Introduction
Cisplatin is a major chemotherapy drug for ovarian
cancer; yet, drug-resistance to cisplatin occurs in many ovarian
cancer patients, leading to an overall 5-year survival rate of
30–40%. To date, a large number of genes appear to contribute to
cisplatin resistance including DNA damage-repair genes,
apoptosis-regulating and survival signal-related genes (1–3). The
effects of these genes on the cellular response to chemotherapy
have been measured by their influence on apoptosis in cancer cells
(4,5). Nonetheless, a large number of
investigations has verified that cancer cells tend to undergo
senescence rather than apoptosis following exposure to DNA-damaging
drugs at low-doses similar to those used in clinical applications.
Recent data have shown that low, clinically relevant doses of
DNA-damaging drugs do not induce apoptosis in epithelial tumor
cells but instead lead to a permanent growth arrest associated with
cellular senescence (6,7), which was primarily detected by the
presence of senescence-associated β-galactosidase (SA-β-gal) and
senescence-associated heterochromatin foci (SAHF) such as
heterochromatin protein 1-γ (HP1-γ) foci (8,9).
Moreover, the presence of SA-β-gal was noted in 41% of specimens
from breast cancer patients who received chemotherapy compared with
10% of specimens from patients who underwent surgery without
chemotherapy (10), suggesting that
chemotherapy induces senescence in vivo as well. The latter
studies also revealed that cellular senescence contributes to
treatment outcome of cancer therapy in vivo (11,12).
Therefore, senescence plays an important role in the in vivo
response to chemotherapy. Furthermore, research has shown that
cisplatin induces tumor cells to exhibit a senescence-like
phenotype and to undergo growth repression (13). However, the mechanisms of action of
cisplatin are still unclear.
Glucose-regulated protein 78 (GRP78), the most
abundant and well-characterized glucose-regulated protein, is a
major stress-induced chaperone localized in the endoplasmic
reticulum (ER) (14). As a
calcium-binding protein in the ER, GRP78 serves as a buffer against
calcium efflux from the ER that could trigger the mitochondrial
apoptotic program (15,16). Moreover, through direct or indirect
interactions with specific caspases and other upstream components
of the pro-apoptotic pathways initiating from the ER, GRP78 is
postulated to regulate the balance between cell survival and
apoptosis (17–19). Notably, previous studies found that
the cisplatin response also involves ER stress and that GRP78
expression is associated with apoptosis-sensitivity of tumor cells
to cisplatin (20–22). However, there is not a clear
relationship between GRP78 and cisplatin-induced senescence. It has
been shown that GRP78 plays an anti-apoptotic role through altered
expression of ataxia telangiectasia mutated (ATM) pathway genes,
which were also found to be associated with chemotherapy-induced
senescence (23). The relationship
between GRP78 and ATM pathway genes in cisplatin-induced senescence
has not yet been explored.
In the present study, we first demonstrated that
GRP78 expression was significantly lower in chemotherapy-sensitive
ovarian tumor sections. We also found that cisplatin-sensitive
A2780 cells tended to undergo senescence easily, while
cisplatin-resistant C13K cells showed no senescence phenotype after
a dose-gradient cisplatin exposure. Inhibition of GRP78 expression
rescued the senescence sensitivity of C13K cells to cisplatin. The
cisplatin response was found to involve the ATM pathway genes,
P53, P21 and CDC2, as well as ER calcium
homeostasis. Collectively, we demonstrated that GRP78 plays a
negative role in cisplatin-induced senescence in ovarian cancer
cells, which may contribute to our understanding of the mechanism
of the drug-resistance of ovarian cancer cells to cisplatin.
Materials and methods
The present study was reviewed and approved by the
Ethics Committee of Tongji Hospital, Tongji Medical College,
Huazhong University of Science and Technology (HUST). All
experimental protocols were approved by the Institutional Animal
Care and Use Committee of HUST, and the study was carried out in
strict accordance with the Animal Research: Reporting of In Vivo
Experiments (ARRIVE) guidelines.
Cell lines
The cisplatin-sensitive human ovarian cancer cell
line A2780 (P53 wild-type) was purchased from the European
Collection of Cell Cultures (ECACC). The cisplatin-resistant human
ovarian cancer cell line C13K (P53 wild-type) was a gift from
Benjamin K. Tsang (Department of Obstetrics and Gynecology and
Cellular and Molecular Medicine, University of Ottawa, Canada).
A2780 and C13K cells were grown in RPMI-1640 medium with 10% fetal
bovine serum (FBS) in a humidified atmosphere of 5% CO2
at 37°C. Cisplatin was added for 1 day at the indicated
concentrations to cells at 30–40% confluency. The cells were washed
twice with phosphate-buffered saline (PBS) and maintained for 6
days in complete medium prior to harvest for analysis. Caffeine was
added 2 h before cisplatin exposure. The drug A23187 was added 1
day before exposure to cisplatin or transfection with
small-interfering RNA.
Reagents
Rabbit polyclonal antibodies against HP1-γ and GRP78
were purchased from PTG Inc. (Wuhan, China). A rabbit polyclonal
antibody against P53 was purchased from Boster Bio-Company (Wuhan,
China). A mouse monoclonal antibody against P21 was purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). A mouse monoclonal
antibody against CDC2 was purchased from BD Company (Franklin
Lakes, NJ, USA). A senescence-associated β-galactosidase staining
kit and a rabbit polyclonal antibody against p-CDC2 were purchased
from Cell Signaling Technology (Beverly, MA, USA).
Carboxyfluorescein succinimidyl ester (CFSE) fluorescent dye was
purchased from Dojin Co. (Tokyo, Japan). Anti-rabbit IgG labeled
with Rhodamine was purchased from Zhongshan Biotech, Co., (Beijing,
China). Caffeine was purchased from Alexion (Lausanne,
Switzerland). Cisplatin and A23187 were purchased from Sigma
Company (Cream Ridge, NJ, USA). RPMI-1640 medium was purchased from
Gibco Company (Billings, MT, USA). Fluo3-AM was purchased from
Invitrogen Company (Grand Island, NY, USA).
Clinical samples
Tissue specimens were obtained from suspect ovarian
tumors during surgery at the Department of Gynecology and
Obstetrics, Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology in China from September 2005
to October 2009. The histological diagnosis of ovarian cancer was
confirmed in all cases, and the clinical International Federation
of Gynecology and Obstetrics (FIGO) disease stage was used. The 6
patients with stage IV ovarian serous adenocarcinoma tumors were
treated with 2–4 cycles of TP (paclitaxel + cisplatin) neoadjuvant
chemotherapy (NAC) before surgery. For each patient, a regression
coefficient was calculated using CA125 levels measured from the day
of NAC (as day 0) until the day their CA125 levels normalized
(<35 IU/ml) or the day of standard surgery. A
chemotherapy-sensitive sample was defined as having a regression
coefficient of ≥−0.039 (3 cases), and a chemotherapy-resistant
sample was defined as having a regression coefficient <-0.039 (3
cases). The method used for the calculation of the regression
coefficient was performed as described previously (24).
Immunohistochemical analyses
The immunohistochemical analyses were carried out as
described previously (25). The
negative control slides were processed similarly but with the
omission of the primary antibody.
SA-β-galactosidase histochemical
staining
Histochemical detection of SA-β-gal was performed
with the Senescence SA-β-Galactosidase Staining kit (Cell
Signaling) according to the manufacturer’s instructions. Briefly,
cultured cells were treated with cisplatin as described above.
Following treatment, the cells were washed twice with PBS and fixed
with a 3.5% paraformaldehyde solution for 15 min at room
temperature. The cells were washed every 5 min 3 times in PBS, and
the SA-β-gal staining solution (pH 6.0) was added and incubated for
16 h at 37°C. The percentage of positively stained cells was
determined by counting 3 random fields of at least 100 cells each.
Images of representative fields were captured under a ×20
magnification.
Cell cycle profiling
For cell cycle profiling, the cells were fixed with
75% ice-cold ethanol and stained with 50 μg/ml propidium iodide
before fluorescence-activated cell sorting analysis (FACS).
Analysis of cell proliferation by CFSE
labeling
CFSE is a green fluorescent dye that is distributed
equally among daughter cells with each cell division. For labeling,
cells were incubated with 3 μM CFSE in serum-free RPMI-1640 medium
for 15 min at 37°C. Excess dye was removed by two rinses in fresh
complete medium. The CFSE-labeled cells were plated in 6-well
flat-bottom plates and cultured in fresh complete medium with or
without cisplatin for the indicated time. The cells were harvested
and examined by flow cytometry. The data were analyzed using the
ModFit software (Becton-Dickinson). The cell proliferation model
was used to calculate the proliferation index, which is the ratio
of the total number of cells analyzed to the calculated number of
parent cells required to produce the observed number of cells.
Immunofluorescence
Cells were plated onto 6-well cell culture plates
(10,000 cells/well) in which slides were placed previously for
immunostaining. Twenty-four hours after plating, the cells were
treated with the indicated doses of cisplatin for 1 day;
subsequently, cells were washed twice with PBS and maintained for 6
days in fresh medium containing 10% FBS until analysis. Cells were
fixed in 75% ethanol for 30 min, permeabilized in 3% Triton X-100
for 5 min, and blocked in 5% normal goat serum in PBS at 37°C for 1
h. Cells were incubated with the HP1-γ polyclonal antibody (1:50)
overnight at 4°C. After washing, the cells were incubated with goat
anti-rabbit immunoglobulin G (IgG)-Rhodamine for 30 min at room
temperature. Slides were observed by fluorescence microscopy using
a Leica DM4000 B microscope (40× lens objective and 0.75 numerical
apertures) with a Qimaging Retiga 1300 camera.
Western blot analysis
Preparation of the protein samples and western blot
analyses were carried out as previously described (26). The western blotting antibodies used
were the following: GRP78 (1:500), P53 (1:500), P21 (1:500), p-CDC2
(1:500), CDC2 (1:500) and β-actin (1:1,000). After washing in PBS,
the membranes were incubated with a secondary antibody at a
dilution of 1:1,000. The proteins were visualized using the
BCIP/NBT immunoblotting detection system.
Transient siRNA transfection
The annealed synthetic small interfering RNAs
(siRNAs) were synthesized by RiboBio Co., Ltd. (Guangzhou, China).
The GRP78-specific siRNA (sense sequence,
5′-GAGUGACAGCUGAAGACAAdTdT-3′ and antisense sequence,
5′-GCCAGCGCACCTdTd-3′) was used to knock down the expression of
GRP78. A2780 and C13K cells were plated onto 6-well culture plates
at 30% density. After 1 day, the cells were transfected with
GRP78-siRNA or control-siRNA with Lipofectamine 2000 (Invitrogen)
according to the manufacturer’s protocol. Briefly, for each well in
a 6-well plate, 5 μl of Lipofectamine 2000 was diluted in 0.25 ml
of serum-free RPMI-1640 medium. This mixture was carefully added to
a solution containing 50 nmol of siRNA in 0.25 ml of serum-free
RPMI-1640 medium. The solution was incubated for 20 min at room
temperature and then gently overlaid onto cells at 40–50%
confluency in 1.5 ml of serum-free RPMI-1640 medium. After 4–6 h,
the cells were cultured with fresh complete medium for 72 h before
being exposed to cisplatin.
Animal treatment protocol
All animal procedures were performed in the animal
facility in accordance with applicable animal welfare regulations
under an approved Institutional Animal Care and Use (IACUC)
protocol and study design. Female athymic Balb/c nu/nu mice (5–8
weeks of age; obtained from the Animal Experimental Center of
Slaccas, Shanghai, China) were implanted subcutaneously in the
right flank with 2×106 A2780 or C13K cells mixed with an
equal volume of saline. After 8 days, mice were randomly sorted
into 2 groups (n=5 for each group) reflecting different treatment
regimens: group 1 received only a saline injection; group 2 was
injected twice with cisplatin at a dose of 10 mg/kg in a 3 day
interval. The mice were monitored twice a week by inspection and
palpitation. Seven days after the first injection, subcutaneous
tumors were freshly frozen in Tissue-Tek cryopreservation medium,
cryostat sectioned and stored at −20°C for SA-β-galactosidase
staining and immunohistochemical analysis.
Detection of relative Ca2+
concentrations
Fluo3-AM exhibits high fluorescence intensity
increases on binding Ca2+ which is visible with
excitation at 488 nm by argon-ion laser sources. Briefly, cells
were plated onto glass bottom dishes with a 35-mm diameter. Twelve
hours after treatment, the control cells and the cells receiving
different types of treatment were washed with D-Hanks for 3 times,
incubated with serum-free RPMI-1640 medium containing 4 μM Fluo3-AM
and 0.05% Pluronic-F127 for 45 min at 37°C, protected from light.
Then the cells were washed twice with Hanks containing 0.2% BSA,
and subsequently washed with Hanks one time for clear up of the
residual Fluo3-AM. The cells were observed by using laser confocal
microscope with a 488-nm excitation wavelength and 505–530 nm
absorption wavelength, 60× objective lens, 1,024×1,024 resolution,
at 37°C. The fluorescent images were collected every 10 sec for 10
times. Ten cells were monitored for each experiment. Three
independent experiments were carried out. The mean intensity of
Ca2+ fluorescence was analyzed with FluoView
software.
Statistical analyses
The results are expressed as means ± SE of 3
independent experiments. Statistical significance between groups
was determined by ANOVA analysis and defined as P<0.05.
Results
GRP78 is related to cisplatin-induced
senescence in vivo
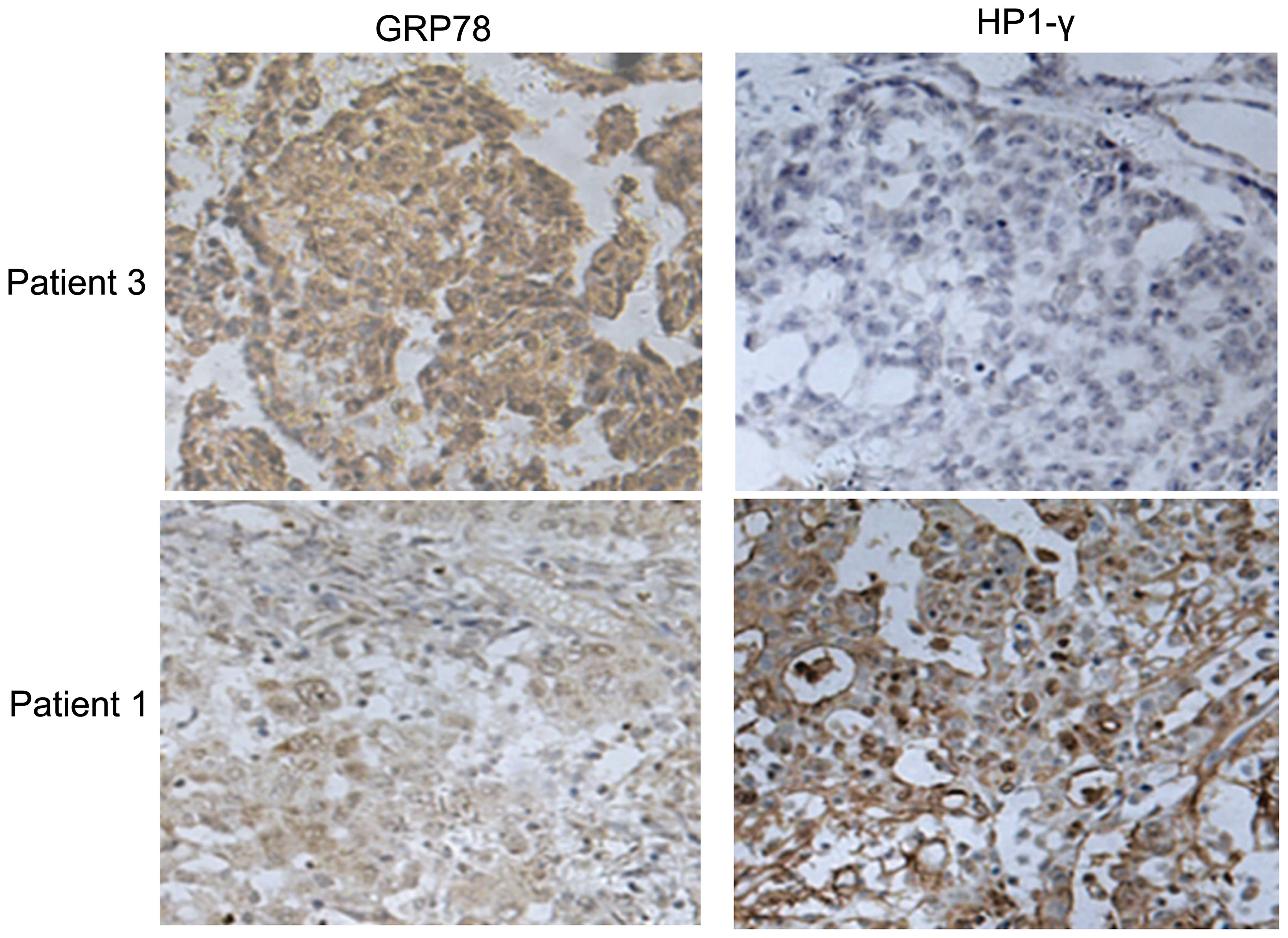
To investigate the relationship between GRP78
expression and senescence induced by cisplatin, tumor sections from
6 ovarian cancer patients who had received 2–4 cycles of
neoadjuvant chemotherapy were stained for GRP78 and
senescence-associated heterochromatin HP1-γ foci. The clinical data
showed that 3 of the patients were chemotherapy-resistant, and the
other 3 patients were chemotherapy-sensitive (Table I). The chemotherapy-sensitive tumor
sections showed strong staining for HP1-γ, but weak staining for
GRP78. We also showed adverse results in the chemotherapy-resistant
tumor sections (Fig. 1). These data
indicate that GRP78 expression was negatively associated with
cisplatin-induced senescence in ovarian cancer.
 | Table IClinical parameters and outcome of
the ovarian cancer patients who underwent neoadjuvant chemotherapy
prior to surgery. |
Table I
Clinical parameters and outcome of
the ovarian cancer patients who underwent neoadjuvant chemotherapy
prior to surgery.
| Patient no. | FIGO stage | Neoajuvant
therapy | GRP78 | HP1-γ | Regression
coefficient |
|---|
| 1 | IV | TPx4 | +/− | ++++ | >−0.039 |
| 2 | IV | TPx2 | + | +++ | >−0.039 |
| 3 | IV | TPx2 | +++ | +/− | <−0.039 |
| 4 | IV | TPx3 | ++ | + | <−0.039 |
| 5 | IV | TPx3 | ++++ | + | <−0.039 |
| 6 | IV | TPx2 | + | +++ | >−0.039 |
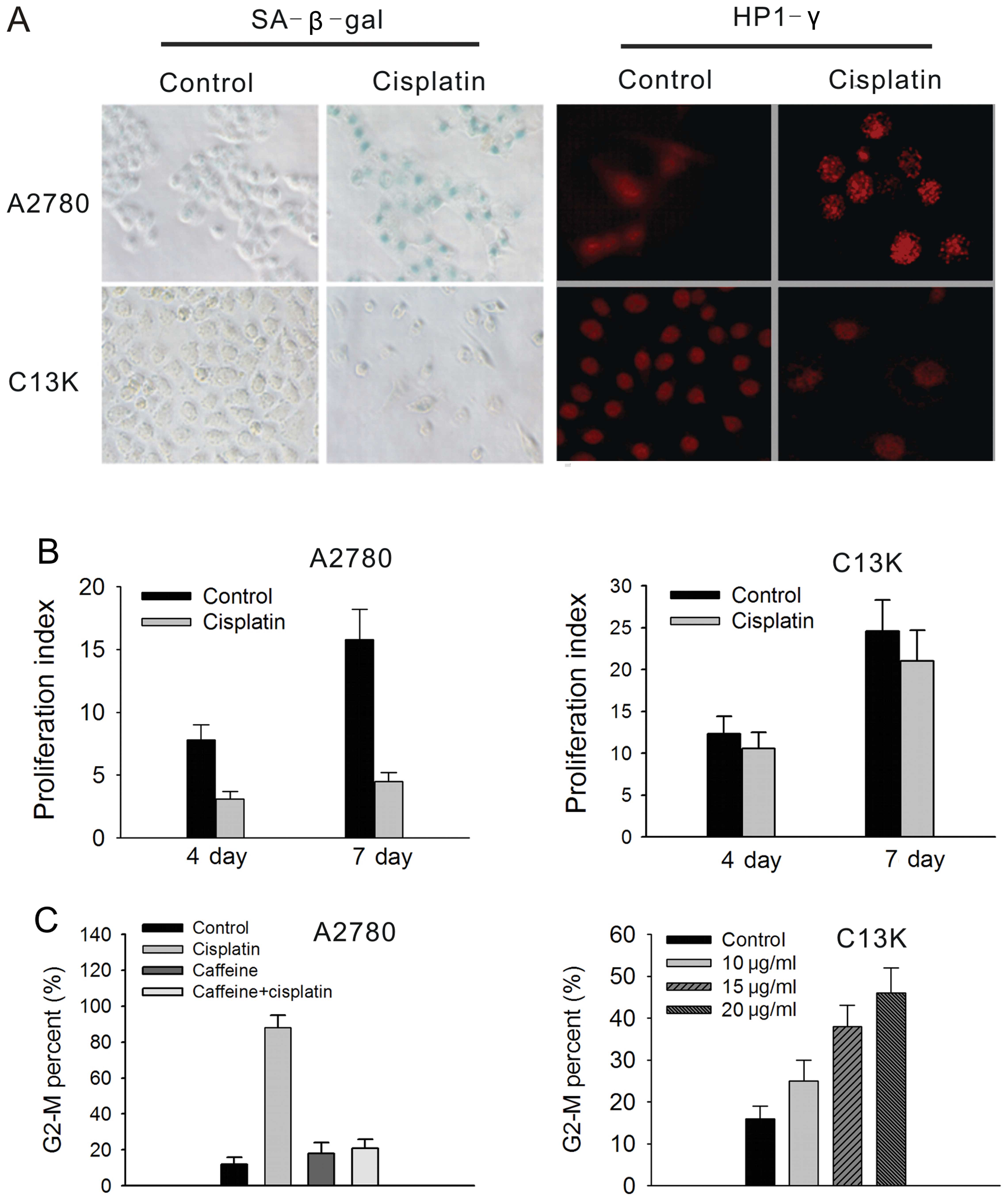
Exhibition of a senescence phenotype in
cisplatin-sensitive A2780 and cisplatin-resistant C13K cells
To study whether cisplatin induces ovarian cancer
cells to exhibit a senescence-like phenotype, cisplatin-sensitive
A2780 cells and cisplatin-resistant C13K cells were both exposed to
a sub-apoptotic dose of cisplatin for 24 h, respectively. The
results showed that A2780 cells exhibited a senescence phenotype at
day 6 following 3 μg/ml cisplatin treatment. Nearly 90% of the
A2780 cells showed an enlarged, flattened morphology, increased
cytoplasmic granularity, positive staining of SA-β-gal and massive
accumulation of HP1-γ foci (Fig.
2A). The senescent A2780 cells did not further divide as
confirmed by the CFSE proliferation index (PI). In the
cisplatin-treated A2780 cells, the PI values were 3.1±0.6 and
4.5±0.7 at day 4 and 7 following treatment, respectively, while the
PI values were 7.8±1.2 and 15.8±2.4, respectively, in the control
cells (Fig. 2B). Furthermore, more
than 80% of A2780 cells were arrested at the G2-M phase at day 4
following cisplatin treatment. These effects were mediated by the
ATM gene as verified by its abrogation with caffeine pretreatment
(5 mmol/l; Fig. 2C).
In contrast to the A2780 cells, C13K cells did not
show any senescent phenotype following treatment with various doses
of cisplatin ranging from 10 to 20 μg/ml. The staining of SA-β-gal
and HP1-γ foci were both negative in the C13K cells treated with a
sub-apoptotics dose of cisplatin (20 μg/ml) (Fig. 2A). In addition, C13K cells showed an
increased CFSE PI following treatment with 20 μg/ml cisplatin. The
PI values of the cisplatin-treated C13K cells were 7.5±1.2 and
14.2±0.5 at day 4 and 7 following treatment, which was similar to
the values for the untreated cells (8.9±1.7 and 16.8±3.2,
respectively) (Fig. 2B). Less than
30% of C13K cells were arrested at the G2-M phase at day 4
following cisplatin treatment (Fig.
2C).
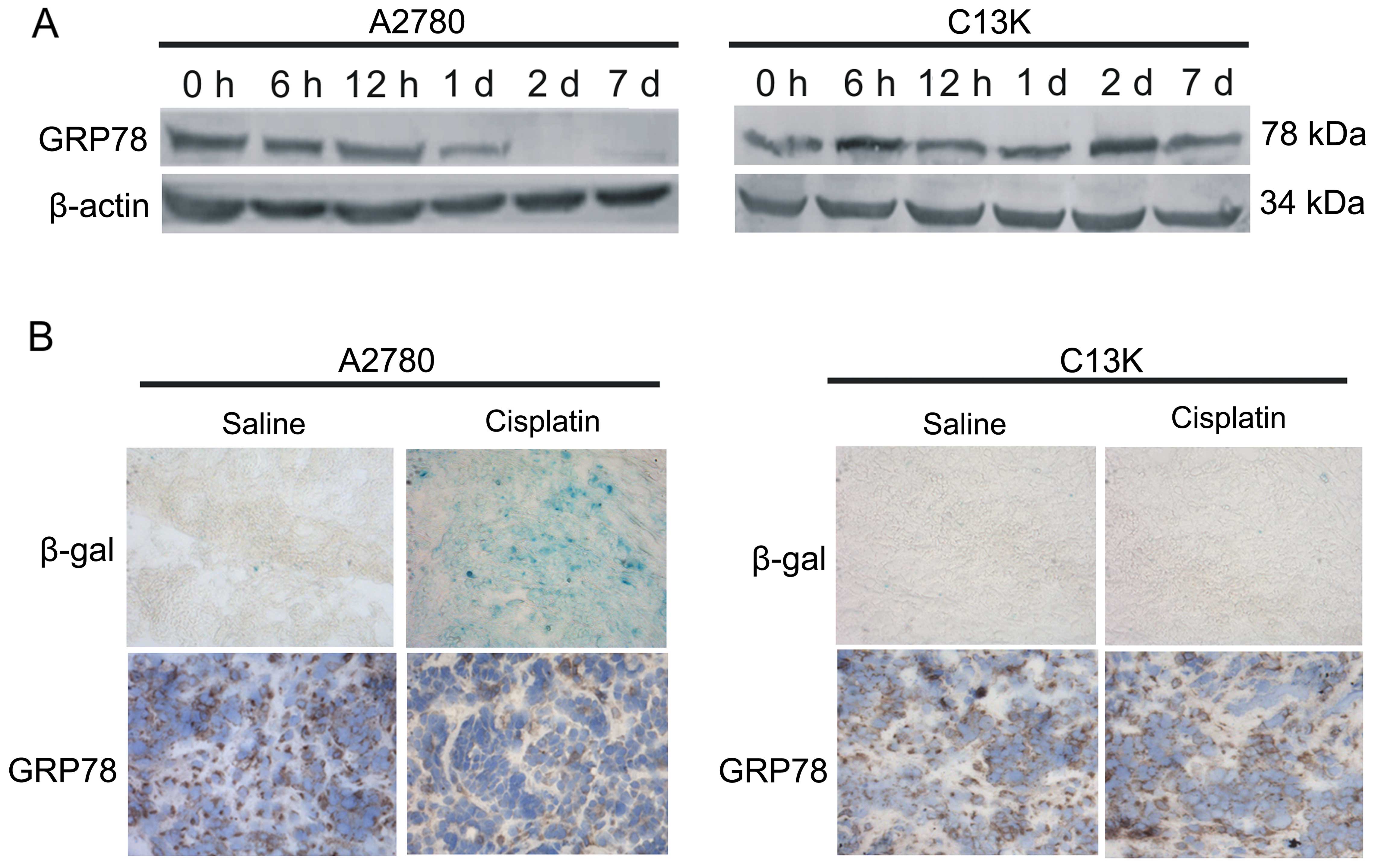
GRP78 expression in ovarian cancer cells
and subcutaneous tumors
To investigate whether GRP78 has an impact on
cisplatin-induced senescence, GRP78 expression was detected in
vitro and in vivo. In the A2780 cells, cisplatin
treatment resulted in decreased GRP78 protein levels at day 1 and
decreased to a minimum at day 2 to 7 (Fig. 3A). In vivo, the
cisplatin-treated subcutaneous tumor sections showed positive
staining for SA-β-gal (Fig. 3B;
upper row), and GRP78 stainning in the cisplatin-treated tumor
sections was significantly weaker than that in the saline-treated
tumor sections (Fig. 3B; lower
row).
In the C13K cells, compared with the untreated
cells, GRP78 protein levels increased at 6 h after exposure to
cisplatin and remained elevated at day 7 (Fig. 3A). SA-β-gal staining in both the
saline-treated and cisplatin-treated subcutaneous tumor sections
was negative (Fig. 3B; upper row),
and there was no difference in the GRP78 expression in these two
groups (Fig. 3B; lower row).
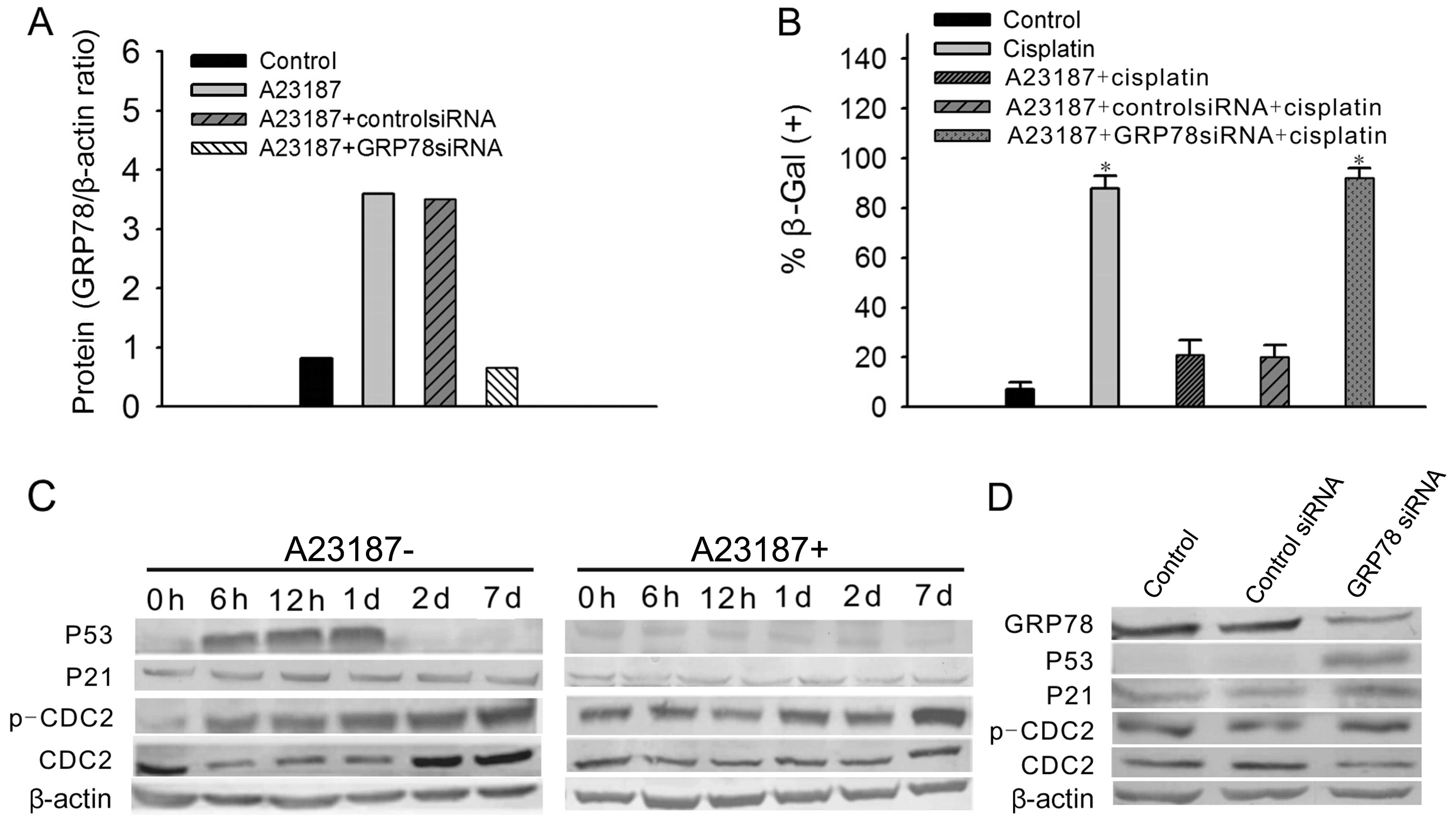
Overexpression of GRP78 results in A2780
cell resistance to cisplatin-induced senescence, which was
accompanied by obvious suppression of P53 expression
A23187 is a mobile ion-carrier that forms stable
complexes with divalent cations and has frequently been used to
upregulate GRP78 expression. In our experiments, A2780 cells were
treated with A23187 at a final concentration of 2.5 mmol/l 1 day
before cisplatin treatment. GRP78 protein levels were significantly
increased following A23187 exposure, but were significantly
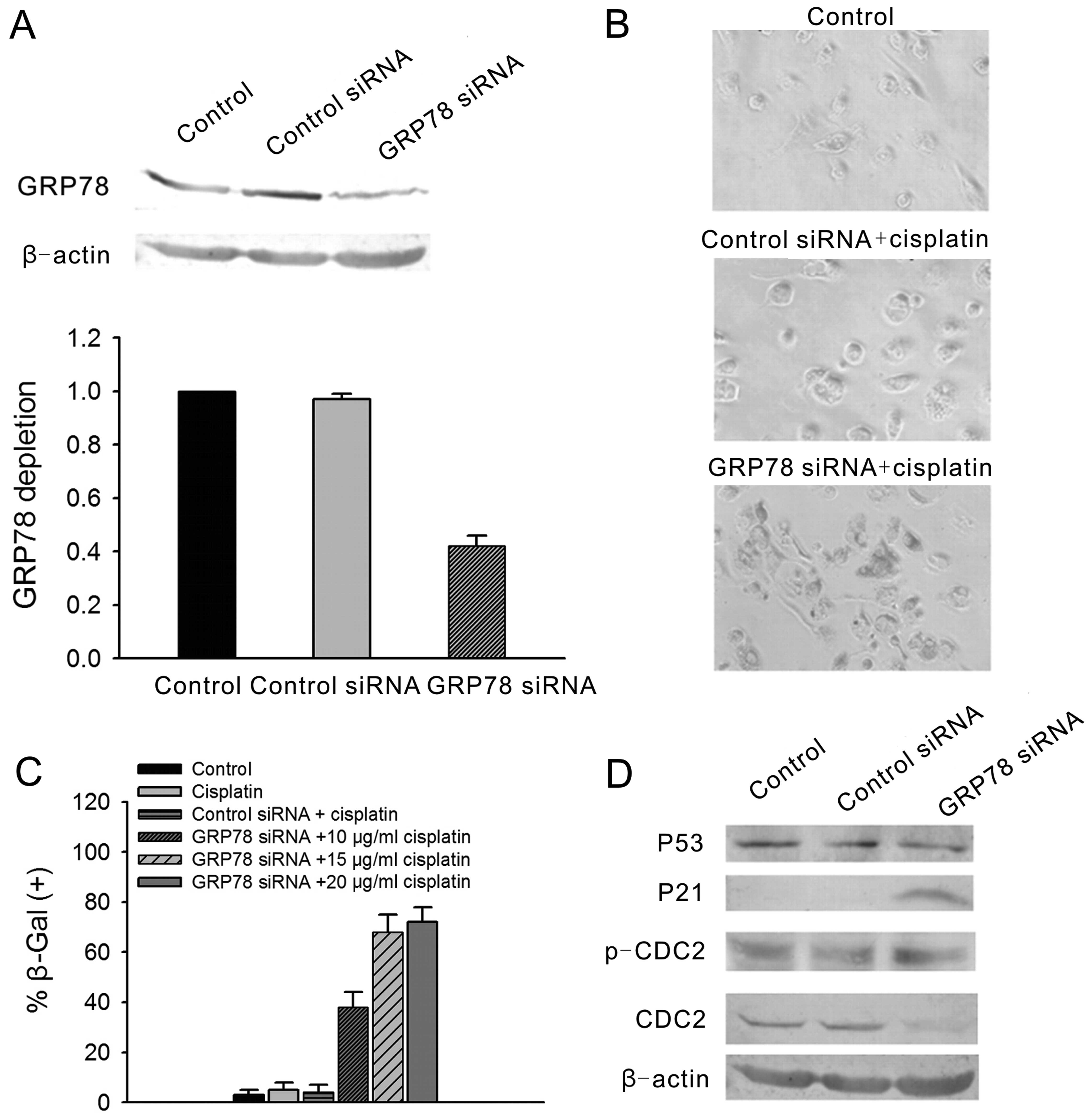
decreased following GRP78-siRNA treatment (Fig. 4A). Seven days after 3 μg/ml
cisplatin treatment, <20% of the A23187-treated A2780 cells
showed positive SA-β-gal staining, whereas cells treated with
cisplatin only were almost all positively stained (Fig. 4B). To confirm the specific
anti-senescence effect of GRP78, GRP78-siRNA was transfected into
the A23187-treated A2780 cells. After 3 days, the treated cells
were exposed to 3 μg/ml cisplatin and incubated with complete
medium. After 6 days, the cells exhibited 90% positive SA-β-gal
staining (Fig. 4B). These data
indicate that overexpression of GRP78 can protect A2780 cells
against cisplatin-induced senescence.
To clarify the mechanisms of the
senescence-resistant effects of GRP78, levels of the ATM pathway
genes in A2780 cells were examined by western blotting at 6 and 12
h; 1, 2 and 7 days following cisplatin treatment with or without
A23187 exposure. In the non-A23187-treated cells, P53 protein
levels were significantly increased from 6 h to 1 day and were
diminished at day 2; P21 levels were slightly decreased following
exposure to cisplatin. CDC2 levels were significantly decreased at
day 1 following exposure to cisplatin but were significantly
increased from day 2 to 7. p-CDC2 levels were significantly
increased from 6 h to 7 days. However, in the A23187-treated cells,
there were no changes in the protein levels of P53 and CDC2
following cisplatin exposure. The changes in the protein levels of
p-CDC2 and P21 were consistent with those in the non-A23187-treated
A2780 cells (Fig. 4C).
Moreover, the ATM pathway gene protein levels were
examined by western blotting after knockdown of GRP78. The results
showed that GRP78 protein levels were significantly decreased after
GRP78-siRNA transfection in the A2780 cells. Accompanied by
knockdown of GRP78, P53 levels were significantly increased and
CDC2 levels were significantly decreased. There were no changes in
the protein levels of P21 and p-CDC2 (Fig. 4D). This suggests that P53 and CDC2
are involved in cisplatin-induced senescence of A2780 cells.
Knockdown of GRP78 rescues the senescence
sensitivity of C13K cells to cisplatin through an increase in P21
expression and a decrease in CDC2 expression
To investigate whether GRP78 rescues the senescence
sensitivity in C13K cells to cisplatin, GRP78 expression was
knocked down by GRP78-siRNA (Fig.
5A). Then, the senescence sensitivity of C13K cells to
cisplatin was detected by β-gal staining. After a 1-day incubation
with 10, 15 or 20 μg /ml cisplatin, the C13K cells transfected with
GRP78-siRNA exhibited a significant senescence phenotype at day 7
following exposure to cisplatin (Fig.
5B and C).
At the same time, we detected the expression of ATM
pathway genes in the C13K cells. The results showed that P21 levels
were significantly increased and CDC2 levels were significantly
decreased following GRP78-siRNA transfection. In contrast, no
changes in P53 and p-CDC2 levels were noted (Fig. 5D).
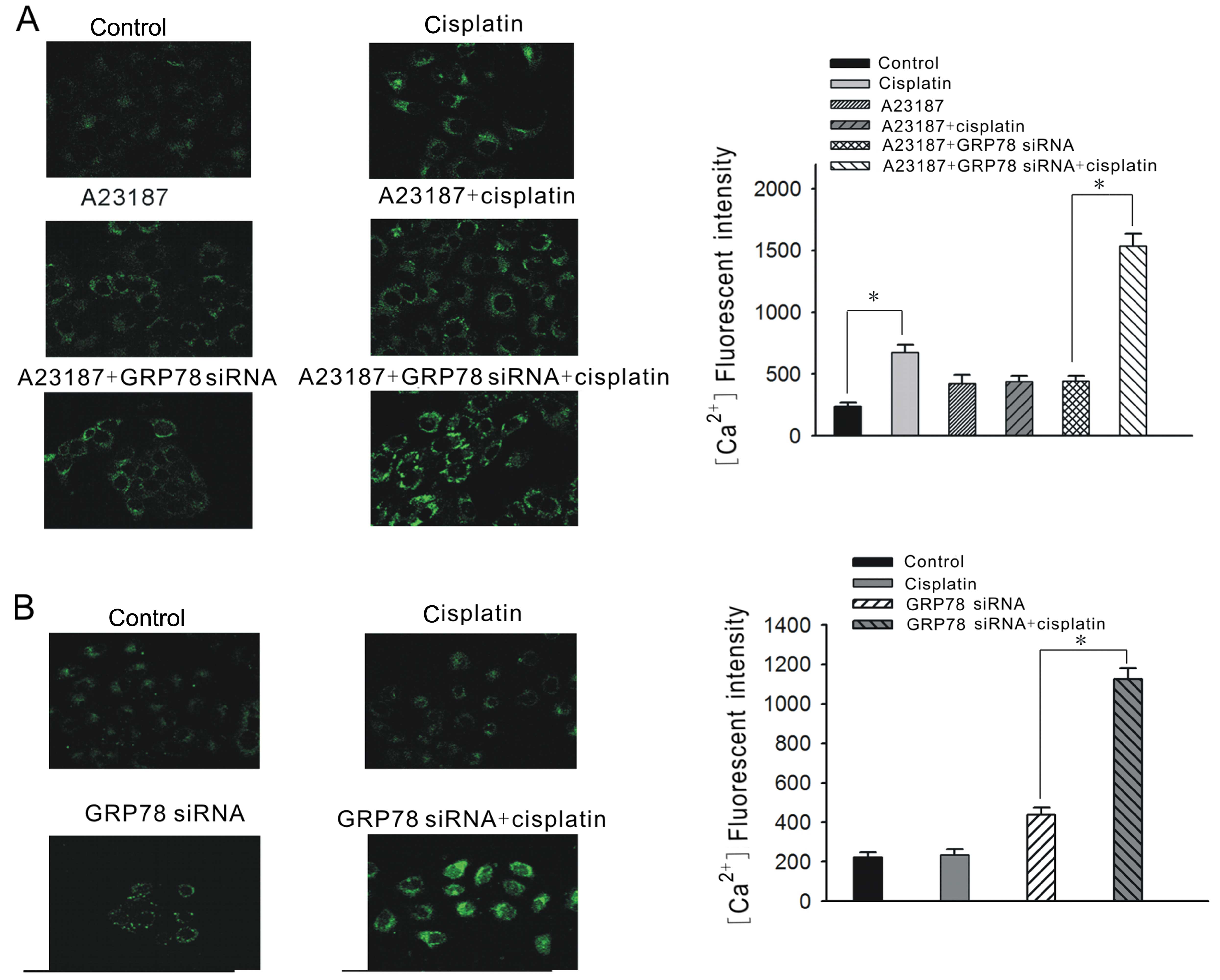
Twisting of Ca2+ release from
ER stores by GRP78 is associated with the sensitivity of
cisplatin-induced senescence in ovarian cancer cell lines
GRP78 is a major ER chaperone with
Ca2+-binding property, which can preserve ER calcium
homeostasis. Therefore, we examined changes in the relative
cellular Ca2+ concentration to ascertain whether it is
relevant to the sensitivity of cisplatin-induced senescence.
In the A2780 cells, the cellular Ca2+
concentration was elevated ~3-fold following a 12-h exposure to 3
μg/ml cisplatin. Nonetheless, with a previous A23187 induction for
24 h, the cellular Ca2+ concentration was not evidently
increased at 12 h following cisplatin treatment. When GRP78
overexpression was suppressed by GRP78-siRNA, the cellular
Ca2+ concentration was significantly elevated again at
12 h following cisplatin treatment (Fig. 6A).
In the C13K cells, the cellular Ca2+
concentration was not obviously elevated following a 12-h exposure
to 20 μg/ml cisplatin. Nonetheless, the cellular concentration
increased nearly 3-fold at 12 h following 20 μg/ml cisplatin
treatment with previous GRP78 suppression by GRP78-siRNA (Fig. 6B). It appears that the capacity of
Ca2+ release from ER stores may be associated with the
sensitivity of cisplatin-induced senescence. GRP78 may influence
the sensitivity through twisting the Ca2+ release from
ER stores.
Discussion
In the present study, we showed that GRP78
expression is negatively associated with cisplatin-induced
senescence in vitro and in vivo. Cisplatin-sensitive
A2780 cells exhibited a senescence phenotype, while
cisplatin-resistant C13K cells showed no senescence phenotype
following a dose-gradient cisplatin exposure. Knockdown of GRP78
expression rescued the senescence sensitivity of C13K cells to
cisplatin. The ATM pathway genes such as P53, P21 and
CDC2, and ER calcium homeostasis were involved in the
cisplatin-induced senescence.
GRP78 is a major stress-induced chaperone localized
to the endoplasmic reticulum. GRP78 has been examined in human
breast carcinomas, and its overexpression has been observed in the
majority of the more aggressive estrogen receptor-negative tumors
(27). Preliminary analysis of
GRP78 in a series of primary and recurrent breast, prostate and
lung cancer samples suggest a correlation between GRP78
overexpression and tumor recurrence and drug resistance (28). Moreover, GRP78 has been gradually
used as a potent target for diagnosis and therapy of different
types of cancers (29). Synthetic
chimeric peptides targeted against GRP78 were found to suppress
tumor growth in xenograft and isogenic mouse models of breast and
prostate cancer (30). Research has
also indicated that GRP78 is associated with the apoptosis of tumor
cell lines (31).
In the present study, we firstly investigated the
relationship between GRP78 expression and chemotherapy-induced
senescence in ovarian cancer patients. The data showed that GRP78
expression was significantly higher in the chemotherapy-resistant
tumor samples with weak HP1-γ staining than that of the
chemotherapy-sensitive samples with obvious HP1-γ staining. It was
suggested that GRP78 may mediate chemotherapy-induced senescence
and play a crucial role in the drug resistance in ovarian
cancer.
Next, we observed the senescence phenotype in
ovarian cancer cells. The results showed that cisplatin-sensitive
A2780 cells exhibited an obvious senescent phenotype following
cisplatin treatment, and most of the A2780 cells were arrested at
the G2/M phase. Accompanied by senescence, GRP78 levels gradually
decreased following cisplatin treatment. Conversely,
cisplatin-resistant C13K cells did not present a senescent
phenotype after a dose-gradient cisplatin exposure, while GRP78
levels increased. Moreover, we also obtained a similar result in
the subcutaneous tumor samples. Therefore, GRP78 expression is
related with the cisplatin-induced senescence in ovarian cancer
cells.
ATM pathway genes are closely associated with
senescence. Therefore, we investigated the relationship between
GRP78 and the ATM pathway genes during cisplatin-induced senescence
in ovarian cancer cells. In the A2780 cells, GRP78 overexpression
protected the cells against cisplatin-induced senescence, which was
mainly through the suppression of P53 expression. In addition,
knockdown of GRP78 resulted in noticeably increased P53 levels.
Studies have shown that wild-type p53 limits cellular proliferation
by inducing senescence, depending on the expression level or
cellular context (32,33). An increase in p53 transcriptional
activity is a molecular signature for cellular senescence. It could
be postulated that P21-independent P53 expression is a requirement
for cisplatin-induced senescence in A2780 cells.
In the C13K cells, knockdown of GRP78 resulted in
significantly enhanced senescence sensitivity to cisplatin, which
occurred mainly through increased P21 levels and decreased CDC2
levels. Nonetheless, there was no change in P53 levels. P21, a
transcriptional target of P53, can inhibit Cdk activity (34). It has been clearly established that
P21 is highly related to senescence, and induces senescence in the
P53-independent pathway (35,36).
CDC2 is the most important gene controlling the cell cycle
transition from the G2 to M phase. Decreased CDC2 expression and
increased expression of its non-active (15-Tyr)-phosphorylated CDC2
often occurs at G2/M phase arrest and senescence (37). It appears that GRP78-suppression
induced senescence was P53-independent in the cisplatin-resistant
C13K cells.
Research has revealed that calcium is related to
apoptosis (38). Nonetheless, the
relationship between calcium and senescence has not been explored.
As GRP78 is a major ER protein controlling the calcium efflux from
the ER to the cytoplasm, we investigated the association between
the change in calcium concentration in the cytoplasm and
cisplatin-induced senescence. In the A2780 cells, GRP78
overexpression induced by A23187 obstructed calcium efflux from the
ER to the cytoplasm and protected against cisplatin-induced
senescence. Conversely, knockdown of GRP78 retrieved the calcium
efflux from the ER to the cytoplasm and recovered the senescence
sensitivity to cisplatin; whereas, there was no calcium efflux from
the ER after cisplatin treatment in the C13K cells, which were
senescence-resistant, and suppression of GRP78 expression led to
the calcium efflux from the ER to the cytoplasm after
cisplatin-treatment and recovered the senescence-sensitivity.
According to the results, calcium efflux from the ER to the
cytoplasm, which is controlled by GRP78, is essential for
cisplatin-induced senescence.
In conclusion, our data showed that GRP78 plays an
anti-senescence role in ovarian cancer cells through changes in the
expression of ATM pathway genes and calcium efflux from the ER to
the cytoplasm. Therefore, targeting against GRP78 may reduce the
drug-resistance of ovarian cancer to cisplatin.
Acknowledgements
This study was supported by grants from the National
Basic Research Program of China 973 Program, and the National
Natural Science Foundation of China (nos. 2013CB911304, 81072132,
81001152, 81101963, 81172466 and 30872751).
Abbreviations:
|
SAHF
|
senescence-associated heterochromatic
foci
|
|
HP1-γ
|
heterochromatin protein 1-γ
|
|
CFSE
|
carboxyfluorescein succinimidyl
ester
|
|
siRNA
|
synthetic small interfering RNA
|
|
SA-β-gal
|
senescence-associated
β-galactosidase
|
|
GRP78
|
glucose-regulated protein 78
|
|
ER
|
endoplasmic reticulum
|
|
ATM
|
ataxia telangiectasia mutated
|
|
NAC
|
neoadjuvant chemotherapy
|
References
|
1
|
Selvakumaran M, Pisarcik DA, Bao R, Yeung
AT and Hamilton TC: Enhanced cisplatin cytotoxicity by disturbing
the nucleotide excision repair pathway in ovarian cancer cell
lines. Cancer Res. 63:11–16. 2003.PubMed/NCBI
|
|
2
|
Yang X, Xing H, Gao Q, Chen G, Lu Y, Wang
S and Ma D: Regulation of HtrA2/Omi by X-linked inhibitor of
apoptosis protein in chemoresistance in human ovarian cancer cells.
Gynecol Oncol. 97:413–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee S, Choi EJ, Jin C and Kim DH:
Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA
mRNA amplification contributes to cisplatin resistance in an
ovarian cancer cell line. Gynecol Oncol. 97:26–34. 2005.PubMed/NCBI
|
|
4
|
Gifford G, Paul J, Vasey PA, Kaye SB and
Brown R: The acquisition of hMLH1 methylation in plasma DNA
after chemotherapy predicts poor survival for ovarian cancer
patients. Clin Cancer Res. 10:4420–4426. 2004.
|
|
5
|
Williams J, Lucas PC, Griffith KA, et al:
Expression of Bcl-xL in ovarian carcinoma is associated with
chemoresistance and recurrent disease. Gynecol Oncol. 96:287–295.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roninson IB: Tumor cell senescence in
cancer treatment. Cancer Res. 63:2705–2715. 2003.PubMed/NCBI
|
|
7
|
Elmore LW, Xu Di, Dumur C, Holt SE and
Gewirtz DA: Evasion of a single-step, chemotherapy-induced
senescence in breast cancer cells: implications for treatment
response. Clin Cancer Res. 11:2637–2643. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee BY, Han JA, Im JS, et al:
Senescence-associated β-galactosidase is lysosomal β-galactosidase.
Aging Cell. 5:187–195. 2006.
|
|
9
|
Bartkova J, Rezaei N, Liontos M, et al:
Oncogene-induced senescence is part of the tumorigenesis barrier
imposed by DNA damage checkpoints. Nature. 444:633–637. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
te Poele RH, Okorokov AL, Jardine L,
Cummings J and Joel SP: DNA damage is able to induce senescence in
tumor cells in vitro and in vivo. Cancer Res. 62:1876–1883.
2002.PubMed/NCBI
|
|
11
|
Schmitt CA, Fridman JS, Yang M, et al: A
senescence program controlled by p53 and p16INK4a
contributes to the outcome of cancer therapy. Cell. 109:335–346.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roninson IB: Tumour senescence as a
determinant of drug response in vivo. Drug Resist Updat. 5:204–208.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang K, Chiu CC, Li CH, Chang YT and Hwang
HT: Cisplatin-induced senescence and growth inhibition in human
non-small cell lung cancer cells with ectopic transfer of
p16INK4a. Oncol Res. 16:479–488. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee AS: The glucose-regulated proteins:
stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ranganathan AC, Zhang L, Adam AP and
Aguirre-Ghiso JA: Functional coupling of p38-induced up-regulation
of BiP and activation of RNA-dependent protein kinase-like
endoplasmic reticulum kinase to drug resistance of dormant
carcinoma cells. Cancer Res. 66:1702–1711. 2006. View Article : Google Scholar
|
|
16
|
Fu Y, Li J and Lee AS: GRP78/BiP inhibits
endoplasmic reticulum BIK and protects human breast cancer cells
against estrogen-starvation induced apoptosis. Cancer Res.
67:3734–3740. 2007. View Article : Google Scholar
|
|
17
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ermakova SP, Kang BS, Choi BY, et al:
(−)-Epigallocatechin gallate overcomes resistance to
etoposide-induced cell death by targeting the molecular chaperone
glucose-regulated protein 78. Cancer Res. 66:9260–9269. 2006.
|
|
19
|
Davidson DJ, Haskell C, Majest S, et al:
Kringle 5 of human plasminogen induces apoptosis of endothelial and
tumour cells through surface-expressed glucose-regulated protein
78. Cancer Res. 65:4663–4672. 2005. View Article : Google Scholar
|
|
20
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee HK, Xiang CL, Cazacu S, et al: GRP78
is overexpressed in glioblastomas and regulates glioma cell growth
and apoptosis. Neuro Oncol. 10:236–243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang CC, Mao ZG, Avery-Kiejda K, Wade M,
Hersey P and Zhang XD: Glucose-regulated protein 78 antagonizes
cisplatin and adriamycin in human melanoma cells. Carcinogenesis.
30:197–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zu K, Bihani T, Lin A, Park YM, Mori K and
Ip C: Enhanced selenium effect on growth arrest by BiP/GRP78
knockdown in p53-null human prostate cancer cells. Oncogene.
25:546–554. 2006.PubMed/NCBI
|
|
24
|
Tate S, Hirai Y, Takeshima N and Hasumi K:
CA125 regression during neoadjuvant chemotherapy as an independent
prognostic factor for survival in patients with advanced ovarian
serous adenocarcinoma. Gynecol Oncol. 96:143–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang W, Wang S, Song X, et al: The
relationship between c-FLIP expression and human papillomavirus
E2 gene disruption in cervical carcinogenesis. Gynecol
Oncol. 105:571–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hawkins LK, Johnson L, Bauzon M, et al:
Gene delivery from the E3 region of replicating human adenovirus:
evaluation of the 6.7 K/gp19 K region. Gene Ther. 8:1123–1131.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fernandez PM, Tabbara SO, Jacobs LK, et
al: Overexpression of the glucose-regulated stress gene GRP78 in
malignant but not benign human breast lesions. Breast Cancer Res
Treat. 59:15–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koomägi R, Mattern J and Volm M:
Glucose-related protein (GRP78) and its relationship to the
drug-resistance proteins P170, GST-pi, LRP56 and angiogenesis in
non-small cell lung carcinomas. Anticancer Res. 19:4333–4336.
1999.PubMed/NCBI
|
|
29
|
Taylor DD, Gercel-Taylor C and Parker LP:
Patient-derived tumor-reactive antibodies as diagnostic markers for
ovarian cancer. Gynecol Oncol. 115:112–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arap MA, Lahdenranta J, Mintz PJ, et al:
Cell surface expression of the stress response chaperone GRP78
enables tumor targeting by circulating ligands. Cancer Cell.
6:275–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou H, Zhang Y, Fu Y, Chan L and Lee AS:
Novel mechanism of anti-apoptotic function of 78-kDa
glucose-regulated protein (GRP78): endocrine resistance factor in
breast cancer, through release of B-cell lymphoma 2 (BCL-2) from
BCL-2-interacting killer (BIK). J Biol Chem. 286:25687–25696. 2011.
View Article : Google Scholar
|
|
32
|
Petroulakis E, Parsyan A, Dowling RJ, et
al: p53-dependent translational control of senescence and
transformation via 4E-BPs. Cancer Cell. 16:439–446. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gannon HS, Donehower LA, Lyle S and Jones
SN: Mdm2-p53 signaling regulates epidermal stem cell senescence and
premature aging phenotypes in mouse skin. Dev Biol. 353:1–9. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vaziri H, West MD, Allsopp RC, et al:
ATM-dependent telomere loss in aging human diploid fibroblasts and
DNA damage lead to the post-translational activation of p53 protein
involving poly(ADP-ribose) polymerase. EMBO J. 16:6018–6033. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen H and Maki CG: Persistent p21
expression after Nutlin-3a removal is associated with
senescence-like arrest in 4N cells. J Biol Chem. 285:23105–23114.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roberson RS, Kussick SJ, Vallieres E, Chen
SY and Wu DY: Escape from therapy-induced accelerated cellular
senescence in p53-null lung cancer cells and in human lung cancers.
Cancer Res. 65:2795–2803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Giorgi C, Ito K, Lin HK, et al: PML
regulates apoptosis at endoplasmic reticulum by modulating calcium
release. Science. 330:1247–1251. 2010. View Article : Google Scholar : PubMed/NCBI
|