Introduction
Pancreatic cancer is an aggressive malignancy, the
fourth leading cause of cancer-related mortality in the United
States, with ~44,980 new diagnoses and ~38,460 deaths predicted in
2013 (1). Owing to pancreatic
cancer characteristics, >80% of patients are diagnosed at an
advanced stage, thereby losing the probability of surgical
resection (2). Despite recent
progress in chemotherapy, radiation therapy, and surgical
resections, the overall survival rate of pancreatic cancer is still
<5% (3), with a median survival
between 3 and 6 months (4,5). Thus, molecular targeted therapy may be
a suitable therapeutic option for human pancreatic cancer
treatment.
Pancreatic tumors usually display a ductal, an
acinar or an endocrine differentiation. Eighty percent of all
pancreatic carcinomas are estimated as pancreatic ductal
adenocarcinoma (PDAC) (6). The
progression model of PDAC is associated with multiple genetic and
epigenetic alterations that result in the deregulation of key
proto-oncogenes, tumor-suppressor genes, and signaling pathways,
including K-ras, p16, p53, BRCA2, Smad4, EGF/EGFR, c-MET/HGF
pathway, Ras/Raf/MAPK pathway, PTEN/PI3K/AKT pathway, JAK/STAT
pathway and Wnt signaling. However, roles of other proto-oncogenes
and tumor suppressor genes in PDAC development remain elusive
(7).
We previously identified Pim-3, a
proto-oncogene with serine/threonine kinase activity, as the gene
selectively expressed in human pancreatic cancer tissues, but not
in the normal pancreas (8).
Pim-3 was originally identified as a depolarization-induced
gene, KID-1, in PC12 cells, a rat pheochromocytoma cell line
(9). Subsequently, Deneen et
al (10) demonstrated that
Pim-3 gene transcription was enhanced in EWS/ETS-induced malignant
transformation of NIH3T3 cells, suggesting the involvement of Pim-3
in tumorigenesis. Consistently, we observed that Pim-3 expression
was enhanced in malignant lesions, but not in normal tissues of
human endoderm-derived organs, including the pancreas (8), liver (11), colon (12) and stomach (13). Moreover, hepatocellular carcinoma
development was accelerated in mice selectively expressing the
Pim-3 transgene in the liver, when these mice were treated with a
hepatocarcinogen (14).
Furthermore, Pim-3 can inactivate a proapoptotic molecule, Bad, and
maintain the expression of an antiapoptotic molecule,
Bcl-XL, and prevent apoptosis of human pancreatic cancer
and colon cancer cells (8,12). In addition, we demonstrated that
TCTP-mediated enhancement in Pim-3 protein stability can be
involved in pancreatic carcinogenesis (15). Thus, Pim-3 is a key player in
pancreatic tumorigenesis.
However, regulatory mechanisms of Pim-3 signaling
networks in vivo are not well understood. In the present
study, we discovered that the incidence of human pancreatic cancer
was significantly decreased from 100% to 46.6% in nude mice
subcutaneously injected with cells stably expressing the inactive
Pim-3 kinase (K69M-Pim-3) compared with mice injected with cells
overexpressing wild-type Pim-3. Moreover, Pim-3 kinase inactivation
reduced the expression of several angiogenesis factors, such as
HGF, EGF, FGF-2, as well as VEGF, and subsequently prevented
neovascularization of nude mice xenografts. These results suggested
that Pim-3 kinase activity played a crucial role in accelerating
human pancreatic cancer development and in promoting tumor
neovascularization and subsequent tumor growth.
Materials and methods
Cell culture and reagents
Human pancreatic cancer cell lines, MiaPaca-2
(16) and PCI55 (17), were maintained in RPMI-1640
(BioWest, Nuaillé, France) containing 10% fetal bovine serum (FBS;
BioWest). Human embryonic kidney HEK293T cells were maintained in
DMEM (Sigma) containing 5% FBS. All cells were cultured in 5%
CO2 at 37°C. The following monoclonal antibodies (mAbs)
and polyclonal antibodies (pAbs) were commercially obtained: rabbit
anti-Pim-3 mAbs, rabbit anti-phospho-Ser112 Bad pAbs,
rabbit anti-phospho-Ser136 Bad pAbs, rabbit
anti-phospho-Ser155 Bad pAbs, rabbit anti-STAT3 mAbs,
rabbit anti-survivin mAbs, rabbit anti-phospho-Ser34
survivin mAbs, mouse anti-phospho-try705 STAT3 mAbs, and mouse
anti-PCNA mAbs (Cell Signaling Technology, Beverly, MA, USA);
rabbit anti-β-actin mAb (Sigma-Aldrich, St. Louis, MO, USA); mouse
anti-Bad mAb and goat anti-mouse HRP-IgG pAbs (Santa Cruz
Biotechnology, Santa Cruz, CA, USA); goat anti-rabbit HRP-IgG pAbs
(Pierce Biotechnology, Rockford, IL, USA); rabbit anti-CD31 mAbs
(Abcam, Cambridge, MA, USA).
Retroviral vector construction and
retrovirus production
Full-length wild-type human Pim-3 cDNA and
kinase-dead mutant human Pim-3 (K69M) cDNA were inserted into the
PmaCI and HpaI sites of the pMEI-5 Neo retroviral
expression vector. Retrovirus was produced by transfecting 293T
cells with retroviral vectors using the Retrovirus Packaging Kit
Ampho (Takara, Dalian, China), according to the manufacturer’s
instructions. Two days post-transfection, the virus-containing
supernatant was collected, passed through 0.45-μm syringe filters,
and the virus titer was determined using the Retrovirus Titer Set
(Takara) according to the manufacturer’s instructions.
Establishment of Pim-3 and K69M-Pim-3
stable cell lines
Human pancreatic cancer cells, MiaPaca-2, as target
cells were incubated with the retroviral supernatant for 24 h.
Subsequently, the infected MiaPaca-2 cells were cultured in 10%
FBS-containing RPMI-1640 in the presence of 800 μg/ml G418 (Gibco)
for 2–3 weeks. The surviving cells were isolated using cloning
rings and analyzed for Pim-3 or K69M-Pim-3 expression using western
blotting. Cells with the highest expression levels of Pim-3 and
K69M-Pim-3 were designated as MiaPaca-2-Pim-3 cells and
MiaPaca-2-Pim-3K69M cells, respectively, and used for subsequent
experiments.
Construction of tetracycline-inducible
Pim-3 shRNA expression vectors
pSingle-tTS-Pim-3shRNA and pSingle-tTS-scramble
shRNA expression constructs were provided by Professor Naofumi
Mukaida (Cancer Research Institute, Kanazawa University, Japan). In
brief, the selected short interfering RNA target sequence in Pim-3
(5′-GCACGUGGUG AAGGAGCGG-3′ corresponding to 642–661 residues) and
non-specific control short interfering RNA duplexes (5′-GCG
CGCUUUGUAGGAUUCG-3′) were designed as previously described
(8), while small hairpin RNA
(shRNA) encoding oligonucleotides were synthesized by Ambion
(Austin, TX, USA). The annealed shRNA was inserted into the
HindIII and XhoI sites of the pSingle-tTS-shRNA
vector (Clontech Laboratories-Takara Bio Inc., Japan) and
designated as pSingle-tTS-Pim-3shRNA and pSingle-tTS-scramble
shRNA, respectively. The pSingle-tTS-shRNA vector expresses the
tetracycline-controlled transcriptional suppressor (tTS), which, in
turn, controls the expression of Pim-3 shRNA or scramble shRNA
inserted into the shRNA cloning site.
Transfection of shRNA expression vectors
and generation of stable cell lines
PCI55 cells were transfected with
pSingle-tTS-Pim-3shRNA or pSingle-tTS-scramble shRNA, which
functioned as the control, using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s protocol. The
transfected PCI55 cells were cultured in 10% FBS-containing
RPMI-1640 in the presence of 800 μg/ml G418 (Gibco) for 2–3 weeks
and monoclonal cells were isolated. Thirty colonies were expanded
and screened for inducible expression using media supplemented with
or without 1 μg/ml tetracycline for 48 h. Cells were analyzed for
Pim-3 expression using western blotting. The stably transfected
Pim-3 shRNA cells with markedly diminished Pim-3 protein expression
under inducible conditions were designated as PCI55-Pim-3shRNA
cells, while the stably transfected scramble shRNA cells
maintaining Pim-3 protein expression under inducible conditions
were designated as PCI55-Scramble shRNA cells. These two stable
cell lines were used for subsequent experiments.
Western blotting
Cells (2×106) were harvested and rinsed
twice with PBS. Cell extracts were prepared with lysis buffer [20
mM Tris (pH 7.5), 0.1% Triton X-100, 0.5% deoxycholate, 1 mM
phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 g/ml
leupeptin] and cleared by centrifugation at 10,000 × g, 4°C for 15
min. Total protein concentration was measured using the BCA assay
kit (Sigma) with BSA as a standard, according to the manufacturer’s
instructions. Cell extracts containing 30 μg of total protein were
subjected to 10% SDS-PAGE, and the resolved proteins were
transferred electrophoretically to polyvinylidene difluoride
membranes (Millipore). Equal protein loading was confirmed by
Coomassie blue (Bio-Rad Laboratories, Hercules, CA, USA) staining
of the gel. After blocking with TBST containing 0.2% BSA for 1 h at
room temperature, membranes were incubated with 3–5 μg/ml
antibodies in PBS containing 0.1% Tween-20 overnight at 4°C,
followed by incubation with ImmunoPure peroxidase-conjugated
anti-rabbit IgG or anti-mouse IgG. Chemiluminescent detection
(Thermo Scientific Pierce, Rockford, IL, USA) was performed in
accordance with the manufacturer’s instructions. The blotted
membrane was then treated with the SuperSignal West Dura Extended
Duration Substrate and signals were detected using the LAS-4000
mini CCD camera. Blots were performed at least three times in
independent experiments. For some experiments, tumor tissues were
prepared with RIPA lysis buffer.
Cell viability assay
Cell viability was determined by the Cell
Proliferation Assay and Cytotoxicity Assay kit (CCK-8; Dojindo
Laboratories, Kumamoto, Japan), according to the manufacturer’s
instructions. Logarithmically growing cells were plated at
2×103 cells/well in 96-well culture plates (Corning),
and allowed to adhere overnight. This time-point was designated as
day 0. The cell viability was determined every day by adding 10 μl
of CCK-8 reagent to each well. After incubation at 37°C for 2 h,
the absorbance at 450 nm was measured and ratios of cell numbers
were determined by comparison of the number of cells at day 0. Each
independent experiment was performed three times.
Cell apoptosis analysis
The cells were trypsinized and 2×105
cells were plated in a 6-well plate. After incubation at 37°C for
24 h, cells were washed and resuspended in 0.5 ml of PBS, 5 μl
Annexin V-FITC (Invitrogen), and 1 μl propidium iodide (100 μg/ml;
Invitrogen). The cells were incubated for 30 min on ice and then
analyzed by flow cytometry (Cytomics™ FC 500; Beckman Coulter,
Miami, USA) for each treatment. The apoptotic fraction was
estimated by dividing the number of apoptotic cells by the total
number of cells (minimum of 104 cells). Data were
analyzed using Cytomics FC 500 with CXP Software (Beckman Coulter).
All observations were reproduced at least three times in
independent experiments.
Xenograft mouse model
Female Balb/c nude mice (6–8 weeks of age, weighing
18–20 g, and specific pathogen free) were obtained from Shanghai
SLAC Laboratory Animal Co. (Shanghai, China). Before the
experiment, mice were divided into six groups (MiaPaca-2,
MiaPaca-2-Pim-3, MiaPaca-2-Pim-3K69M, PCI55, PCI55-Pim-3shRNA and
PCI55-scramble shRNA) according to body weight, and each cell line
(4×106/site) was injected subcutaneously into the right
flank of the nude mice. After establishment of the nude mice
xenograft model, tumor dimensions were measured every 3–4 days
using micrometer calipers. Tumor volumes were calculated using the
following formula: Volume = 1/2 a × b2, where a and b
represent the larger and smaller tumor diameters, respectively. At
30 days after the tumor cell injection, tumor tissues were removed
and subjected to immunohistochemical analysis. All animal
experiments were performed in compliance with the Guideline for the
Care and Use of Laboratory Animals of Fudan University. The
protocol was approved by the Committee on the Ethics of Animal
Experiments of Fudan University (Permit Number,
SYXK(Hu)2009–0082).
Immunohistochemical analysis
Paraffin-embedded tissue sections were
deparaffinized in xylene and rehydrated through graded
concentrations of ethanol (70–100%). Following incubation with 0.3%
hydrogen peroxide, sections were incubated with 3% normal goat
serum (DakoCytomation, Glostrup, Denmark). Subsequently, the slides
were treated with rabbit anti-PCNA IgG (3 μg/ml), anti-CD31 IgG (3
μg/ml), and anti-VEGF IgG (3 μg/ml), followed by incubation with
goat anti-rabbit IgG at room temperature for 1 h. PCNA and CD31
immunoreactivity was visualized by using the Vectastain Elite ABC
kit and the Vectastain DAB substrate kit (Vector Laboratories,
Burlingame, CA, USA). The slides were counterstained with ChemMate
Hematoxylin (DakoCytomation), mounted and observed under a
microscope (BX-50; Olympus, Tokyo, Japan). The PCNA-positive cell
numbers in each animal were determined in 10 randomly chosen fields
at ×400 magnification by an examiner blinded to the experimental
procedures. The CD31-positive vascular areas were determined as
previously described (18).
Immunofluorescence analysis of apoptotic
cells in xenograft specimens
Frozen tumor xenograft specimens were stained using
a fluorescent terminal deoxynucleotidyl transferase-mediated nick
end labeling-based apoptosis detection kit (In Situ Cell Death Kit;
Takara), in accordance with the manufacturer’s instructions.
Fluorescence microscopy was performed using a ×40 objective (Zeiss
Plan-Neofluar) on an Olympus Eclipse TE2000-S inverted phase
microscope (Olympus, Melville, NY, USA). Images were analyzed using
Image-Pro Plus software version 4.0. The apoptosis-positive cell
numbers in each animal were determined in 10 randomly chosen fields
at ×400 magnification by an examiner blinded to the experimental
procedures.
Real-time RT-PCR
Total RNA was extracted using the TRIzol LS reagent
(Invitrogen). mRNA was reverse-transcribed using the SuperScript
First-Strand Synthesis System (Invitrogen). Real-time PCR was
performed using the Applied Biosystems 7900HT PCR system with 2×
QuantiFast SYBR-Green PCR Master Mix (Qiagen), 1 μM primers
(Table I), and <100 ng cDNA in a
25 μl reaction mixture. Relative expression of target genes was
analyzed by the ΔΔCt method. Results are expressed as means ±
SD.
 | Table IPrimer sequences for quantitative
PCR. |
Table I
Primer sequences for quantitative
PCR.
| Sense (5′-3′) | Antisense
(5′-3′) |
|---|
| MMP-2 |
TACAGGATCATTGGCTACACACC |
GGTCACATCGCTCCAGACT |
| MMP-9 |
TGTACCGCTATGGTTACACTCG |
GGCAGGGACAGTTGCTTCT |
| EGF |
TGGATGTGCTTGATAAGCGG |
ACCATGTCCTTTCCAGTGTGT |
| FGF-2 |
AGAAGAGCGACCCTCACATCA |
CGGTTAGCACACACTCCTTTG |
| VEGFA |
AGGGCAGAATCATCACGAAGT |
AGGGTCTCGATTGGATGGCA |
| PDGFA |
GCAAGACCAGGACGGTCATTT |
GGCACTTGACACTGCTCGT |
| PDGFB |
CTCGATCCGCTCCTTTGATGA |
CGTTGGTGCGGTCTATGAG |
| HGF |
GCTATCGGGGTAAAGACCTACA |
CGTAGCGTACCTCTGGATTGC |
Statistical analysis
The means ± SD were calculated for all parameters
determined. Statistical significance was evaluated by one-way
ANOVA, followed by the Tukey-Kramer test, using SPSS 10 software
(IBM, Inc., Chicago, IL, USA). P-values <0.05 were considered to
indicate a statistically significant result.
Results
Establishment of overexpression of Pim-3
or K69M-Pim-3 cells and Tet-inducible Pim-3 shRNA or scramble shRNA
expressing cells
To demonstrate the role of Pim-3 in human pancreatic
carcinogenesis, we established MiaPaca-2 cells overexpressing the
Pim-3 or K69M-Pim-3 gene using retroviral vectors, which inserted
the wild-type human Pim-3 cDNA or kinase-dead mutant (K69M) Pim-3
cDNA. Furthermore, we also established Pim-3 gene silenced cell
lines using a Tet-inducible Pim-3 shRNA in PCI55 cells, which
express high levels of Pim-3 protein compared with MiaPaca-2 cells,
as previously described (8). We
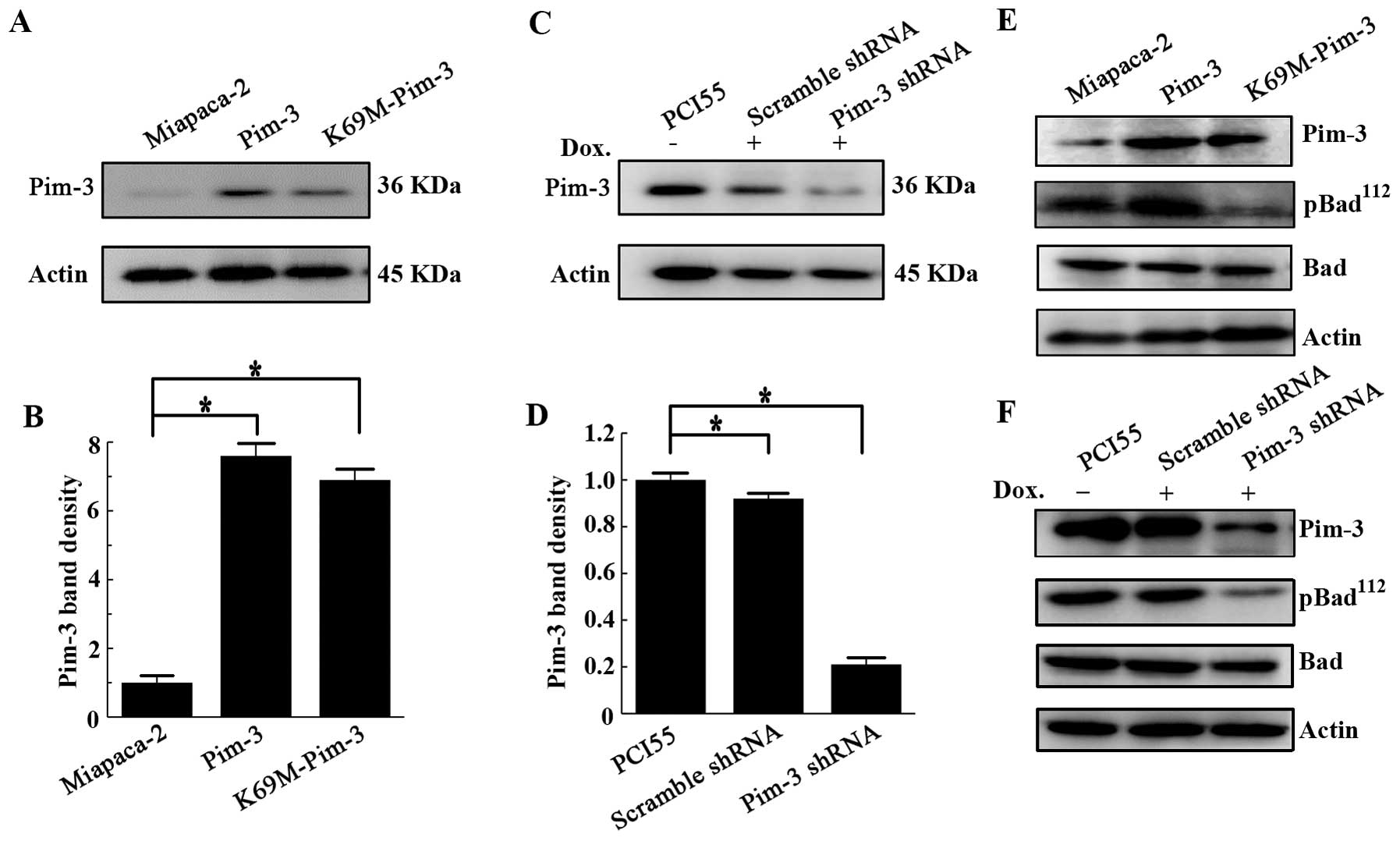
confirmed the overexpression of Pim-3 or K69M-Pim-3 protein in
MiaPaca-2 cells (Fig. 1A and B),
and that Pim-3 shRNA markedly diminished Pim-3 protein expression
compared to control scramble shRNA under inducible conditions in
PCI55 cells (Fig. 1C and D). Since
Pim-3 can phosphorylate a proapoptotic molecule Bad at
Ser112, we examined the phosphorylation states of Bad to
confirm the functionality of these stable pancreatic cancer cell
lines. Bad was constitutively phosphorylated at Ser112
in MiaPaca-2 and PCI55 cell lines. Pim-3 kinase inactivation or
silencing of Pim-3 expression diminished the amount of
phospho-Ser112-Bad, without influencing the expression
of total Bad protein (Fig. 1E and
F). These observations indicate that overexpression of Pim-3
was functional in terms of its capacity to phosphorylate its
substrate Bad, and Pim-3 inactivation by its kinase dead mutant or
knockdown of Pim-3 expression by Pim-3 shRNA functionally decreased
the amount of phospho-BadSer112.
Pim-3 kinase inactivation inhibits cell
proliferation and promotes apoptosis in pancreatic cancer cells in
vitro
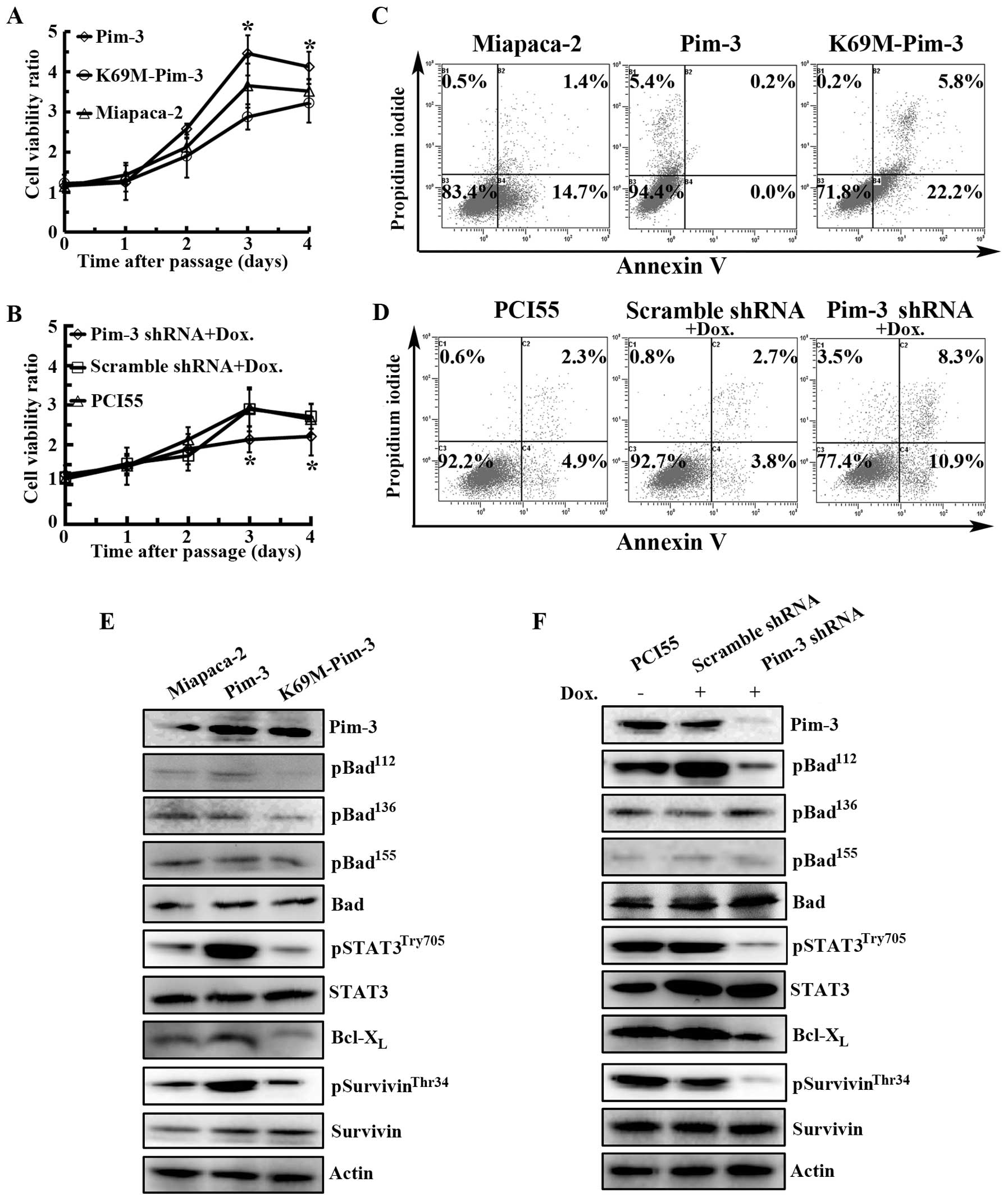
We previously observed that Pim-3 shRNA treatment
decreases the in vitro proliferation of various types of
cancer cells by enhancing their apoptosis (8,11,12).
Consistent with our previous observation, Pim-3 kinase inactivation
or silencing of Pim-3 expression decreased the proliferation of
human pancreatic cancer cell lines, MiaPaca-2 and PCI55 (Fig. 2A and B), together with an increase
in the proportion of both early apoptotic cells (Annexin V-positive
and PI-negative) and late apoptotic cells (Annexin V-positive and
PI-positive) (Fig. 2C and D), when
compared with cells overexpressing Pim-3 or cells stably expressing
the scrambled shRNA. To assess the effects of Pim-3 kinase
inactivation on the apoptotic process, we conducted western
blotting. As shown in Fig. 2E and
F, Bad was constitutively phosphorylated at Ser112
in MiaPaca-2 and PCI55 cell lines; however, Pim-3 kinase
inactivation or silencing of Pim-3 expression diminished the amount
of phospho-BadSer112, without any effects on the
expression of the total Bad protein. Concomitantly, Pim-3 kinase
inactivation or silencing of Pim-3 expression decreased
Bcl-XL expression, which confirmed our previous results
(8). Moreover, compared with Pim-3
overexpression, Pim-3 kinase inactivation decreased
phospho-survivinThr34 and
phospho-STAT3Try705, that are an upstream of
Bcl-XL, with only few effects on total survivin and
STAT3. Collectively, these results suggested that the Pim-3
overexpression might increase phosphorylation of Stat3, survivin
and Bad, resulting in reduced apoptosis, and eventually promoting
human pancreatic carcinogenesis. Inversely, Pim-3 kinase
inactivation reverses this effect.
Pim-3 kinase inactivation suppresses
pancreatic tumorigenesis in nude mice
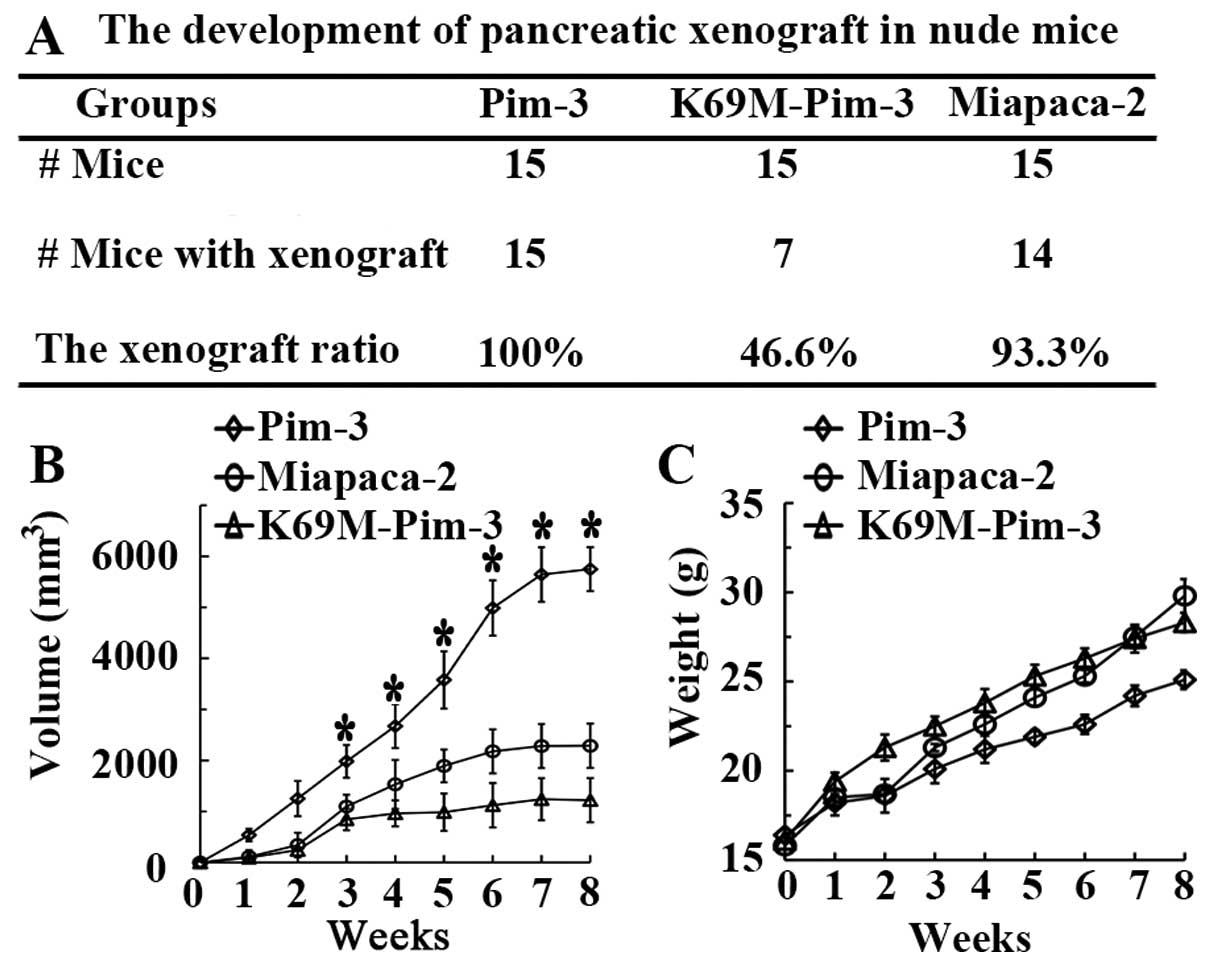
Results from preliminary experiments demonstrated
that nude mice subcutaneously inoculated with 4 million PCI55 cells
failed to develop tumors. Thus, to establish the crucial role of
Pim-3 in pancreatic carcinogenesis, we injected the same number of
stable Pim-3-MiaPaca-2, K69M-Pim-3-MiaPaca-2 or parent MiaPaca-2
cells subcutaneously into nude mice. At 30 days following tumor
inoculation, we found that 93.3% (14/15) of parental MiaPaca-2
group mice and 100% (15/15) of Pim-3-MiaPaca-2 group mice developed
subcutaneous tumors (Fig. 3A). In
contrast, only 46.6% (7/15) of K69M-Pim-3 group mice developed
tumors following subcutaneous injection of tumor cells (Fig. 3A). Moreover, mice injected with
Pim-3 overexpressing cells exhibited progressive tumor growth
compared with parental MiaPaca-2 cells, whereas the growth rate of
K69M-Pim-3 tumor cells in nude mice was significantly decreased
(Fig. 3B). Throughout the trial
period, none of the mice presented with loss in body weight
(Fig. 3C). These results indicated
that Pim-3 plays a crucial role in subcutaneous pancreatic
carcinogenesis in nude mice, and Pim-3 kinase inactivation
suppresses tumor growth in vivo.
Pim-3 kinase inactivation inhibits
proliferation and promotes apoptosis in pancreatic cancer in
vivo
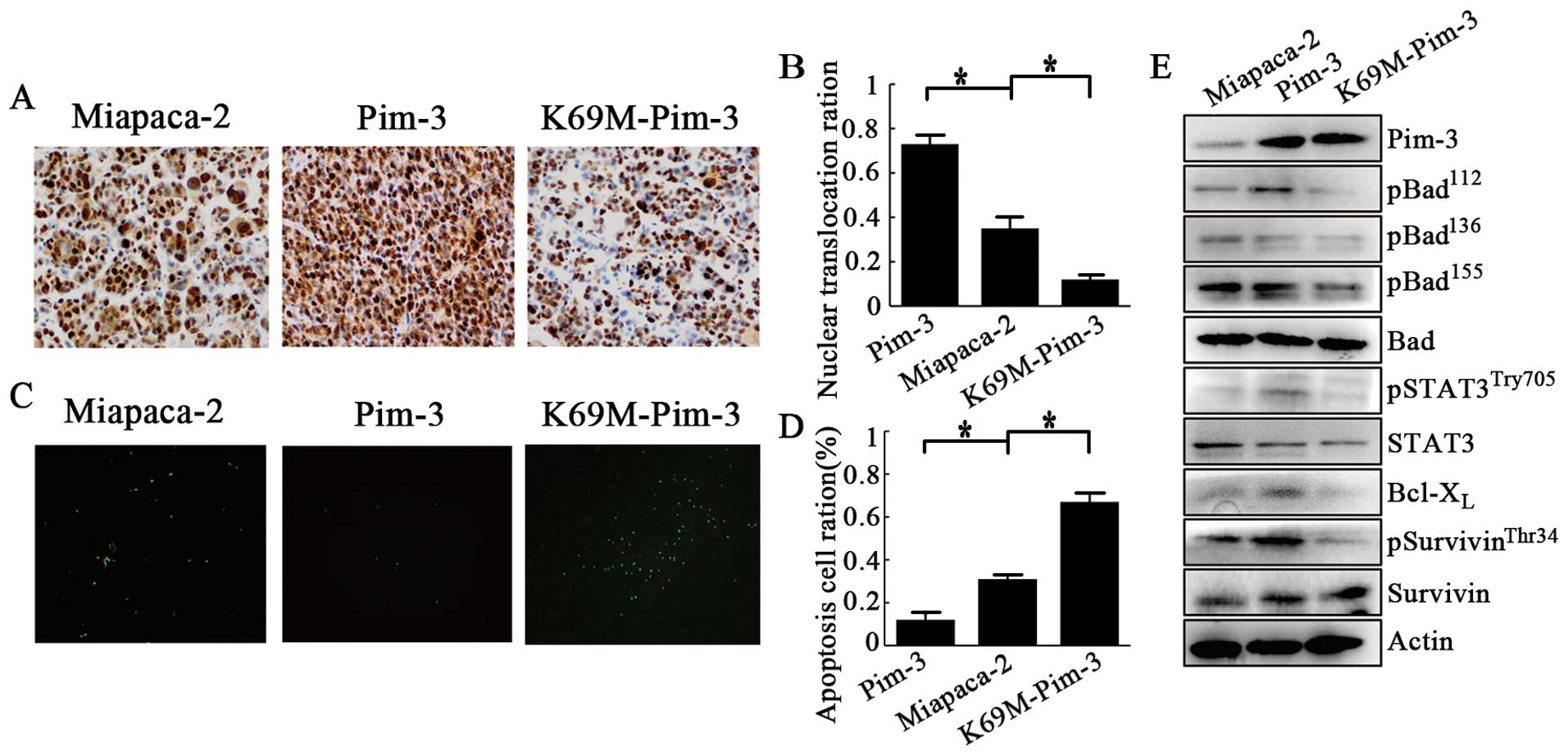
We next examined the effects of Pim-3 kinase
inactivation on pancreatic cancer cell proliferation and apoptosis
in vivo. Histological analysis revealed that Pim-3 kinase
inactivation significantly decreased PCNA-positive proliferating
cell numbers while increasing TUNEL-positive apoptotic cell numbers
(Fig. 4A–D). In contrast, Pim-3
overexpression significantly increased PCNA-positive proliferating
cell numbers and reciprocally decreased TUNEL-positive apoptotic
cell numbers, when compared with the parent MiaPaca-2 cells
(Fig. 4A–D). Moreover, in the
xenograft tumor tissues, overexpression of Pim-3 increased the
amount of phospho-Stat3Try705,
phospho-survivinThr34, and phospho-BadSer112,
whereas Pim-3 inactivation decreased the expression of these
phosphorylated proteins (Fig. 4E).
However, the expression of unphosphorylated Stat3, survivin, and
Bad proteins were unchanged (Fig.
4E). These observations were consistent with the results of our
in vitro experiment.
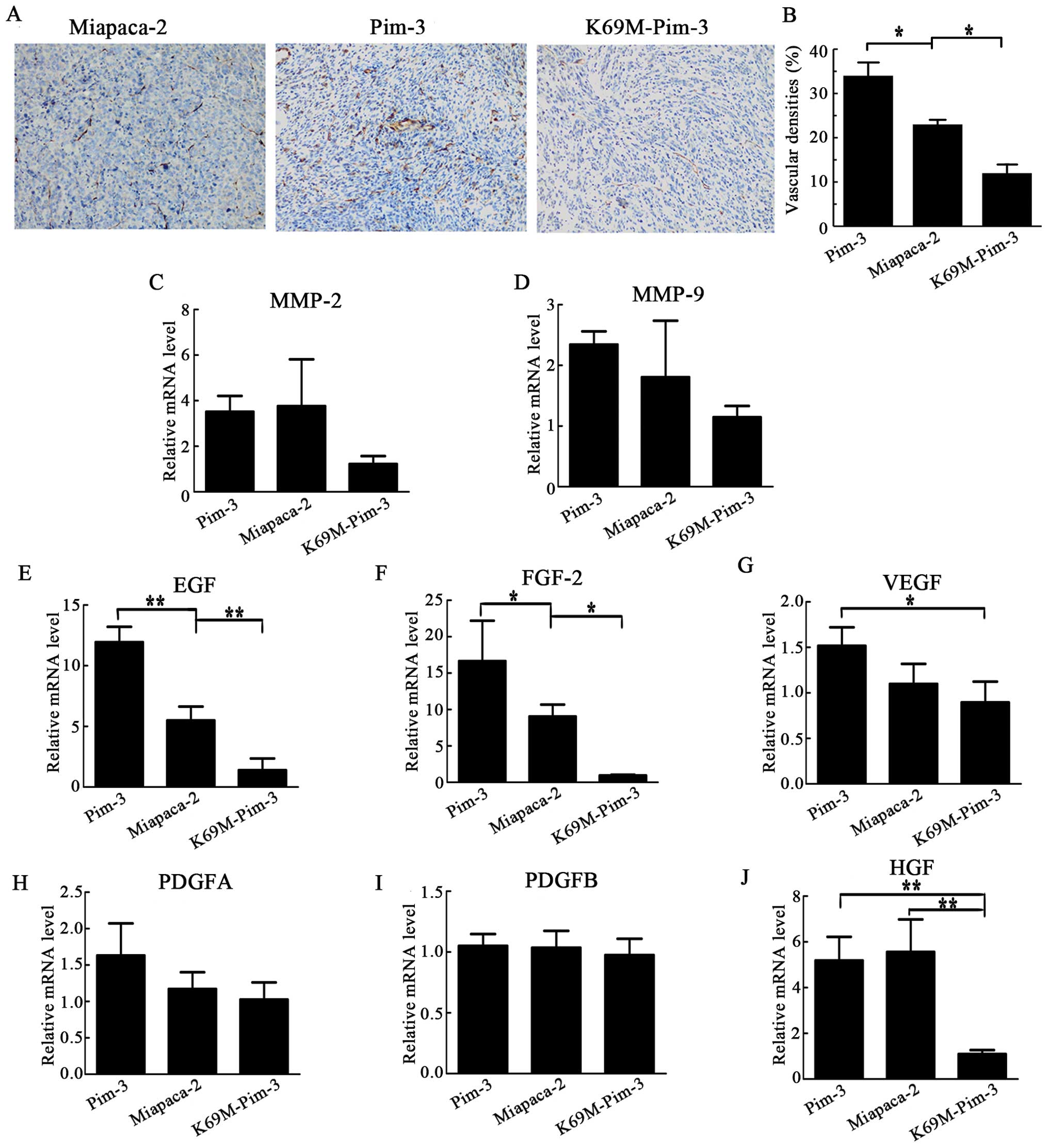
Reduced neovascularization
Neovascularization is required for tumor growth and
accumulating evidence has proved an important role for VEGF in
angiogenesis. We recently demonstrated that Pim-3 could promote
tumor growth and angiogenesis by stimulating the VEGF pathway
(19). Herein, we detected that the
intratumoral CD31-positive vascular areas increased following the
injection of Pim-3-MiaPaca-2 cells, but significantly decreased
following injection of K69M-Pim-3-MiaPaca-2 cells (Fig. 5A and B). Hence, we examined VEGF
mRNA expression together with other angiogenesis factors, including
HGF, platelet-derived growth factor (PDGF), fibroblast growth
factor (FGF), epithelial growth factor (EGF), matrix
metalloproteinases (MMP)-2 and -9. The intratumoral mRNA expression
of VEGF, EGF, FGF-2 and HGF, but not MMP-2 and MMP-9 with
gelatinase activity was significantly increased in mice injected
with Pim-3 overexpressing cells, while their expression was
markedly reduced in mice injected with Pim-3 kinase inactive cells,
to the parental MiaPaca-2 cells (Fig.
5C–J). These observations may mirror the fact that
neovascularization, an essential process for pancreatic
carcinogenesis, was augmented in Pim-3 overexpression compared with
that observed for Pim-3 kinase inactivation, as demonstrated by
increasing in CD31-positive areas in the tumor tissue.
Discussion
The diagnosis of human pancreatic cancer is often
difficult, and in most patients the tumor is already disseminated
when discovered. We previously observed that Pim-3, a
proto-oncogene with serine/threonine kinase activity, was
aberrantly expressed in cancer cells but not in the normal cells of
the pancreas. Moreover, the Ser112 phosphorylation and
inactivation of Bad by Pim-3 maintains the expression of
Bcl-XL and, thus, prevents apoptosis of human pancreatic
cancer cells (8). Silencing Pim-3
expression can retard in vitro cell proliferation of
pancreatic cancer by promoting apoptosis (8). A recent report demonstrated that Pim-3
suppression can sensitize pancreatic cancer cells to gemcitabine
(20). Thus, Pim-3 might be a novel
target for the treatment of refractory pancreatic cancer. Since the
regulatory mechanisms of Pim-3 signaling networks in vivo
are not well understood, we established stable cell lines that
overexpress wild-type or the kinase-dead form of Pim-3
(K69M-Pim-3), and utilized a nude mouse tumor xenograft model to
assess the regulatory mechanisms of Pim-3 in human pancreatic
carcinogenesis in vivo.
Gene delivery in cancer cells can be achieved using
transient transfection and stable transfection methods. Stable
transfection methods are more appealing and enable continuing
expression of the transgene. In the present study, we established
overexpression of Pim-3 or K69M-Pim-3, in which Lys-69 in the ATP
binding domain was replaced by methionine, rendering its kinase
domain non-functional in MiaPaca-2 cells. We also established
expression of the Pim-3 shRNA or scramble shRNA in a
tetracycline-inducible manner in PCI55 cells, which contain high
levels of Pim-3 protein compared with MiaPaca-2 cells, as
previously described (8). Kinase
activation generally requires a post-translational modification, in
particular, phosphorylation in its regulatory domain. However,
other members of the Pim kinase family, Pim-1 and Pim-2, are
constitutively active without any further alteration in their
conformation, as they lack any regulatory domain (21), as does Pim-3 (11). Consistently, stable expression of
Pim-3 exhibited enhanced phosphorylation of Bad at
Ser112, whereas stable expression of K69M-Pim-3
significantly attenuated phosphorylation of Bad at its
Ser112 in MiaPaca-2 cells. Moreover, Pim-3 shRNA
expression in PCI55 cells, but not in parental PCI55 cells or
scramble shRNA-PCI55 cells, markedly reduced the expression of
phospho-BadSer112. Thus, Pim-3 inactivation or knockdown
of Pim-3 expression by Pim-3 shRNA can functionally decrease the
amount of phospho-BadSer112. Moreover, Pim-3 kinase
inactivation or silencing of Pim-3 expression decreased the cell
proliferation of human pancreatic cancer cell lines, MiaPaca-2 and
PCI55, together with increasing the apoptotic cells compared with
that observed for cells overexpressing Pim-3 or cells expressing
the scrambled shRNA.
Deneen et al (10) demonstrated that Pim-3 gene
transcription was enhanced in EWS/ETS-induced malignant
transformation of NIH3T3 cells, suggesting the involvement of Pim-3
in tumorigenesis. In line with these observations, we demonstrated
that the development of hepatocellular carcinoma was accelerated in
mice expressing the Pim-3 transgene selectively in the liver, when
these mice were treated with a hepatocarcinogen (14). Moreover, forced expression of Pim-3
can promote anchorage-independent growth and co-expression of a
kinase-deficient Pim-3 mutant can attenuate EWS/FLI-mediated NIH3T3
tumorigenesis in nude mice (22).
Our data demonstrated that overexpression of Pim-3 in the MiaPaca-2
human pancreatic cancer cells developed 100% (15/15) of
subcutaneous tumors and exhibited progressive tumor growth, whereas
Pim-3 kinase inactivation decreased tumorigenicity to 46.6% (7/15)
and inhibited tumor growth. However, subcutaneous inoculation with
4×106 PCI55 cells into nude mice did not develop tumors.
These observations prompted us to investigate the mechanism of
Pim-3 kinase inactivation on decreasing pancreatic carcinogenesis,
including cell apoptosis and angiogenesis.
An elevated activity of Stat3 has been frequently
observed in a wide variety of human tumors including pancreatic
cancer (23–26). Several lines of evidence
demonstrated that the gene expression of Pim-1 and Pim-2 could be
regulated by IL-6-gp130-mediated signal transducers and activators
of transcription (STAT) family (27,28).
Moreover, Pim-3 expression is enhanced in murine embryonic stem
cells by leukemia inhibitory factor/gp130-dependent signaling and
Stat3 transcription factor (29).
However, transfection of a dominant negative form of Stat3 failed
to inhibit the promoter activity of the Pim-3 gene in human
pancreatic cancer cells (30). The
excessive activation of Stat3 can promote anti-apoptotic gene
expression, such as Bcl-XL and mc-1, as well as
promoting cell proliferation in a variety of tumor cells (31–33).
We previously showed that Pim-3 can maintain the expression of
Bcl-XL, but the mechanism was not clear. Recently, Chang
et al (34) demonstrated
that the knockdown of Pim-3, but not of Pim-1 or Pim-2, in prostate
cancer cell line DU-145 results in a significant downregulation of
pStat3Try705, indicating Pim-3 kinase is a positive
regulator of Stat3 signaling. In line with these observations, we
detected constitutive Stat3 and phosphorylated
pStat3Try705 expression in human pancreatic cancer
cells. Overexpression of Pim-3 increased the expression levels of
phospho-Stat3Try705 and Bcl-XL, whereas Pim-3
kinase inactivation or ablation of Pim-3 protein expression
significantly reduced the expression levels of
pStat3Try705 and Bcl-XL, while the expression
levels of total Stat3 remained unchanged. However, the mechanism of
apoptosis-related Bcl-XL induction by Stat3 remains to
be elucidated.
Survivin is a member of the ‘inhibitor of apoptosis’
(IAP) gene family of proteins that is barely detected in normal
tissues (35,36). However, survivin appears to be
selectively expressed in transformed cells and in most human
cancers, including pancreatic carcinomas (31,37).
It has been previously reported that inhibition of Stat3 signaling
blocked the expression of survivin protein and induced apoptosis in
breast cancer cells (38). However,
Pim-3 kinase inactivation or ablation of Pim-3 protein expression
reduced the phosphorylated levels of STAT3 at Try705, but did not
influence the expression of total survivin in human pancreatic
cancer cells. Several recent studies reported that the therapeutic
modulation of survivin is critically regulated by interaction with
prominent cell-signaling pathways, such as HIF-1α, HSP90, PI3K/AKT,
mTOR, ERK, tumor suppressor genes (p53, PTEN), oncogenes (Bcl-2,
Ras), and a wide range of growth factors (EGFR, VEGF) (39). Thus, it is likely that other
pathways in human pancreatic cancer cells regulate the expression
of survivin. The suppression of apoptotic cell death by survivin
requires phosphorylation at Thr34 (40). Survivin can be phosphorylated by
cyclin-dependent kinase-1, cyclic AMP, and protein kinase C as well
as AKT (40–42). Similarly, overexpression of Pim-3
increased the levels of phospho-survivinThr34, whereas
Pim-3 kinase inactivation or ablation of Pim-3 protein expression
decreased the levels of phospho-survivinThr34, similar
to Akt (42).
Angiogenesis is widely recognized as a hallmark of
cancer (43), and potent
neovascularization that contributes to tumor progression was
observed in a variety of aggressive malignant tumors (44). We identified a role for the
kinase-dead Pim-3 mutant in reducing the CD31-positive vascular
regions in the tumor, whereas vascularity was increased by the
overexpression of Pim-3 compared with the parental MiaPaca-2,
consistent with our previous report (19).
Folkman (45) first
developed a theory regarding tumor angiogenesis in 1971, in which
he proposed that a tumor produces its own new vasculature from
existing blood vessels. Following the introduction of angiogenesis
by Folkman in 1971, numerous studies have indicated that tumor
cells overexpress a variety of angiogenic genes and secrete various
angiogenic factors, including VEGF, PDGF, FGF, EGF and MMP, which
recruit vascular ECs into tumor tissues and induce potent
angiogenesis. We detected that Pim-3 overexpression increased VEGF
content consistent with our previous results (19). Moreover, a loss of Pim-3 kinase
activity significantly decreased EGF and FGF mRNA, but not MMP-2
and MMP-9 expression, whereas Pim-3 overexpression markedly
increased EGF and FGF expression compared with parental cells.
Consequently, the EGF- and FGF-mediated signals may account for
neovascularization and subsequently promote tumor growth.
Tumor-associated fibroblasts can produce HGF and are
presumed to be crucial in tumor progression (46). We also observed the decreasing mRNA
expression of HGF in the intratumoral tissues expressing an
inactive Pim-3 kinase. This may mirror the fact that fibroblasts
participate in pancreatic carcinogenesis.
Antiangiogenic therapy has shown promise as a
treatment for several cancers, such as colon cancer and non-small
cell lung cancer (43,44,47).
However, the antitumor effects of current angiostatic drugs are
short-lived in most patients. Moreover, the overall survival rates
for most cancer patients are not significantly prolonged (48,49).
Hence, there is a need for other tumor-selective proangiogenic
molecules that can be used in combination with or without
conventional antiangiogenic drugs. The genetic deficiency of Pim-3
gene does not result in apparent changes in phenotypes, suggesting
that Pim-3 may be physiologically dispensable. Unlike other
survival kinases, such as the Akt kinases, Pim kinases are not
localized downstream of the insulin receptor signaling pathway and,
therefore, the inhibition of Pim kinases has few effects on insulin
receptor pathway. Thus, Pim-3 would be a preferred target molecule
for the development of anticancer drugs against solid tumor
angiogenesis, in which Pim-3 is aberrantly expressed. Thus,
targeting Pim-3 may play a dual role in halting tumor progression,
by promoting tumor cell death and blocking angiogenesis.
Acknowledgements
The authors would like to express their sincere
gratitude to Professor Naofumi Mukaida (Cancer Research Institute,
Kanazawa University) for his critical comments on the manuscript.
The authors gratefully acknowledge grant support from The National
Science Foundation of China (NSFC) (30973476, 812727), the Shanghai
Pujiang Program (KW201028464), the Fudan University ‘985 Project’
Phase III Cancer Research Projects II (985III-YFX0102), and the
Shanghai Committee of Science and Technology (12DZ2260100).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Winter JM, Cameron JL, Campbell KA, et al:
1423 pancreaticoduodenectomies for pancreatic cancer: a
single-institution experience. J Gastrointest Surg. 10:1199–1211.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Z, Li Y, Ahmad A, et al: Pancreatic
cancer: understanding and overcoming chemoresistance. Nat Rev
Gastroenterol Hepatol. 8:27–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chu D, Kohlmann W and Adler DG:
Identification and screening of individuals at increased risk for
pancreatic cancer with emphasis on known environmental and genetic
factors and hereditary syndromes. JOP. 11:203–212. 2010.PubMed/NCBI
|
|
5
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
6
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
7
|
Zavoral M, Minarikova P, Zavada F, Salek C
and Minarik M: Molecular biology of pancreatic cancer. World J
Gastroenterol. 17:2897–2908. 2011. View Article : Google Scholar
|
|
8
|
Li YY, Popivanova BK, Nagai Y, Ishikura H,
Fujii C and Mukaida N: Pim-3, a proto-oncogene with
serine/threonine kinase activity, is aberrantly expressed in human
pancreatic cancer and phosphorylates bad to block bad-mediated
apoptosis in human pancreatic cancer cell lines. Cancer Res.
66:6741–6747. 2006. View Article : Google Scholar
|
|
9
|
Feldman JD, Vician L, Crispino M, et al:
KID-1, a protein kinase induced by depolarization in brain. J Biol
Chem. 273:16535–16543. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deneen B, Welford SM, Ho T, Hernandez F,
Kurland I and Denny CT: PIM3 proto-oncogene kinase is a common
transcriptional target of divergent EWS/ETS oncoproteins. Mol Cell
Biol. 23:3897–3908. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fujii C, Nakamoto Y, Lu P, et al: Aberrant
expression of serine/threonine kinase Pim-3 in hepatocellular
carcinoma development and its role in the proliferation of human
hepatoma cell lines. Int J Cancer. 114:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Popivanova BK, Li YY, Zheng H, et al:
Proto-oncogene, Pim-3 with serine/threonine kinase activity, is
aberrantly expressed in human colon cancer cells and can prevent
Bad-mediated apoptosis. Cancer Sci. 98:321–328. 2007. View Article : Google Scholar
|
|
13
|
Zheng HC, Tsuneyama K, Takahashi H, et al:
Aberrant Pim-3 expression is involved in gastric
adenoma-adenocarcinoma sequence and cancer progression. J Cancer
Res Clin Oncol. 134:481–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Y, Wang YY, Nakamoto Y, et al:
Accelerated hepatocellular carcinoma development in mice expressing
the Pim-3 transgene selectively in the liver. Oncogene.
29:2228–2237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang F, Liu B, Wang Z, et al: A novel
regulatory mechanism of Pim-3 kinase stability and its involvement
in pancreatic cancer progression. Mol Cancer Res. 11:1508–1520.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yunis AA, Arimura GK and Russin DJ: Human
pancreatic carcinoma (MIA PaCa-2) in continuous culture:
sensitivity to asparaginase. Int J Cancer. 19:128–135. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yano T, Ishikura H, Kato H, et al:
Vaccination effect of interleukin-6-producing pancreatic cancer
cells in nude mice: a model of tumor prevention and treatment in
immune-compromised patients. Jpn J Cancer Res. 92:83–87. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang YY, Taniguchi T, Baba T, Li YY,
Ishibashi H and Mukaida N: Identification of a phenanthrene
derivative as a potent anticancer drug with Pim kinase inhibitory
activity. Cancer Sci. 103:107–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang C, Li HY, Liu B, Huang S, Wu L and Li
YY: Pim-3 promotes the growth of human pancreatic cancer in the
orthotopic nude mouse model through vascular endothelium growth
factor. J Surg Res. 185:595–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu D, Cobb MG, Gavilano L, et al:
Inhibition of oncogenic Pim-3 kinase modulates transformed growth
and chemosensitizes pancreatic cancer cells to gemcitabine. Cancer
Biol Ther. 14:492–501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qian KC, Wang L, Hickey ER, et al:
Structural basis of constitutive activity and a unique nucleotide
binding mode of human Pim-1 kinase. J Biol Chem. 280:6130–6137.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang XY, Ren CP, Wang L, et al:
Identification of differentially expressed genes in metastatic and
non-metastatic nasopharyngeal carcinoma cells by suppression
subtractive hybridization. Cell Oncol. 27:215–223. 2005.
|
|
23
|
Byers LA, Sen B, Saigal B, et al:
Reciprocal regulation of c-Src and STAT3 in non-small cell lung
cancer. Clin Cancer Res. 15:6852–6861. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He M and Young CY: New approaches to
target the androgen receptor and STAT3 for prostate cancer
treatments. Mini Rev Med Chem. 9:395–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim DY, Cha ST, Ahn DH, et al: STAT3
expression in gastric cancer indicates a poor prognosis. J
Gastroenterol Hepatol. 24:646–651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scholz A, Heinze S, Detjen KM, et al:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hirano T, Ishihara K and Hibi M: Roles of
STAT3 in mediating the cell growth, differentiation and survival
signals relayed through the IL-6 family of cytokine receptors.
Oncogene. 19:2548–2556. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shirogane T, Fukada T, Muller JM, Shima
DT, Hibi M and Hirano T: Synergistic roles for Pim-1 and c-Myc in
STAT3-mediated cell cycle progression and antiapoptosis. Immunity.
11:709–719. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aksoy I, Sakabedoyan C, Bourillot PY, et
al: Self-renewal of murine embryonic stem cells is supported by the
serine/threonine kinases Pim-1 and Pim-3. Stem Cells. 25:2996–3004.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li YY, Wu Y, Tsuneyama K, Baba T and
Mukaida N: Essential contribution of Ets-1 to constitutive Pim-3
expression in human pancreatic cancer cells. Cancer Sci.
100:396–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mackenzie GG, Huang L, Alston N, et al:
Targeting mitochondrial STAT3 with the novel phospho-valproic acid
(MDC-1112) inhibits pancreatic cancer growth in mice. PLoS One.
8:e615322013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
You W, Tang Q, Zhang C, et al: IL-26
promotes the proliferation and survival of human gastric cancer
cells by regulating the balance of STAT1 and STAT3 activation. PLoS
One. 8:e635882013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thoennissen NH, Iwanski GB, Doan NB, et
al: Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–5884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang M, Kanwar N, Feng E, et al: PIM
kinase inhibitors downregulate STAT3(Try705) phosphorylation. Mol
Cancer Ther. 9:2478–2487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Talbot DC, Ranson M, Davies J, et al:
Tumor survivin is downregulated by the antisense oligonucleotide
LY2181308: a proof-of-concept, first-in-human dose study. Clin
Cancer Res. 16:6150–6158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sah NK, Khan Z, Khan GJ and Bisen PS:
Structural, functional and therapeutic biology of survivin. Cancer
Lett. 244:164–171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu BB and Wang WH: Survivin and
pancreatic cancer. World J Clin Oncol. 2:164–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diaz N, Minton S, Cox C, et al: Activation
of stat3 in primary tumors from high-risk breast cancer patients is
associated with elevated levels of activated SRC and survivin
expression. Clin Cancer Res. 12:20–28. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kanwar JR, Kamalapuram SK and Kanwar RK:
Targeting survivin in cancer: the cell-signalling perspective. Drug
Discov Today. 16:485–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
O’Connor DS, Grossman D, Plescia J, et al:
Regulation of apoptosis at cell division by p34cdc2 phosphorylation
of survivin. Proc Natl Acad Sci USA. 97:13103–13107.
2000.PubMed/NCBI
|
|
41
|
Wheatley SP and McNeish IA: Survivin: a
protein with dual roles in mitosis and apoptosis. Int Rev Cytol.
247:35–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Y, Park TS and Gidday JM: Hypoxic
preconditioning protects human brain endothelium from ischemic
apoptosis by Akt-dependent survivin activation. Am J Physiol Heart
Circ Physiol. 292:H2573–H2581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cao Z, Shang B, Zhang G, et al: Tumor
cell-mediated neovascularization and lymphangiogenesis contrive
tumor progression and cancer metastasis. Biochim Biophys Acta.
1836:273–286. 2013.PubMed/NCBI
|
|
45
|
Folkman J: Tumor angiogenesis: therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar
|
|
47
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Johannessen TC, Wagner M, Straume O,
Bjerkvig R and Eikesdal HP: Tumor vasculature: the Achilles’ heel
of cancer? Expert Opin Ther Targets. 17:7–20. 2013.
|
|
49
|
De Bock K, Mazzone M and Carmeliet P:
Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or
not? Nat Rev Clin Oncol. 8:393–404. 2011.
|