Introduction
The incidence of esophageal adenocarcinoma (EA) has
risen rapidly in the last two decades (1–3). Since
EA is associated with poor prognosis, recent studies have mainly
focused on prevention. Barrett’s esophagus (BE) is the precursor
lesion of EA, associated with gastroesophageal reflux disease
(GERD) which heralds the sequence: GERD, inflammation, BE,
low-grade dysplasia, high-grade dysplasia, carcinoma in situ
and invasive adenocarcinoma (4).
Numerous epidemiological studies have demonstrated a
protective effect of non-steroidal anti-inflammatory drugs
(NSAIDs), including acetylsalicylic acid (ASA), against several
types of gastrointestinal tumors and EA (5). The main effect of NSAIDs and ASA
appears to be inhibition of cyclooxygenase enzymes (COX-1 and
COX-2) which are involved in the synthesis of prostaglandins.
Several in vivo animal studies in experimental models of
chronic esophagitis and EA have shown that COX-2 is overexpressed
during the GERD-EA sequence. In these studies, the administration
of COX inhibitors reduced the incidence of esophageal cancer. In
addition, upregulation of COX-2 has been described at both the mRNA
and protein levels in humans with GERD, BE or EA (6).
Although the protective effect of NSAIDs against
several gastrointestinal (GI) tumors has been well demonstrated,
the long-term use of these drugs or the newer and safer COX-2
selective inhibitors as chemopreventive agents has been dismissed
due to their adverse cardiovascular effects (7,8).
Aspirin is the only drug that combines a protective effect on the
cardiovascular system and the prevention of some types of GI cancer
(9,10).
The aim of the present study was to ascertain
whether the administration of aspirin prevents the development of
EA in a rat model of gastroesophageal reflux.
Materials and methods
Animals
All animal studies were carried out in the Animal
Research Unit of the University of Zaragoza, an officially
recognized research establishment that practices adequate
husbandry; all research animals were individually housed and used
in accordance with the Good Laboratory Practice guidelines. All
procedures were approved by the University of Zaragoza Ethics
Committee for Animal Experiments. The care and use of animals were
performed in accordance with the Spanish Policy for Animal
Protection RD1201/05, which meets the European Union Directive
86/609 on the protection of animals used for experimental and other
scientific purposes. Throughout the experiment, all rats were
housed in specifically designed cages (EU Dim; IFFA Credo) and kept
in computer-controlled conditions of light (12-h light/dark cycle),
noise (45–50 dB), temperature (21±2°C) and humidity (60–70%). The
animals were cared for by veterinary staff. A total of 50
5-week-old female Wistar rats (Harlan Laboratories, Inc.,
Barcelona, Spain) weighing 220–300 g were used for the present
study.
Experimental model
Solid food was withdrawn for 24 h and water for 6–8
h before surgery. Anesthesia was induced and maintained with an
isoflurane-air mixture. Before surgery, an intramuscular injection
of 100 mg/kg body weight cefazoline (Kefol; Lilly, Madrid, Spain)
was administered as antibiotic prophylaxis. Rats underwent
esophagojejunostomy with gastric preservation, allowing the
gastroduodenal content to flow back into the esophagus. Surgery was
performed according to a previously described protocol (11). This model is widely used since it
reproduces the sequence of histological changes that occur in human
esophageal adenocarcinogenesis and shows morphological and
phenotypic characteristics similar to those observed in human
Barrett’s esophagus and associated adenocarcinoma, including mucin
features and expression of differentiation markers and markers of
neoplastic progression (12,13).
After surgery, the rats were housed in hanging cages to prevent bed
ingestion and allowed to drink water ad libitum, with
fasting until day 3. Water with 0.3 mg/ml buprenorphine (Buprex;
Esteve, Barcelona, Spain) was provided during the first 72
postoperative hours. An additional intramuscular injection of
cefazoline was administered 24 h after surgery. Rats received 50
mg/kg iron dextran (Difortin; Econatura, Madrid, Spain)
intramuscularly once every 4 weeks until the end of the experiment.
Rats were sacrificed by carbon dioxide inhalation 4 months after
surgery. A blood sample was obtained for drug analysis from all of
the animals immediately before death. Immediately after death, the
entire esophagus with 0.5 cm of the jejunum (including the
anastomosis) was removed, and the specimen was opened
longitudinally. In all cases, biochemical determinations were
performed in the area of the esophagus proximal to the anastomosis,
where the sequence inflammation-intestinal
metaplasia-dysplasia-adenocarcinoma takes place in this model. For
these purposes, two 1-mm-wide longitudinal slices were cut out of
the lower half of the esophagus and frozen immediately in liquid
nitrogen. These were stored at −80°C until biochemical studies were
performed. The rest of the esophageal specimen was prepared for
pathological study.
General design of the experiment
Two weeks after surgery, the 50 surviving rats were
randomly divided into three groups: Group I: n=17, control
(vehicle); Group II: n=16, treatment with ASA 50 mg/kg body weight
per day; Group III: n=17, treatment with ASA 5 mg/kg body weight
per day. Only those rats that were sacrificed at the time
designated by the protocol were finally included in the study. As
we observed in a previous study (11), reactive changes and intestinal
metaplasia in continuity to the anastomotic site appeared at high
rates in the first month, while the prevalence of metaplasia
distant from the anastomosis increased considerably in month 2.
High rates of AC were found in month 4; thus, we considered this
time of follow-up to be sufficient for our study.
Drug administration and dosage
selection
Acetylsalicylic acid (ASA) (Sigma, Munich, Germany)
was administered daily at two different doses: 5 mg/kg body weight
per day (low dose) and 50 mg/kg body weight per day (high dose).
These doses match those prescribed to patients in different
clinical settings and have also been shown to be effective in
previous studies of colon adenocarcinoma chemoprevention in rats
and intestinal adenoma chemoprevention in mice (14–16).
ASA was added to drinking water, replenished on a daily basis.
Histopathological analysis
Rat esophagi were examined by an experienced
pathologist (C.C.) who was unaware of the experimental conditions.
Changes in the squamous epithelium were classified into the
following five categories. i) Reactive changes: characterized by
the presence of basal cell hyperplasia, increased length of
papillae, and hyperkeratosis with areas of inflammation and
ulceration. ii) Columnar-lined metaplasia (intestinal metaplasia):
squamous epithelium was replaced with columnar-lined epithelium
comprising occasional and incompletely differentiated goblet cells;
the length of the epithelial transformation was >3 mm starting
from the anastomosis site or was intercalated, or it could be
observed beyond the surgical anastomosis, and was surrounded by
squamous epithelium. iii) Dysplasia: characterized by nuclear
atypia, partial loss of mucosecretory function and cell polarity
and an increase in mitotic figures. Dysplasia was classified as
low-grade dysplasia (LGD) and high-grade dysplasia (HGD) according
to the criteria proposed by Haggit (17), as previously described in this
experimental model (13). In LGD,
the crypt architecture is preserved or minimally distorted; the
nuclei may be stratified but without reaching the apical surface of
glands, nuclei are enlarged, crowed and hyperchromatic, mitotic
figures may be found in the upper portion of the crypt; goblet and
columnar cell mucus is usually diminished or absent, but goblet
cells in which the mucous droplet does not communicate with the
luminal surface may be observed. The abnormalities extend to the
mucosal surface. In HGD, distortion of crypt architecture is
usually present and may be pronounced. It is composed of branching
and lateral budding of crypts, a villiform configuration of the
mucosal surface or intraglandular bridging of epithelium to form a
cribriform pattern of ‘back-to-back’ glands. Nuclear abnormalities
are present as in LGD, and stratification reaches the crypt luminal
surface. There may be a loss of nuclear polarity, and nuclei often
vary markedly in size, shape and staining characteristics. Goblet
and columnar cell mucus is usually absent. The abnormalities extend
to the mucosal surface. iv) Adenocarcinoma: glandular structures of
dysplastic columnar epithelial with stromal invasion and deep
infiltration. v) Squamous carcinoma: characterized by accumulation
of atypical cells with nuclear hyperchromasia, abnormally clumped
chromatin, and loss of polarity (13).
Measurement of prostaglandin levels in
esophageal mucosa
All biochemical analyses were performed in the lower
half of the esophagus. Tissue samples were introduced in 1 ml of
cold PBS containing indomethacin (10 μM). They were homogenized
(Heidolph DIAX 900) on ice and centrifuged 15 min at 1,200 rpm and
4°C. Supernatants were collected in 2-ml tubes. Prostaglandin
extraction was performed as previously described by Powell et
al (18). The concentration of
PGE2 was measured using the Enzyme immunoassay kit from
Amersham (Amersham Pharmacia Biotech, Inc., Piscataway, NJ,
USA).
Measurement of 15-epi-lipoxin
A4 levels in esophageal mucosa
Tissue samples were homogenized in ethanol (5 ml/g)
and centrifuged to obtain the supernatant. One milliliter of the
supernatant was diluted with 5 ml of deionized water and acidified
to a pH of 3.5 with HCl. 15-Epi-lipoxin A4 extraction
and determination were performed according to the manufacturer’s
instructions (Oxford Biomedical Research, Rochester Hills, MI,
USA).
Plasma drug concentration
Aspirin and salicylic acid plasma levels were
measured by high performance liquid chromatography on a 4-μm
ultrasphere C18 silica column (150 × 3.9 mm, Waters; Millipore
Iberica, Madrid, Spain) according to a previously described method
(19). Blood samples were placed in
an ice water bath, treated with 10 μl/ml of 50% (v/v) potassium
fluoride (an enzyme inhibitor) to avoid aspirin hydrolysis, and
centrifuged for 3 min. The supernatant was then acidified and
extracted with diethyl ether (1:3; v/v) for 20 min, which allows
recoveries over 80%. The organic phase of the extract was then
evaporated under nitrogen and stored at −20°C. Samples were
reconstituted in 500 μl of mobile phase (acetonitrile/water/TFA;
30/70/0,01; v:v), and 20 μl aliquots of the reconstituted sample
were analyzed using a reverse phase high performance liquid
chromatography (Waters 600E; Millipore). Aspirin and its metabolite
absorbances were measured at 237 nm. Toluic acid (Sigma, St. Louis,
MO, USA) was used as the internal standard (20 μg/ml). Aspirin and
salicylic acid concentrations were determined by comparison of
standards.
Statistical analysis
Data management and statistical analysis were
performed using SPSS software. Results from the biochemical assays
are expressed as mean ± standard error (SE). Data were compared
between groups by non-parametric tests (Kruskall-Wallis and
Mann-Whitney); differences with a P-value <0.05 were considered
to indicate statistically significant results. Fisher’s exact test
was used to analyze the qualitative variables. Multiple comparisons
were corrected by means of the Bonferroni method.
Results
Twenty-seven rats survived to the end of the
experiment (54%). The main cause of death was asphyxia provoked by
bedding intake.
Anatomopathological analysis
Macroscopically, all rats with esophagojejunostomy
showed dilatation and thickening of the mid and lower esophagus,
and the distal esophagus showed ulceration. Macroscopically, 2 rats
from the high-dose aspirin group presented esophageal tumors. All
esophagi presented inflammatory infiltrate. In the upper third of
the esophagus rats presented some erosion, and ulceration was
observed in the proximity of the anastomosis. In most animals, the
upper and the middle parts of the esophagus showed squamous
hyperplasia. All operated-rat esophagi showed columnar-lined
metaplasia in continuity with the jejunal epithelium. In addition,
columnar-lined metaplasia was found far above the anastomosis in
some animals. This metaplasia consisted of isolated or grouped
goblet cells, forming glands in some cases. This type of metaplasia
could only be identified microscopically.
Neoplastic transformation of the epithelium was
always near the anastomosis, surrounded by columnar epithelium.
Dysplasic changes were present in all rats to some degree.
Histologically, all tumors were adenocarcinomas, and all but one
(adenosquamous carcinoma in a low-dose aspirin rat) were of the
mucinous type.
Effect of ASA treatment on lesions
Inflammation, ulceration, intestinal metaplasia and
dysplasia were present in all rats (Table I). Treatment with either low- or
high-dose aspirin did not modify the grade of inflammation or the
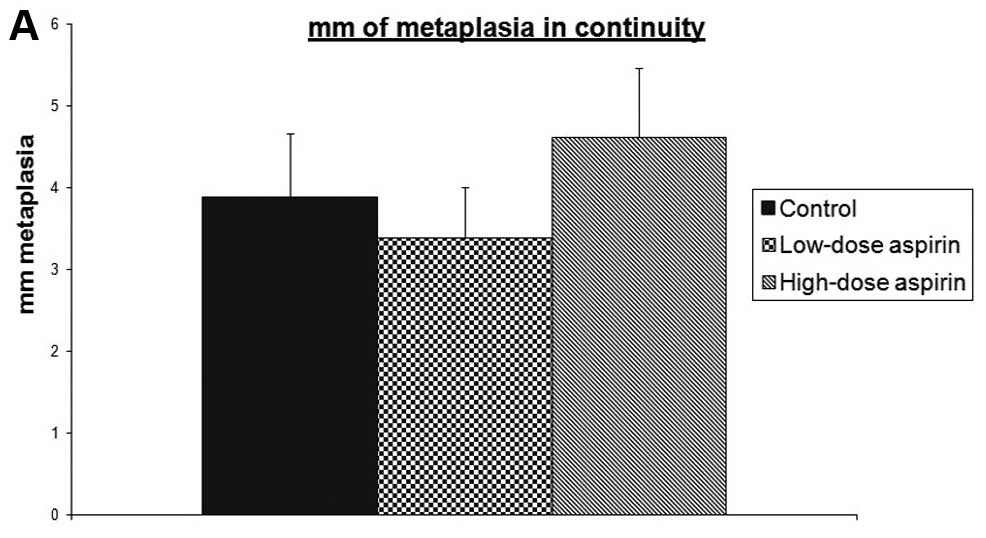
extent of ulcerated mucosa. Metaplasia in continuity with
anastomosis length was not affected by aspirin treatment at any
dose (Fig. 1). The majority (62.5%)
of control (vehicle) rats developed metaplasia beyond the
anastomosis. This type of metaplasia was also found in the rats
treated with aspirin: 45.5% of rats treated with high-dose ASA and
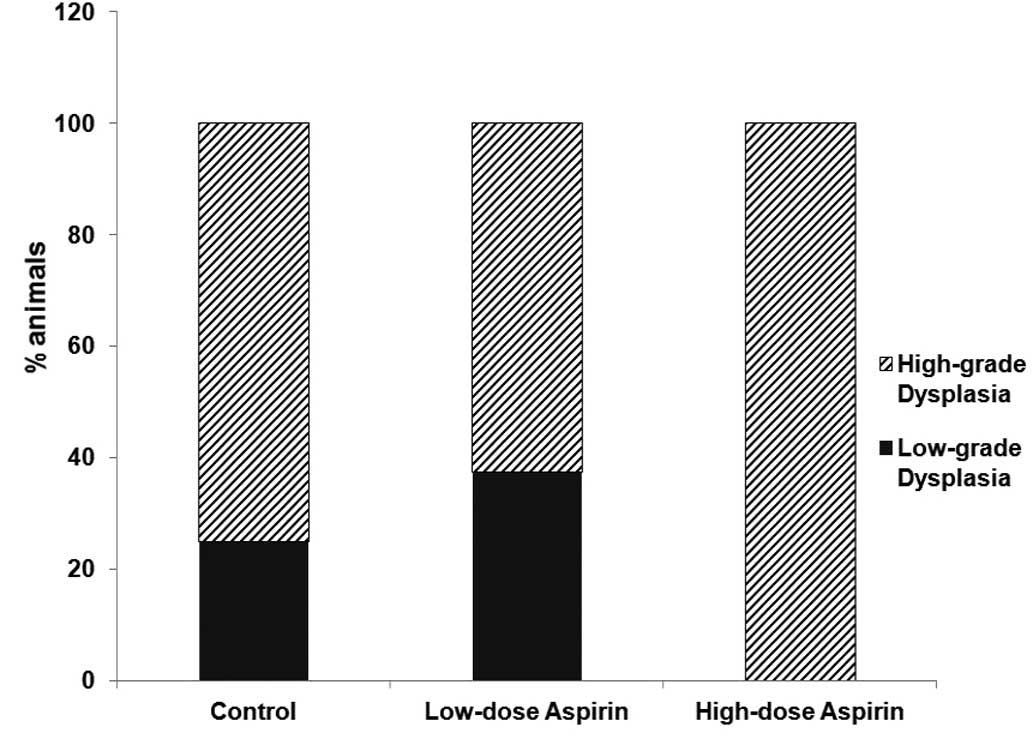
75% of rats treated with low-dose ASA. All rats developed
dysplasia. No differences between groups were observed including
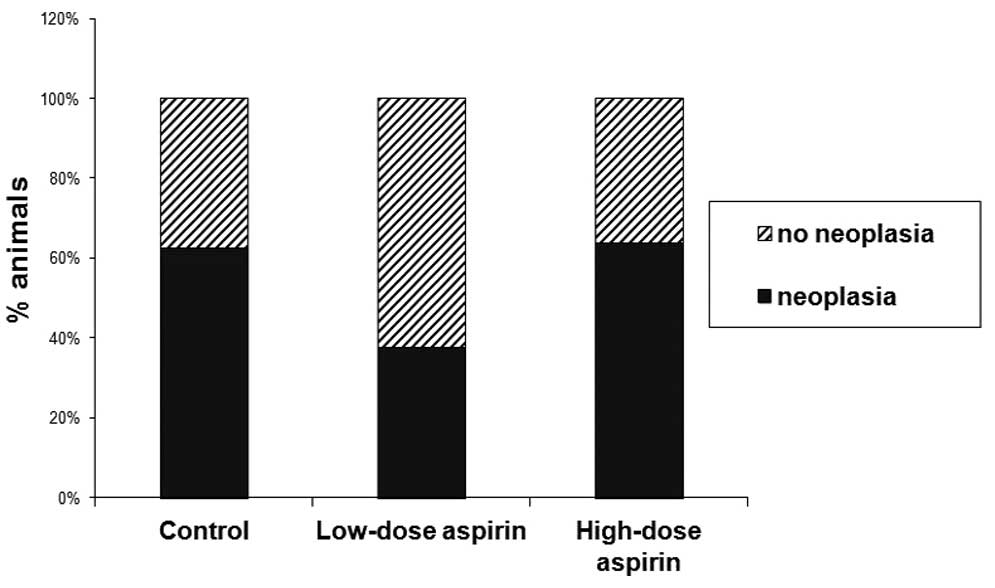
differences in low- vs. high-grade dysplasia (Fig. 2). The number of rats that presented
neoplasia in the low-dose aspirin group was lower than the numbers
in the other two groups, but these differences were not significant
(Fig. 3).
 | Table IEffect of ASA treatment on lesions in
surviving rats. |
Table I
Effect of ASA treatment on lesions in
surviving rats.
| Group | I (n=8) | II (n=8) | III (n=11) |
|---|
| Inflammation | 8 | 8 | 11 |
| Ulcerated mucosa
(%) | 35 | 37.5 | 38.18 |
| Squamous
hyperplasia (%) | 87.5 | 87.5 | 100 |
| Intestinal
metaplasia in continuity with anastomosis (mm) | 3.875 | 3.375 | 4.611 |
| Intestinal
metaplasia beyond anastomosis (%) | 62.5 | 75 | 45.5 |
| Dysplasia | 8 | 8 | 11 |
| High-grade
dysplasia/low-grade dysplasia | 2/6 | 2/6 | 0/11 |
| Carcinoma | 5 | 4 | 7 |
Prostaglandin E2 and
15-epi-lipoxin A4 levels
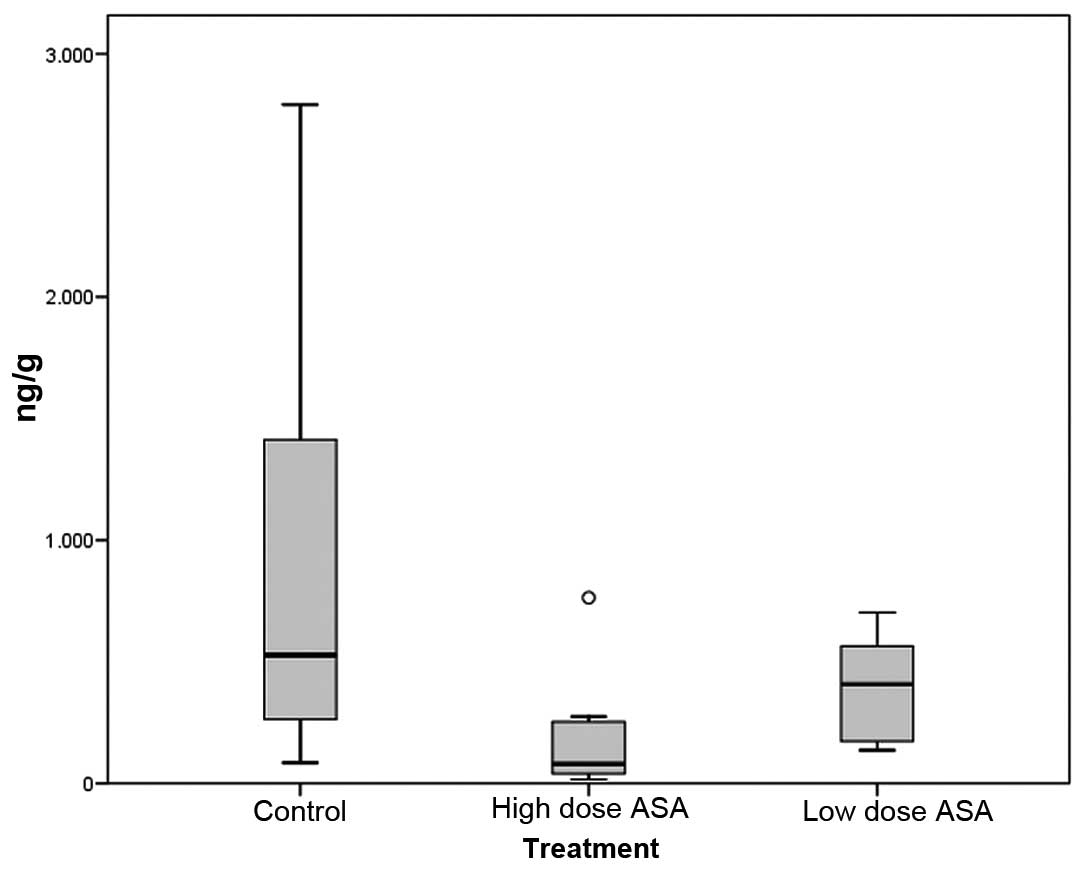
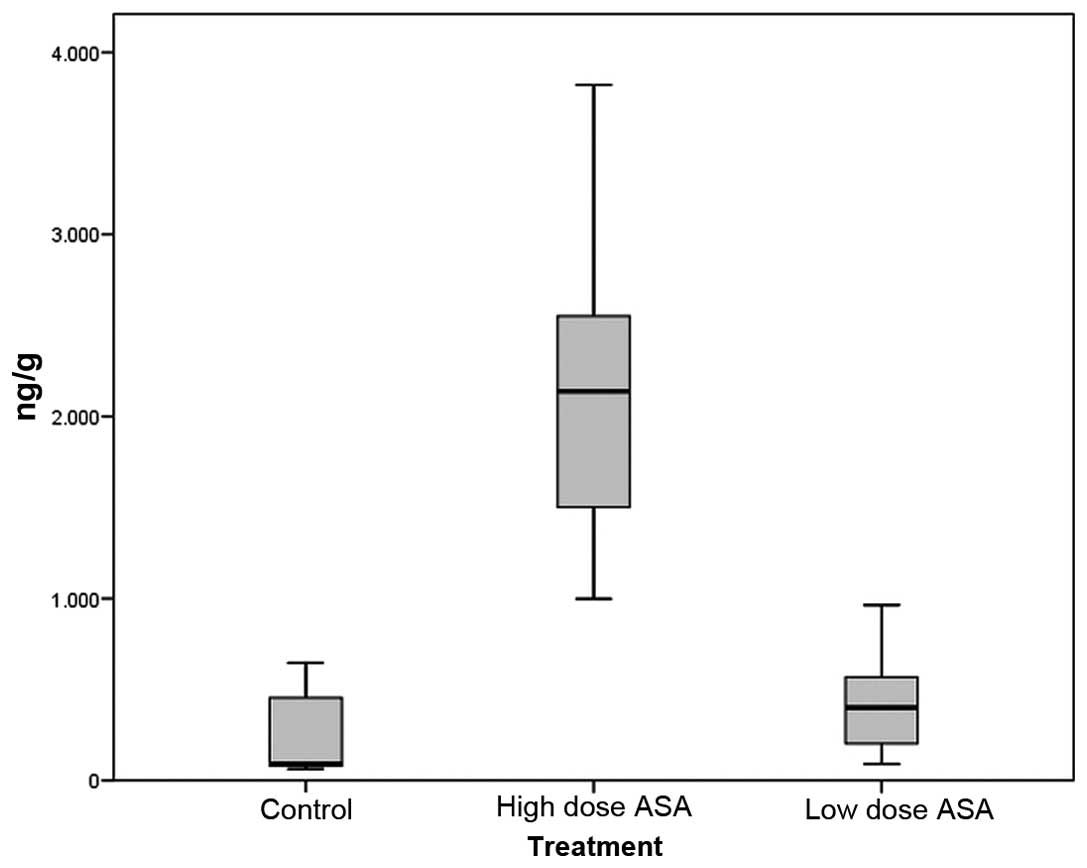
We observed a decrease in PGE2 levels in
the esophageal tissue in both the high-dose and low-dose aspirin
groups (183.17±65.68 and 391.02±76.40 ng/g tissue; mean ± SEM,
respectively) compared to the control group (910.80±324.98 ng/g;
P=0.08 and P=0.401, respectively) (Fig.
4).
Regarding the impact of aspirin on lipoxin
A4 production in this model, in both the high-dose and
low-dose aspirin groups we observed an increase in 15-epi-lipoxin
A4 in the esophagi (2096.38 ng/g ± 820.40 and 424.53
ng/g ± 281.00; mean ± SEM, respectively) when compared to the
controls (267.60 ng/g ± 250.83; P=0.01 and P=0.203, respectively)
(Fig. 5).
Plasma drug concentrations
All rats receiving ASA showed detectable levels of
salicylic acid (SA) in the plasma. Plasma drug concentration of SA
in rats receiving 50 mg/kg was 57.28±1.61 μg/ml. Rats receiving 5
mg/kg had a plasma level of SA of 3.94±0.29 μg/ml (mean ± SE). As
expected, the rats in the control group did not have detectable ASA
or SA levels.
Discussion
In the present study, we attempted to assess whether
the administration of aspirin prevents the development of
esophageal adenocarcinoma in vivo. The surgical model of
Barrett’s esophagus and esophageal adenocarcinoma in the rat
appears to be a suitable model of human disease (12). Regarding the COX pathway, COX2,
prostaglandin E synthase, prostaglandin receptors E2 and
E4 overexpression and PGE2 overproduction
have been observed during rat esophageal adenocarcinogenesis
(20,21), mimicking the expression profiles of
these markers in both human Barrett’s and esophageal adenocarcinoma
(22,23). Administration of different COX
inhibitors has been shown to provoke a significant reduction in
PGE2 production as well as to prevent the development of
adenocarcinoma in this experimental model (24–27).
The best known molecular target of aspirin is COX.
ASA, but not other NSAIDs, can cause irreversible inactivation of
COX isozymes through the acetylation of a specific serine moiety
(Ser 529 of COX-1 and Ser 516 of COX-2) (28). In contrast to other NSAIDs,
ASA-mediated acetylation of COX-2 provokes a change in the active
enzyme site leading to incomplete reaction of arachidonic acid. The
product of this reaction is 15 R hydroxyeicosatetraenoic
acid (15R-HETE), which is rapidly metabolized by
lipoxygenase enzyme to 15-epi-lipoxin A4, also called
aspirin-triggered lipoxin (ATL) (29). ATLs have been shown to exert potent
anti-inflammatory effects (30).
Additionally, lipoxin A4 (LXA4) has been
shown to reduce cell proliferation, inhibit tumor cell invasion,
and suppress experimental tumor growth (31,32).
The ability of aspirin to inhibit COX-2 activity
in vivo depends mainly on the dose administered. It is
noteworthy that systemic concentrations of aspirin reached after
the administration of low doses are inadequate to significantly
acetylate COX-2. At higher doses, aspirin may affect COX-2 in a
dose-dependent fashion. It can be hypothesized that local aspirin
concentrations in the digestive system, including the colon and
esophagus, after dosing with the drug, are sufficiently high to
acetylate epithelial COX isozymes (33). In one study in humans, inhibition of
PGE2 was detected in rectal biopsies performed after 1
month of treatment with three aspirin doses (81, 325 and 650 mg)
(34). Unexpectedly, the 81-mg
daily aspirin dose also suppressed PGE2 levels to the
same extent as did the 650-mg dose. In the esophagus, it has been
shown that administration of aspirin at a dose of 325 mg daily in
conjunction with esomeprazole administered for 10 days resulted in
lower esophageal mucosal content in patients with Barrett’s
esophagus, whereas esomeprazole alone or in combination with
rofecoxib did not reduce PGE2 production (35). Recently, a randomized, double-blind,
placebo controlled phase-II 28-day trial compared the effect of 2
dose levels of aspirin in combination with esomeprazole on tissue
PGE2 concentration in patients with Barrett’s esophagus
(36). The results revealed that
325 mg aspirin daily, but not 81 mg, significantly reduced tissue
concentrations of PGE2 compared with a placebo. In the
present study, we used two different doses of aspirin, the lower
dose (5 mg/kg/day), which would be equivalent to 300–350 mg/day for
a body weight of 60–70 kg, and a higher dose of 50 mg/kg/day, which
would correspond to a high anti-inflammatory dose in humans. The
rationale for the use of such a high dose was to test whether the
lower dose of aspirin was equally effective in inhibiting COX
activity. Thus, although PGE2 levels showed a tendency
to decrease in the low-dose aspirin group, the differences were not
statistically significant. The lack of a significant effect might
be due to a lack of power due to the high variability of
PGE2 in the non-treated control group and not a true
lack of efficacy of this dose of aspirin to inhibit PGE2
synthesis in the esophageal mucosa. As expected, a profound and
significant decrease in PGE2 content was observed in the
high-dose aspirin group, indicating that administration of aspirin
indeed led to an inhibition of COX activity at the esophageal
level.
The present study also investigated the generation
of ATL, the other lipid mediator generated by aspirin-acetylated
COX-2. Our data showed the generation of 15(R) epi-LXA4
in the absence of aspirin in this experimental model. Furthermore,
production of aspirin-triggered 15-epi-lipoxin A4 in rat
esophagi was increased in a dose-dependent manner in animals
receiving aspirin, but only in the high-dose group were these
levels significantly higher than that in the control group. These
data indicate that high doses of ASA are necessary to acetylate
COX-2 isoenzyme in the esophagus and argue against the idea that
ATLs are produced mainly with low-dose rather than high-dose
aspirin (37). There are no
previous studies exploring the role of lipoxins in the esophagus,
but previous reports have shown that LXA4 exerts potent
protective effects in the stomach. Thus, in an experimental model
of gastritis induced by iodoacetamide, Souza et al (38) observed that, after the
administration of aspirin (50 or 100 mg/kg), the levels of
LXA4 in gastric mucosa of rats with gastritis were
significantly higher than in healthy rats. Moreover, the presence
of LXA4 reduced the mucosal injury provoked by aspirin
intake. Consistently with these results, Fiorucci et al
(39) showed that LXA4
production in the stomach was significantly increased in rats
treated with aspirin (50 mg/kg) in an experimental model of acute
gastric damage provoked by aspirin. In that rat model,
intraperitoneal pretreatment with LXA4 reduced the
severity of aspirin-induced gastric injury in a dose-dependent
manner. In addition to this, the intraperitoneal administration of
an LXA4 receptor antagonist led to a significantly
increased aspirin-induced gastric damage.
In the present study, we observed the expected
effect of aspirin in esophageal tissues at the biochemical level,
which was a dose-dependent decrease in PGE2 content and
an increase in ATL production. However, this effect was not
accompanied by any improvement at the histological level at any
dose. Thus, administration of aspirin did not lead to any reduction
in the area of ulcerated mucosa, the length of metaplasia in
continuity with anastomosis, the incidence of metaplasia beyond the
anastomosis, the severity of dysplasia, or the incidence of
adenocarcinoma. These results are consistent with previous reports.
Pawlik et al (40), in an
experimental model of acute reflux esophagitis, showed that aspirin
from 12.5 mg/kg/day to 100 mg/kg/day significantly inhibited
prostaglandin synthesis. In parallel, aspirin provoked a
significant decrease in esophageal blood flow and an increase in
acute esophagitis. In another study, using the same experimental
model as we employed, Rizvi et al (41) found no differences in the rate of
esophageal adenocarcinoma evaluated 8 months after
esophagojejunostomy in rats receiving aspirin in the diet compared
with a control group. The reasons for the absence of a correlation
between the biochemical and histological effects of aspirin in this
model warrant further investigation. One possible explanation is
that in Barrett’s carcinogenesis it would be necessary to target
not only the consequence of injury (inflammation-associated
pathway) but also the cause of injury. This hypothesis is supported
by Rizvi et al (41), who
demonstrated that the combination of aspirin with ursodeoxycolic
acid (which reduces the concentration of bile salts), but not each
agent individually, reduced the risk of esophageal adenocarcinoma
in a rat model of esophagojejunostomy. On the other hand, we cannot
ignore the fact that, in addition to its anti-inflammatory effect,
aspirin can also induce gastrointestinal damage through a variety
of mechanisms. Some clinical trials have observed that even
low-dose aspirin treatment provokes esophageal damage (42,43).
It has been shown that aspirin makes the esophageal mucosa more
sensitive to the injurious action of acid and pepsin (44). In another study, intra-gastric
administration of aspirin induced dilated intercellular spaces in
the rat esophageal epithelium (45). In addition, some studies have
observed that treatment with high doses of ASA provokes both
significantly increased production of reactive oxygen species (ROS)
(46) and inhibition of ROS
scavenger activity (47). Since the
involvement of ROS in the development of esophageal adenocarcinoma
has been previously demonstrated in this experimental model
(11), another possibility could be
that the potential benefits derived from inhibition of COX activity
are counterbalanced by a deleterious effect through ROS
generation.
In conclusion, the present study demonstrated that
aspirin, at a high dose, affects COX activity, hence producing a
profound inhibition of PGE2 content and the generation
of aspirin-triggered lipoxins in esophageal tissues, but failed to
demonstrate any potential benefit in the prevention of esophageal
adenocarcinoma in this rat model of gastroesophageal reflux.
Further studies are necessary to elucidate how aspirin acts at the
esophagus and the mechanisms underlying the protective role that
epidemiological studies have shown for aspirin against esophageal
adenocarcinoma.
References
|
1
|
Pera M, Manterola C, Vidal O and Grande L:
Epidemiology of esophageal adenocarcinoma. J Surg Oncol.
92:151–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blot WJ, Devesa SS, Kneller RW and
Fraumeni JF Jr: Rising incidence of adenocarcinoma of the esophagus
and gastric cardia. JAMA. 265:1287–1289. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Devesa SS, Blot WJ and Fraumeni JF Jr:
Changing patterns in the incidence of esophageal and gastric
carcinoma in the United States. Cancer. 83:2049–2053. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mutoh M, Watanabe K, Kitamura T, Shoji Y,
Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi
K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T and Wakabayashi
K: Involvement of prostaglandin E receptor subtype EP4
in colon carcinogenesis. Cancer Res. 62:28–32. 2002.PubMed/NCBI
|
|
5
|
Husain SS, Szabo IL and Tamawski AS: NSAID
inhibition of GI cancer growth: clinical implications and molecular
mechanisms of action. Am J Gastroenterol. 97:542–553. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Piazuelo E, Jimenez P and Lanas A: COX-2
inhibition in esophagitis, Barrett’s esophagus and esophageal
cancer. Curr Pharm Des. 9:2267–2280. 2003.
|
|
7
|
Al-Saeed A: Gastrointestinal and
cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman
Med. 26:385–391. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patrignani P, Tacconelli S, Bruno A,
Sostres C and Lanas A: Managing the adverse effects of nonsteroidal
anti-inflammatory drugs. Expert Rev Clin Pharmacol. 4:605–621.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hennekens CH: Update on aspirin in the
treatment and prevention of cardiovascular disease. Am Heart J.
137:S9–S13. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai Y and Ge J: Clinical use of aspirin in
treatment and prevention of cardiovascular disease. Thrombosis.
2012:2450372012.PubMed/NCBI
|
|
11
|
Piazuelo E, Cebrián C, Escartín A, Jiménez
P, Soteras F, Ortego J and Lanas A: Superoxide dismutase prevents
development of adenocarcinoma in a rat model of Barrett’s
esophagus. World J Gastroenterol. 11:7436–7443. 2005.PubMed/NCBI
|
|
12
|
Hindmarsh A, Belshaw N, Mehta S, Johnson
IT and Rhodes M: Can the rat be used as a valid model of human
esophageal adenocarcinoma? Dis Esophagus. 25:159–165. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su Y, Chen X, Klein M, Fang M, Wang S,
Yang CS and Goyal RK: Phenotype of columnar-lined esophagus in rats
with esophagogastroduodenal anastomosis: similarity to human
Barrett’s esophagus. Lab Invest. 84:753–765. 2004.PubMed/NCBI
|
|
14
|
Reddy BS, Rao CV, Rivenson A and Kelloff
G: Inhibitory effect of aspirin on azoxymethane-induced colon
carcinogenesis in F344 rats. Carcinogenesis. 14:1493–1497. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barnes CJ and Lee M: Chemoprevention of
spontaneous intestinal adenomas in the adenomatous polyposis coli
Min mouse model with aspirin. Gastroenterology. 114:873–877. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bousserouel S, Gosse F, Bouhadjar M, Soler
L, Marescaux J and Rau F: Long-term administration of aspirin
inhibits tumour formation and triggers anti-neoplastic molecular
changes in a pre-clinical model of colon carcinogenesis. Oncol Rep.
23:511–517. 2010.
|
|
17
|
Haggitt RC: Barrett’s esophagus,
dysplasia, and adenocarcinoma. Hum Pathol. 25:982–993. 1994.
|
|
18
|
Powell WS: Reversed-phase high-pressure
liquid chromatography of arachidonic acid metabolites formed by
cyclooxygenase and lipoxygenases. Anal Biochem. 148:59–69. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lanas AI, Arroyo MT, Esteva F, Cornudella
R, Hirschowitz BI and Sáinz R: Aspirin related gastrointestinal
bleeders have an exaggerated bleeding time response due to aspirin
use. Gut. 39:654–660. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jang TJ, Min SK, Bae JD, Jung KH, Lee JI,
Kim JR and Ahn WS: Expression of cyclooxygenase 2, microsomal
prostaglandin E synthase 1, and EP receptors is increased in rat
oesophageal squamous cell dysplasia and Barrett’s metaplasia
induced by duodenal contents reflux. Gut. 53:27–33. 2004.PubMed/NCBI
|
|
21
|
Piazuelo E, Santander S, Cebrián C,
Jiménez P, Pastor C, García-González MA, Esteva F, Esquivias P,
Ortego J and Lanas A: Characterization of the prostaglandin E2
pathway in a rat model of esophageal adenocarcinoma. Curr Cancer
Drug Targets. 12:132–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wilson KT, Fu S, Ramanujam KS and Meltzer
SJ: Increased expression of inducible nitric oxide synthase and
cyclooxygenase-2 in Barrett’s esophagus and associated
adenocarcinomas. Cancer Res. 58:2929–2934. 1998.
|
|
23
|
Jimenez P, Piazuelo E, Cebrian C, Ortego
J, Strunk M, García-Gonzalez MA, Santander S, Alcedo J and Lanas A:
Prostaglandin EP2 receptor expression is increased in Barrett’s
oesophagus and oesophageal adenocarcinoma. Aliment Pharmacol Ther.
31:440–451. 2010.
|
|
24
|
Esquivias P, Morandeira A, Escartín A,
Cebrián C, Santander S, Esteva F, García-González MA, Ortego J,
Lanas A and Piazuelo E: Indomethacin but not a selective
cyclooxygenase-2 inhibitor inhibits esophageal adenocarcinogenesis
in rats. World J Gastroenterol. 18:4866–4874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buttar NS, Wang KK, Leontovich O, Westcott
JY, Pacifico RJ, Anderson MA, Krishnadath KK, Lutzke LS and Burgart
LJ: Chemoprevention of esophageal adenocarcinoma by COX-2
inhibitors in an animal model of Barrett’s esophagus.
Gastroenterology. 122:1101–1112. 2002.PubMed/NCBI
|
|
26
|
Oyama K, Fujimura T, Ninomiya I, Miyashita
T, Kinami S, Fushida S, Ohta T and Koichi M: A COX-2 inhibitor
prevents the esophageal inflammation-metaplasia-adenocarcinoma
sequence in rats. Carcinogenesis. 26:565–570. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen X, Wang S, Wu N, Sood S, Wang P, Jin
Z, Beer DG, Giordano TJ, Lin Y, Shih WC, Lubet RA and Yang CS:
Overexpression of 5-lipoxygenase in rat and human esophageal
adenocarcinoma and inhibitory effects of zileuton and celecoxib on
carcinogenesis. Clin Cancer Res. 10:6703–6709. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Patrono C and Rocca B: Aspirin: promise
and resistance in the new millennium. Arterioscler Thromb Vasc
Biol. 28:s25–s32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Claria J and Serhan CN: Aspirin triggers
previously undescribed bioactive eicosanoids by human endothelial
cell-leukocyte interactions. Proc Natl Acad Sci USA. 92:9475–9479.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morris T, Stables M, Hobbs A, de Souza P,
Colville-Nash P, Warner T, Newson J, Bellingan G and Gilroy DW:
Effects of low-dose aspirin on acute inflammatory responses in
humans. J Immunol. 183:2089–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou XY, Li YS, Wu P, Wang HM, Cai ZY, Xu
FY and Ye DY: Lipoxin A4 inhibited hepatocyte growth
factor-induced invasion of human hepatoma cells. Hepatol Res.
39:921–930. 2009.PubMed/NCBI
|
|
32
|
Chen Y, Hao H, He S, Cai L, Li Y, Hu S, Ye
D, Hoidal J, Wu P and Chen X: Lipoxin A4 and its
analogue suppress the tumor growth of transplanted H22 in mice: the
role of antiangiogenesis. Mol Cancer Ther. 9:2164–2174.
2010.PubMed/NCBI
|
|
33
|
Dovizio M, Tacconelli S, Sostres C,
Ricciotti E and Patrignani P: Mechanistic and pharmacological
issues of aspirin as an anticancer agent. Pharmaceuticals.
5:1346–1371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sample D, Wargovich M, Fischer SM, Inamdar
N, Schwartz P, Wang X, Do KA and Sinicrope FA: A dose-finding study
of aspirin for chemoprevention utilizing rectal mucosal
prostaglandin E2 levels as a biomarker. Cancer Epidemiol
Biomarkers Prev. 11:275–279. 2002.PubMed/NCBI
|
|
35
|
Triadafilopoulos G, Kaur B, Sood S,
Traxler B, Levine D and Weston A: The effects of esomeprazole
combined with aspirin or rofecoxib on prostaglandin E2
production in patients with Barrett’s oesophagus. Aliment Pharmacol
Ther. 23:997–1005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Falk GW, Buttar NS, Foster NR, Ziegler KL,
Demars CJ, Romero Y, Marcon NE, Schnell T, Corley DA, Sharma P,
Cruz-Correa MR, Hur C, Fleischer DE, Chak A, Devault KR, Weinberg
DS, Della’Zanna G, Richmond E, Smyrk TC, Mandrekar SJ and Limburg
PJ; Cancer Prevention Network. A combination of esomeprazole and
aspirin reduces tissue concentrations of prostaglandin
E2in patients with Barrett’s esophagus.
Gastroenterology. 143:917–926.e1. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chiang N, Bermudez EA, Ridker PM, Hurwitz
S and Serhan CN: Aspirin triggers antiinflammatory 15-epi-lipoxin
A4 and inhibits thromboxane in a randomized human trial.
Proc Natl Acad Sci USA. 101:15178–15183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Souza MH, de Lima OM Jr, Zamuner SR,
Fiorucci S and Wallace JL: Gastritis increases resistance to
aspirin-induced mucosal injury via COX-2-mediated lipoxin
synthesis. Am J Physiol Gastrointest Liver Physiol. 285:G54–G61.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fiorucci S, de Lima OM Jr, Mencarelli A,
Palazzetti B, Distrutti E, McKnight W, Dicay M, Ma L, Romano M,
Morelli A and Wallace JL: Cyclooxygenase-2-derived lipoxin
A4 increases gastric resistance to aspirin-induced
damage. Gastroenterology. 123:1598–1606. 2002.PubMed/NCBI
|
|
40
|
Pawlik M, Pajdo R, Kwiecien S,
Ptak-Belowska A, Sliwowski Z, Mazurkiewicz-Janik M, Konturek SJ,
Pawlik WW and Brzozowski T: Nitric oxide (NO)-releasing aspirin
exhibits a potent esophagoprotection in experimental model of acute
reflux esophagitis. Role of nitric oxide and proinflammatory
cytokines. J Physiol Pharmacol. 62:75–86. 2011.
|
|
41
|
Rizvi S, Demars CJ, Comba A, Gainullin VG,
Rizvi Z, Almada LL, Wang K, Lomberk G, Fernández-Zapico ME and
Buttar NS: Combinatorial chemoprevention reveals a novel
smoothened-independent role of GLI1 in esophageal carcinogenesis.
Cancer Res. 70:6787–6796. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sugimoto M, Nishino M, Kodaira C, Yamade
M, Ikuma M, Tanaka T, Sugimura H, Hishida A and Furuta T:
Esophageal mucosal injury with low-dose aspirin and its prevention
by rabeprazole. J Clin Pharmacol. 50:320–330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawai T, Watanabe M and Yamashina A:
Impact of upper gastrointestinal lesions in patients on low-dose
aspirin therapy: preliminary study. J Gastroenterol Hepatol.
25(Suppl 1): S23–S30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lanas A, Jiménez P, Ferrández A, Escartín
A, Arenas J, Esteva F and Ortego J: Selective COX-2 inhibition is
associated with decreased mucosal damage induced by acid and pepsin
in rabbit esophagitis. Inflammation. 27:21–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang DH, Zhou LY, Dong XY, Cui RL, Xue Y
and Lin SR: Factors influencing intercellular spaces in the rat
esophageal epithelium. World J Gastroenterol. 16:1063–1069. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pohle T, Brzozowski T, Becker JC, Van der
Voort IR, Markmann A, Konturek SJ, Moniczewski A, Domschke W and
Konturek JW: Role of reactive oxygen metabolites in aspirin-induced
gastric damage in humans: gastroprotection by vitamin C. Aliment
Pharmacol Ther. 15:677–687. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Raza H, John A and Benedict S:
Acetylsalicylic acid-induced oxidative stress, cell cycle arrest,
apoptosis and mitochondrial dysfunction in human hepatoma HepG2
cells. Eur J Pharmacol. 668:15–24. 2011. View Article : Google Scholar
|