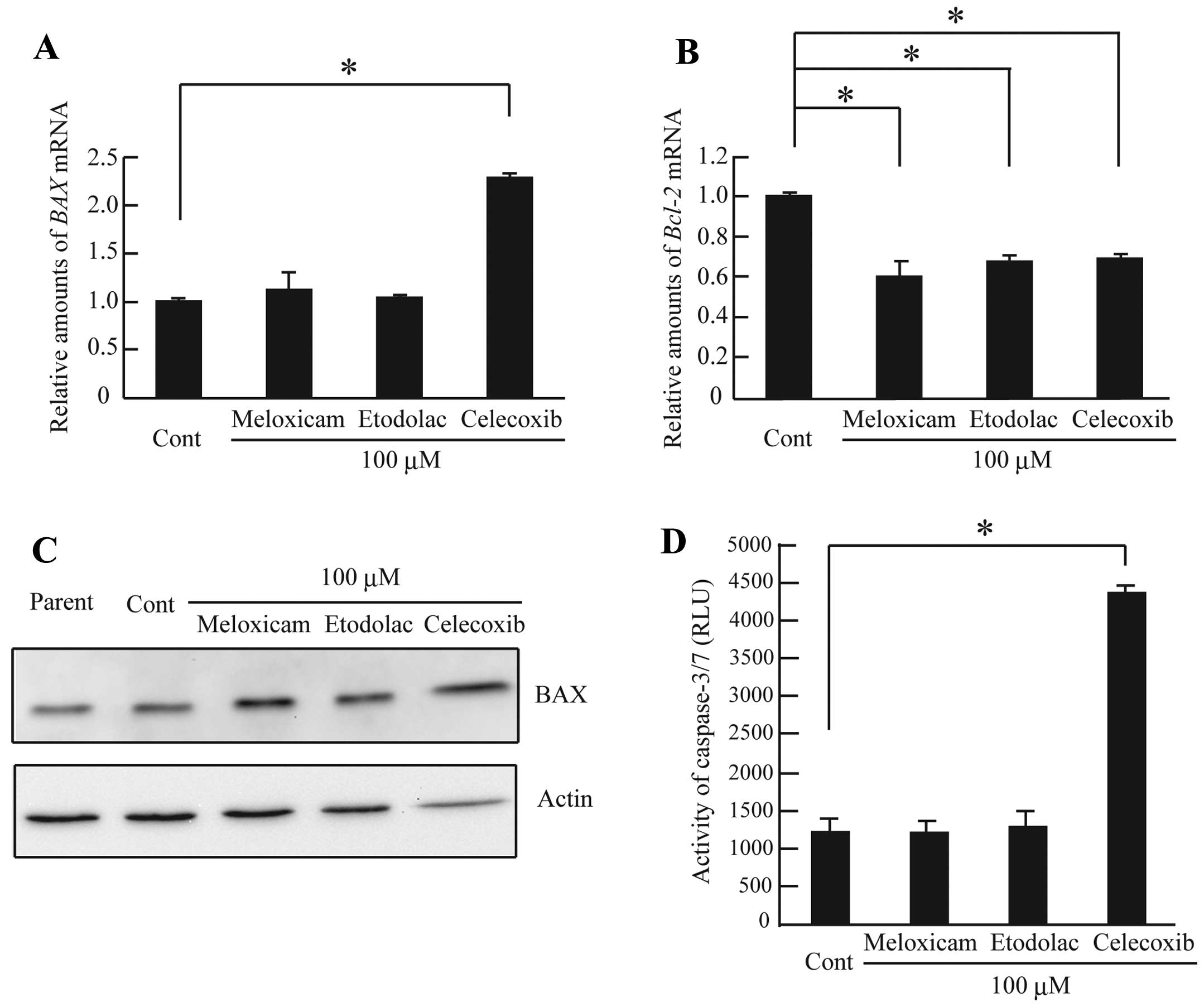
using real-time RT-PCR and western blotting. Celecoxib treatment
caused a marked increase in BAX mRNA expression and a
decrease in Bcl-2 mRNA expression compared with the control
cells (Fig. 5A and B). In addition,
we observed that celecoxib treatment caused a significant increase
in BAX protein levels compared with that in the other cells
(Fig. 5C). However, the
Bcl-2 mRNA levels were also downregulated in the CF33 cells
treated with the other selective COX-2 inhibitors (Fig. 5B). The activation of the effectors
caspase-3 and caspase-7 following the activation of the initiator
caspase-9 or caspase-8 is important for the induction of apoptosis
(18). To further clarify the
effect of selective COX-2 inhibitors on apoptosis, we analyzed the
degree of caspase-3/7 activity. Only the celecoxib-treated cells
induced the activation of caspase-3 and caspase-7 (Fig. 5D). To further confirm the induction
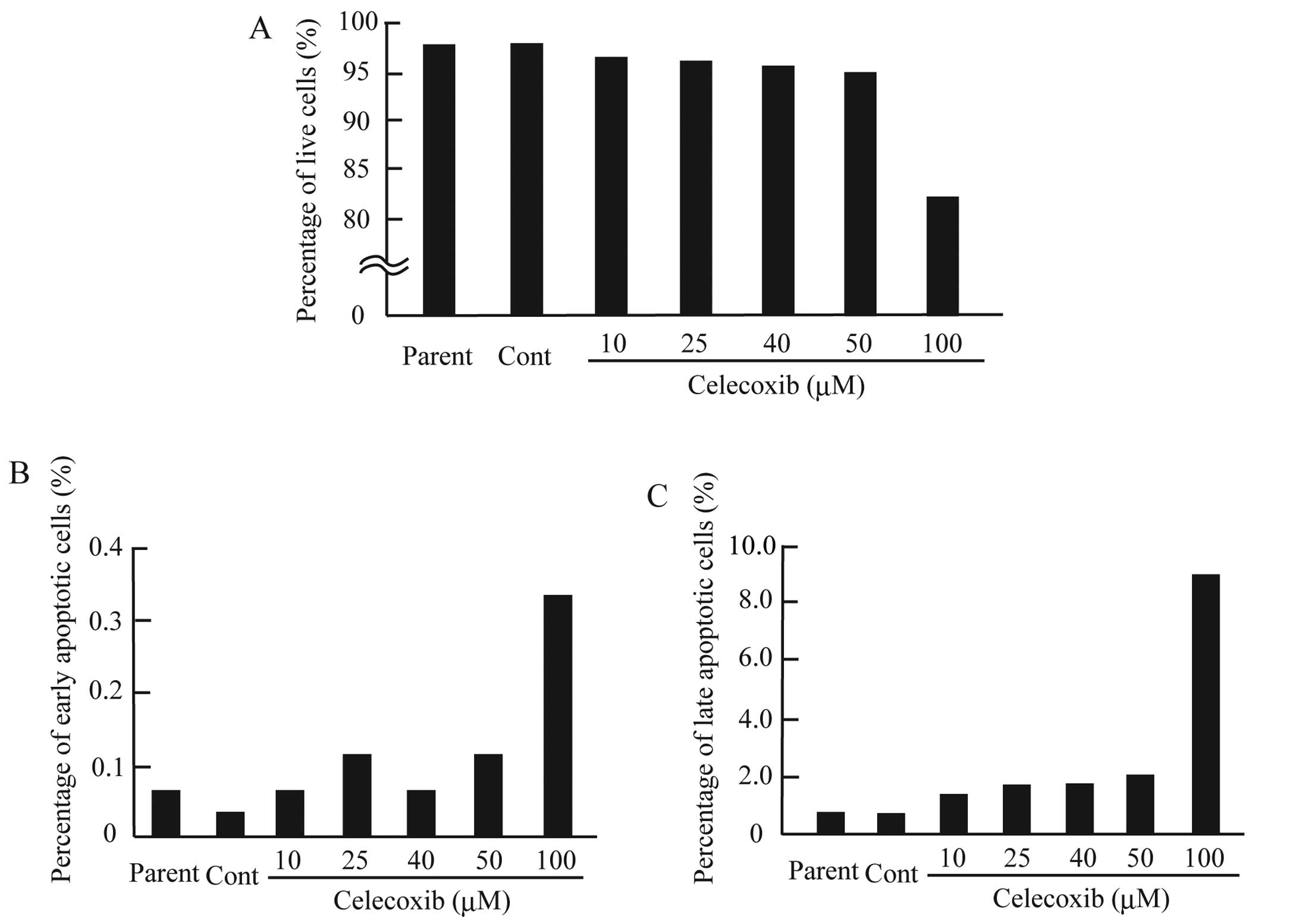
of apoptosis in the CF33 cells treated with celecoxib, we assessed
apoptosis using Annexin V and PI staining. Our results demonstrated
that celecoxib (100 μM) treatment decreased the CF33 cell survival
rate and increased the percentages of both early and late apoptotic
CF33 cells (Fig. 6). These results
may indicate that celecoxib induces strong cell proliferation
inhibition in canine mammary tumor cells by activating the
intrinsic apoptosis pathway. Our data indicate that celecoxib and
etodolac may induce apoptosis in canine mammary tumor cells.
However, meloxicam did not induce an apoptotic effect in canine
mammary tumor cells in the present study.
Discussion
Recent studies have demonstrated that NSAIDs,
especially selective COX-2 inhibitors, are useful for chemotherapy
or chemoprevention of various human cancers via the induction of
apoptosis and the inhibition of cell growth (19,20).
However, the mechanism of action of selective COX-2 inhibitors
against canine mammary tumors remains unclear; therefore, the aim
of the present study was to elucidate the mechanism of action of
selective COX-2 inhibitors against canine mammary tumor cells. Our
experiments revealed that selective COX-2 inhibitors inhibited the
proliferation of canine CF33 mammary tumor cells. Specifically, our
findings demonstrated that celecoxib induced CF33 apoptosis more
robustly than meloxicam and etodolac treatment in CF33 cells.
Furthermore, our findings indicated that celecoxib-induced
apoptosis is mediated by the activation of the intrinsic apoptosis
pathway. These results suggest that celecoxib has a strong
antitumor potency in canine mammary tumor cells.
Compared with the findings for normal tissue, COX-2
expression has been observed to be elevated in various human
premalignant and malignant tumors, such as colorectal, breast and
lung cancers (21). Similarly, in
canines, COX-2 is overexpressed in breast and prostate cancers
(22). Compared with the findings
for canine mammary adenoma tumors, COX-2 expression is increased in
cases of canine mammary adenocarcinoma with malignant phenotypes
(9). Furthermore, metastatic
lesions of malignant mammary tumor tissue exhibited intense
immunohistochemical COX-2 staining (23). These previous reports indicate that
COX-2 plays an important role in the initiation and promotion of
mammary tumors and the maintenance of the malignant phenotype of
canine mammary tumors. The present results demonstrated that
celecoxib induced apoptosis, which was associated with a
downregulation of COX-2 protein expression levels in canine mammary
tumor cells. Therefore, this finding supports the hypothesis that
celecoxib is a useful therapeutic agent for COX-2-positive canine
mammary tumors.
In vertebrate cells, caspase-dependent apoptosis is
divided into two pathways, the intrinsic and extrinsic pathways,
which are induced by different initiation cascades (24–26).
The intrinsic apoptosis pathway (also known as the mitochondrial
pathway or the stress pathway) is initiated by a variety of
chemical or physical stressors, such as DNA damage by UV
irradiation and endoplasmic reticulum stress (27). However, the extrinsic apoptosis
pathway is initiated via the interaction of a death receptor (FAS
and TNF-α) with its ligand (FAS-L and TNF-α L) (25,26).
Whether the mechanism of celecoxib-induced apoptosis occurs through
the intrinsic pathway or the extrinsic pathway remains
controversial. Some studies have proposed that celecoxib initiates
the extrinsic apoptosis pathway mediated by activating caspase-8
via the induction of DR5 (TRAIL receptor 2) expression (28). However, several studies have
reported that celecoxib induces apoptosis by activating the
intrinsic apoptosis pathway (29,30).
Our observation indicates that celecoxib activated the intrinsic
apoptosis pathway in canine mammary tumor cells by activating
caspase-3/7 via the downregulation of Bcl-2 and the upregulation of
BAX. Furthermore, PGE2 treatment has been shown to
inhibit the induction of apoptosis in human colon cancer cells with
selective COX-2 inhibitor-mediated Bcl-2 upregulation (31). The present study also suggests that
celecoxib directly affects the expression levels of Bcl-2 in canine
mammary tumor cells.
In patients with a history of colorectal neoplasms,
the chronic administration of celecoxib increases the risk for
serious cardiovascular side-effects compared to that for the
placebo group (32), and other
coxib-class drugs can produce similar side-effects. This phenomenon
may be explained by an imbalance between prostacyclin
(PGI2) and thromboxane A2 (TXA2),
which is caused by the inhibition of COX-2-derived PGI2
in endothelial cells without the inhibition of COX-1-dependent
TXA2 production in platelets (33). To determine the inhibition of COX-2
activity by selective COX-2 inhibitors, we analyzed the alteration
of COX-2 activity and PGE2 production of CF33 cells.
However, we did not observe any changes in CF33 COX-2 activity and
PGE2 production after treatment with selective COX-2
inhibitors (meloxicam, etodolac and celecoxib) (data not shown).
Celecoxib analogs have previously been shown to exhibit an
antitumor effect through a COX-2-independent mechanism (34). Our results raise the possibility of
selective COX-2 inhibitor-mediated CF33 cell proliferation
inhibition via a COX-2-independent manner. Furthermore, since COX-2
activity was not inhibited, the chronic administration of selective
COX-2 inhibitors in canines may not cause the severe cardiovascular
side-effects observed in humans.
In conclusion, our results indicate that selective
COX-2 inhibitors may be a viable option for chemotherapy or
chemoprevention against canine mammary tumors. In canine mammary
tumors, celecoxib would function as a chemotherapeutic agent by
inducing apoptosis. Furthermore, our findings also provide
additional evidence that COX-2 is a suitable for therapeutic and
preventative target in canine mammary tumors. In the future, it may
be possible to use a combination of other antitumor drugs and
selective COX-2 inhibitors as a treatment protocol for canine
mammary tumors.
Acknowledgements
We thank H. Sugiya and T. Narita for the critical
discussions. The present study was supported in part by a
Grant-in-Aid from Nihon University (to T.S.) and funds from the
Laboratory of Veterinary Pharmacology, Nihon University College of
Bioresource Sciences.
References
|
1
|
Chandrasekharan NV, Dai H, Roos KL,
Evanson NK, Tomsik J, Elton TS and Simmons DL: COX-3, a
cyclooxygenase-1 variant inhibited by acetaminophen and other
analgesic/antipyretic drugs: cloning, structure, and expression.
Proc Natl Acad Sci USA. 99:13926–13931. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dennis LK, Lynch CF and Torner JC:
Epidemiologic association between prostatitis and prostate cancer.
Urology. 60:78–83. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Howe LR: Inflammation and breast cancer.
Cyclooxygenase/prostaglandin signaling and breast cancer. Breast
Cancer Res. 9:2102007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liao X, Lochhead P, Nishihara R, et al:
Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival.
N Engl J Med. 367:1596–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Holmes MD, Chen WY, Li L, Hertzmark E,
Spiegelman D and Hankinson SE: Aspirin intake and survival after
breast cancer. J Clin Oncol. 28:1467–1472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stratmann N, Failing K, Richter A and
Wehrend A: Mammary tumor recurrence in bitches after regional
mastectomy. Vet Surg. 37:82–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Doré M, Lanthier I and Sirois J:
Cyclooxygenase-2 expression in canine mammary tumors. Vet Pathol.
40:207–212. 2003.
|
|
10
|
Queiroga FL, Alves A, Pires I and Lopes C:
Expression of Cox-1 and Cox-2 in canine mammary tumours. J Comp
Pathol. 136:177–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klopfleisch R, von Euler H, Sarli G, Pinho
SS, Gartner F and Gruber AD: Molecular carcinogenesis of canine
mammary tumors: news from an old disease. Vet Pathol. 48:98–116.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saito T, Tamura D, Nakamura T, et al:
4-methylumbelliferone leads to growth arrest and apoptosis in
canine mammary tumor cells. Oncol Rep. 29:335–342. 2013.PubMed/NCBI
|
|
13
|
Saito T, Dai T and Asano R: The hyaluronan
synthesis inhibitor 4-methylumbelliferone exhibits antitumor
effects against mesenchymal-like canine mammary tumor cells. Oncol
Lett. 5:1068–1074. 2013.
|
|
14
|
Saito T, Kawana H, Azuma K, Toyoda A,
Fujita H, Kitagawa M and Harigaya K: Fragmented hyaluronan is an
autocrine chemokinetic motility factor supported by the
HAS2-HYAL2/CD44 system on the plasma membrane. Int J Oncol.
39:1311–1320. 2011.PubMed/NCBI
|
|
15
|
Zhang GS, Liu DS, Dai CW and Li RJ:
Antitumor effects of celecoxib on K562 leukemia cells are mediated
by cell-cycle arrest, caspase-3 activation, and downregulation of
Cox-2 expression and are synergistic with hydroxyurea or imatinib.
Am J Hematol. 81:242–255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shishodia S, Koul D and Aggarwal BB:
Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced
NF-κB activation through inhibition of activation of IκBα kinase
and Akt in human non-small cell lung carcinoma: correlation with
suppression of COX-2 synthesis. J Immunol. 173:2011–2022.
2004.PubMed/NCBI
|
|
17
|
Chipuk JE and Green DR: How do BCL-2
proteins induce mitochondrial outer membrane permeabilization?
Trends Cell Biol. 18:157–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
William WN Jr, Heymach JV, Kim ES and
Lippman SM: Molecular targets for cancer chemoprevention. Nat Rev
Drug Discov. 8:213–225. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harris RE: Cyclooxygenase-2 (COX-2)
blockade in the chemoprevention of cancers of the colon, breast,
prostate, and lung. Inflammopharmacology. 17:55–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dannenberg AJ and Subbaramaiah K:
Targeting cyclooxygenase-2 in human neoplasia: rationale and
promise. Cancer Cell. 4:431–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doré M: Cyclooxygenase-2 expression in
animal cancers. Vet Pathol. 48:254–265. 2011.
|
|
23
|
Dias Pereira P, Lopes CC, Matos AJ, Santos
M, Gärtner F, Medeiros R and Lopes C: COX-2 expression in canine
normal and neoplastic mammary gland. J Comp Pathol. 140:247–253.
2009.PubMed/NCBI
|
|
24
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Igney FH and Krammer PH: Death and
anti-death: tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tait SW and Green DR: Mitochondria and
cell death: outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu X, Yue P, Zhou Z, Khuri FR and Sun SY:
Death receptor regulation and celecoxib-induced apoptosis in human
lung cancer cells. J Natl Cancer Inst. 96:1769–1780. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jendrossek V, Handrick R and Belka C:
Celecoxib activates a novel mitochondrial apoptosis signaling
pathway. FASEB J. 17:1547–1549. 2003.PubMed/NCBI
|
|
30
|
Jendrossek V: Targeting apoptosis pathways
by Celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sheng H, Shao J, Morrow JD, Beauchamp RD
and DuBois RN: Modulation of apoptosis and Bcl-2 expression by
prostaglandin E2 in human colon cancer cells. Cancer
Res. 58:362–366. 1998.PubMed/NCBI
|
|
32
|
Solomon SD, McMurray JJ, Pfeffer MA, et
al: Cardiovascular risk associated with celecoxib in a clinical
trial for colorectal adenoma prevention. N Engl J Med.
352:1071–1080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fitzgerald GA: Coxibs and cardiovascular
disease. N Engl J Med. 351:1709–1711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schönthal AH, Chen TC, Hofman FM, Louie SG
and Petasis NA: Celecoxib analogs that lack COX-2 inhibitory
function: preclinical development of novel anticancer drugs. Expert
Opin Investig Drugs. 17:197–208. 2008.PubMed/NCBI
|