Introduction
Angiogenesis is a physiological process through
which new blood vessels are formed from the existing ones,
requiring coordinated proliferation, migration, invasion, adhesion,
and tube formation of endothelial cells (1). However, angiogenesis also plays a
vital role in tumor growth and metastasis since tumor expansion
depends on blood supply for the delivery of essential nutrients and
oxygen (2,3). Although several angiogenesis
inhibitors are in clinical use, they often have adverse effects
such as dysfunction of endothelial cells, cardiovascular toxicity,
and treatment resistance (4–6).
Therefore, new antiangiogenic agents are still required to
effectively and safely treat angiogenesis-related diseases,
including cancer.
Vascular endothelial growth factor (VEGF) is one of
the most important regulators of angiogenesis, and anti-VEGF
therapies have been approved by the US Food and Drug Administration
(7–11). VEGF receptor 2 (VEGFR2), the primary
receptor of VEGF, acts as the major mediator of VEGF-induced
angiogenesis signaling pathways such as AKT, mitogen-activated
protein kinase (MAPK), and nuclear factor (NF)-κB. Consequently,
the activation of these pathways leads to endothelial cell
proliferation, migration, and eventually capillary tube formation
(12). VEGFR2-dependent activation
of AKT signaling regulates the survival, proliferation,
permeability, and anti-apoptotic functions of endothelial cells
(13–15). In addition, the members of MAPK
cascades, extracellular signal-regulated kinase (ERK1/2), c-Jun
NH2-terminal kinase (JNK), and stress-activated protein kinase-2
(p38) regulate endothelial cell proliferation, migration, and
differentiation when stimulated by VEGF (16–18).
In addition, VEGFR2 signaling induces NF-κB activation in
endothelial cells, and the NF-κB pathway sequentially upregulates
proangiogenic and proinflammatory gene expression such as VEGF and
tumor necrosis factor (TNF)-α (19). The binding of VEGF to VEGFR2 also
activates the secretion of matrix metalloproteinase (MMP)-2 and
MMP-9 for extracellular matrix (ECM) degradation in endothelial
cells (20).
Moreover, rapid tumor expansion induces a hypoxic
state inside the tumor, and in turn stimulates hypoxia-inducible
factor (HIF)-1α, which is considered a master regulator of
angiogenesis in hypoxia (21).
HIF-1 is a heterodimeric transcription factor composed of two
subunits, HIF-1α and HIF-1β. HIF-1α is the key regulatory component
of hypoxic responses. The HIF-1 complex recognizes a consensus
hypoxia response element (HRE) in the promoter region of a broad
range of target genes that mediate hypoxic response, including
angiogenesis. However, the aberrant activation of HIF-1α causes the
overexpression of VEGF and tumor angiogenesis (22,23).
Therefore, dual inhibition of VEGFR2 and HIF-1α activities could be
a major strategy for the treatment of hypervascular tumors.
Microorganism-derived products, including proteins,
enzymes, immunotoxins, and secondary metabolites, have been studied
for cancer and other angiogenesis-related diseases (24–26).
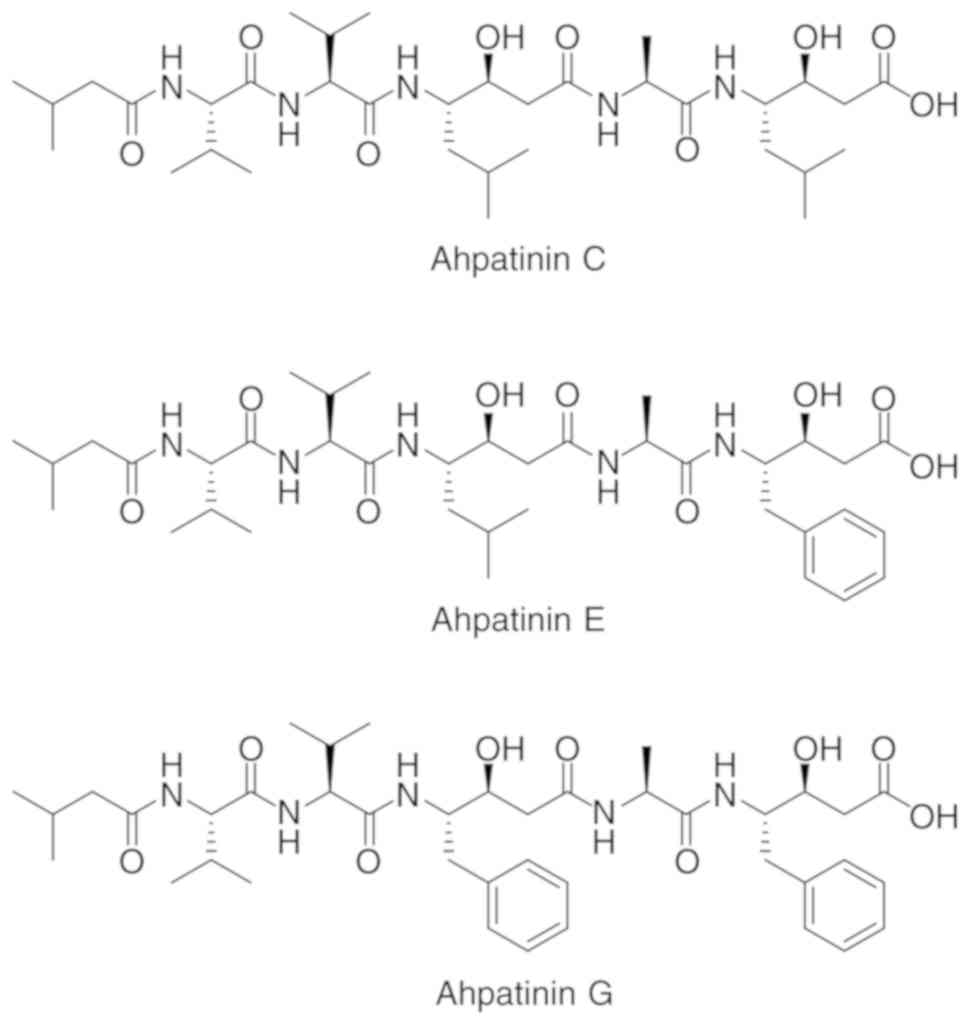
Recently, ahpatinins C, E, and G were isolated from a soil-derived
Streptomyces sp. 15JA150 (Fig.
1), which are known to have pepsin and renin inhibitory
activities (27,28). Ahpatinin C, also called pepstatin A,
is a potent inhibitor of aspartic proteinases, such as pepsin and
cathepsins D and E (29). Previous
studies have reported that several natural protease inhibitors have
antiangiogenic activities. PIVL, a serine protease inhibitor
isolated from Macrovipera lebetina venom, exhibited a strong
antiangiogenic effect both in vitro and in vivo by
blocking the adhesive function of integrins and increasing
microtubule dynamic instability in human microvascular endothelial
cells (30). Amblyomin-X, a serine
protease inhibitor from Amblyomma cajennense tick, impaired
VEGF-A-induced angiogenesis by interfering with
platelet-endothelial cell adhesion molecule-1 (PECAM-1) expression
(31). Furthermore, a cystatin F
homolog obtained from the buccal glands of Lampetra morii,
which can suppress the activity of C1 cysteine proteases, was
revealed to inhibit the key steps of angiogenesis, such as
proliferation, adhesion, migration, invasion, and tube formation of
human umbilical vein endothelial cells (HUVECs) (32). However, the antiangiogenic
activities and underlying molecular mechanisms of ahpatinins C, E,
and G have not been fully elucidated. In the present study, the
antiangiogenic effects and the mechanisms of action of ahpatinins
C, E, and G were investigated. The present study demonstrated that
ahpatinins C, E, and G potently inhibited VEGF-induced angiogenesis
by suppressing both VEGFR2 signal transduction and HIF-1α
expression, indicating that these new natural products could be
used in the treatment of angiogenesis-related diseases, including
cancer.
Materials and methods
Materials
Ahpatinins C, E, and G were prepared at a
concentration of 50 mM using dimethyl sulfoxide (DMSO). In all
experiments, the negative control groups were treated with equal
volumes of DMSO. Fetal bovine serum (FBS) and minimum essential
medium (MEM) were purchased from Invitrogen (Thermo Fisher
Scientific, Inc.), and endothelial growth medium-2 (EGM-2) and
antibiotics were obtained from Lonza Group, Ltd. Recombinant human
VEGF, Matrigel, gelatin, and Transwell chamber systems were
obtained from Koma Biotech, BD Biosciences, Sigma-Aldrich (Merck
KGaA), and Corning Costar, Inc., respectively. Antibodies against
VEGFR2 (230 kDa; cat. no. 2479), phospho-VEGFR2 (Tyr1175, 230 kDa;
cat. no. 2478), AKT (60 kDa; cat. no. 9272), phospho-AKT (Ser473,
60 kDa; cat. no. 9271), ERK1/2 (42, 44 kDa; cat. no. 9102),
phospho-ERK1/2 (Thr202/Tyr204, 42, 44 kDa; cat. no. 9101), JNK (46
kDa; cat. no. 9252), phospho-JNK (Thr183/Tyr185, 46 kDa; cat. no.
9251), p38 (43 kDa; cat. no. 9212), phospho-p38 (Thr180/Tyr182, 43
kDa; cat. no. 9211), NF-κB (65 kDa; cat. no. 3034), phospho-NF-κB
(Ser536, 65 kDa; cat. no. 3033), MMP-2 (64, 72 kDa; cat. no. 4022),
MMP-9 (84, 92 kDa; cat. no. 3852), HIF-1α (120 kDa; cat. no.
79233), β-actin (42 kDa; cat. no. 4967), rabbit IgG (cat. no.
7074), and mouse IgG (cat. no. 7076) were purchased from Cell
Signaling Technology, Inc.
Fermentation, extraction, and
purification of ahpatinins C, E, and G
Streptomyces sp. 15JA150 was cultured in a
250-ml Erlenmeyer flask containing 50 ml of seed culture medium for
3 days at 28°C on a rotary shaker with agitation at 125 rpm. For a
large culture, 1% of the preculture broth was inoculated into
40×1,000 ml baffled Erlenmeyer flasks containing 250 ml of modified
YMG broth (glucose 1%, soluble starch 2%, malt extract 0.5%, yeast
extract 0.5%, and CaCO3 0.05%) and cultured for 7 days
at 28°C on a rotary shaker with agitation at 125 rpm. The residue
was partitioned with EtOAc three times and evaporated to remove the
EtOAc. The crude extract was fractionated by reversed phase
C18 vacuum column chromatography with a stepwise solvent
system of MeOH:H2O (20:80-100:0, each × 1 l). The 70%
fraction was further separated by RP-HPLC with
CH3CN:H2O (70:30) to yield ahpatinins C, E,
and G.
Cell culture and hypoxic
conditions
HUVECs (ATCC® CRL- 1730™) and human
glioblastoma U87MG cells (Korean Cell Line Bank No. 30014;
glioblastoma of unknown origin) were grown in EGM-2 and MEM
supplemented with 10% FBS, respectively. The identity of the U87MG
cell line was confirmed by STR profiling (D3S1358: 16,17; vWA:
15,17; FGA: 18,24; Amelogenin: X,Y; TH01: 9.3; TPOX: 8; CSF1PO:
10,11; D5S818: 11,12; D13S317: 8,11; D7S820: 8,9). All cells were
maintained at 37°C in a humidified 5% CO2 incubator. For
hypoxic conditions, the cells were incubated in a hypoxic chamber
(Forma Scientific) under 5% CO2 and 1% O2
balanced with N2.
Cell viability assay
HUVECs (1×105 cells/well) were seeded in
a 12-well culture plate. Ahpatinins C, E, and G (1–25 µM) were
added to each well, and the cells were incubated for 72 h. After 72
h, the cells were stained with trypan blue and counted by a
hemocytometer using an optical microscope (Olympus Corporation) at
an ×200 magnification.
Cell proliferation assay
HUVECs were seeded at a density of 3×103
cells/well in a 96-well culture plate and then treated with various
concentrations (1–25 µM) of ahpatinins C, E, and G in the presence
of VEGF (30 ng/ml) for 72 h. The cell proliferation was measured
using a 3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium
bromide (MTT) colorimetric assay.
Chemoinvasion assay
The invasiveness of HUVECs was examined using a
Transwell chamber system with polycarbonate filter inserts with a
pore size of 8.0 µm. The lower surface of the filter was coated
with 10 µl of gelatin (1 mg/ml) for 1 h and the upper surface was
coated with 10 µl of Matrigel (3 mg/ml) for 2 h. The serum-starved
HUVECs (6×104 cells) were seeded in the upper chamber of
the filter, and ahpatinins C, E, and G (10 and 20 µM) were added to
the lower chamber in the presence of VEGF (30 ng/ml). The chamber
was incubated at 37°C for 6 h, and then the cells were fixed with
70% methanol at room temperature for 5 min and stained with
hematoxylin and eosin (H&E) at room temperature for 5 min,
respectively. The total number of cells that invaded the lower
chamber of the filter was observed and counted using an optical
microscope at an ×400 magnification.
Adhesion assay
The cell-matrix adhesion assay was performed in a
24-well culture plate coated with gelatin overnight at 4°C. The
HUVECs (5×104 cells/well) were cultured in each well in
EGM-2 containing 1% FBS and treated with ahpatinins C, E, and G (10
and 20 µM) in the presence of VEGF (30 ng/ml). After 3 h, the
unbound cells were carefully removed, and the attached cells were
observed and counted under an optical microscope at an ×200
magnification.
Capillary tube formation assay
The serum-starved HUVECs (8×104 cells)
were placed on a surface containing Matrigel (10 mg/ml) from an
angiogenesis kit (Ibidi GmbH) and incubated with ahpatinins C, E,
and G (10 and 20 µM) for 8 h in the presence of VEGF (30 ng/ml).
The morphological changes of the cells and tube formation were
visualized under an optical microscope and photographed at an ×200
magnification.
Chorioallantoic membrane (CAM)
assay
The fertilized chick eggs were incubated in a
humidified incubator at 37°C and 50% humidity for 3 days.
Approximately 6–8 ml of egg albumin was removed with a hypodermic
needle, enabling the CAM and yolk sac to drop away from the shell
membrane. After 2 days, the shell was punched out and peeled away.
Thermanox coverslips (Nalge Nunc International) with or without
ahpatinin E (10 µg/egg) were air-dried and applied to the CAM
surface. Two days later, 2 ml of 10% fat emulsion (Sigma-Aldrich;
Merck KGaA) was injected into the chorioallantois and the vascular
images were observed by an optical microscope at an ×200
magnification.
Tumor cell-induced chemoinvasion
assay
A tumor cell-induced chemoinvasion assay was
performed using an in vitro co-culture system based on the
chemoinvasion assay. The U87MG cells were plated on the lower
chamber and treated with ahpatinins C, E, and G (10 and 20 µM) for
24 h. The medium in each lower chamber was then replaced with fresh
medium without ahpatinins C, E, and G, and serum-starved HUVECs
(1×105 cells) were placed in the upper chamber. The
chamber was incubated at 37°C for 20 h, and the HUVECs that invaded
the lower chamber of the filter were analyzed using the same
procedure as described in the chemoinvasion assay.
Tumor cell-induced capillary tube
formation assay
To assess the effects of ahpatinins C, E, and G on
tumor cell-induced capillary tube formation, a conditioned medium
was obtained from the U87MG cells and used as the angiogenic
stimulus for the tube formation of HUVECs. Briefly, the U87MG cells
were treated with ahpatinins C, E, and G (10 and 20 µM) for 24 h,
and then the medium was replaced with fresh medium without
ahpatinins C, E, and G. The conditioned medium was used in the
in vitro tube formation assay.
Western blot analysis
Cells were lysed using RIPA buffer (Sigma-Aldrich;
Merck KGaA) supplemented with a protease inhibitor cocktail (Roche
Diagnostics) on ice. Protein concentrations of the extracts were
determined using a BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of cell lysate (40 µg/lane) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and the separated proteins were
transferred to polyvinylidene difluoride (PVDF) membranes (EMD
Millipore) using standard electroblotting procedures. The blots
were blocked in Tris-buffered saline with Tween-20 (TBST)
containing 5% skim milk at room temperature for 1 h and
immunolabeled with primary antibodies against phospho-VEGFR2
(dilution 1:2,000), VEGFR2 (dilution 1:2,000), phospho-AKT
(dilution 1:2,000), AKT (dilution 1:2,000), phospho-ERK1/2
(dilution 1:2,000), ERK1/2 (dilution 1:2,000), phospho-JNK
(dilution 1:2,000), JNK (dilution 1:2,000), phospho-p38 (dilution
1:2,000), p38 (dilution 1:2,000), phospho-NF-κB (dilution 1:2,000),
NF-κB (dilution 1:2,000), MMP-2 (dilution 1:2,000), MMP-9 (dilution
1:2,000), HIF-1α (dilution 1:2,000), and β-actin (dilution 1:2,000)
overnight at 4°C. After washing with TBST three times, the
membranes were incubated with horseradish peroxidase-conjugated
anti-rabbit (dilution 1:3,000) or anti-mouse (dilution 1:3,000)
secondary antibody for 1 h at room temperature. Immunolabeling was
detected with an enhanced chemiluminescence (ECL) kit (Bio-Rad
Laboratories, Inc.) according to the manufacturer's instructions.
The band density was analyzed using ImageJ software (version 1.5;
NIH).
Measurement of VEGF by enzyme-linked
immunosorbent assay (ELISA)
VEGF concentration in the media containing cells
treated with ahpatinins C, E, and G (5, 10 µM) was detected using a
VEGF immunoassay kit (R&D Systems, Inc.) according to the
manufacturer's instructions. The results were expressed as the
concentration of VEGF relative to the total amount of protein
obtained from each well.
Statistical analysis
The data were presented as the mean ± standard error
(SE) of three independent experiments. Differences among groups
were analyzed using the analysis of variance (ANOVA) with SPSS
statistics package (SPSS 9.0; SPSS Inc.). Post hoc analysis was
carried out by Tukey's test. A P-value of <0.05 was considered
to indicate a statistically significant difference.
Results
Effects of ahpatinins C, E, and G on
the growth of HUVECs
Prior to assessing the antiangiogenic activities of
ahpatinins C, E, and G, the cytotoxic effects of ahpatinins C, E,
and G on HUVECs were first investigated using the trypan blue
exclusion method. As revealed in Fig.
2A, treatment with 1–25 µM of ahpatinins C, E, and G did not
affect the viability of HUVECs. Thus, the effects of ahpatinins C,
E, and G on in vitro angiogenesis at sub-toxic doses were
evaluated. To assess the endothelial cell growth inhibitory
activities of ahpatinins C, E, and G, the proliferation assay was
performed using the MTT colorimetric method. Treatment with VEGF
(30 ng/ml) increased the proliferation of HUVECs up to 121.3%,
whereas ahpatinins C, E, and G inhibited the VEGF-induced
proliferation of HUVECs with different sensitivity to growth
inhibition (Fig. 2B). Ahpatinin C
inhibited cell growth at 5–25 µM, and ahpatinin G exhibited a
relatively weak proliferation inhibitory activity. Among them,
ahpatinin E most effectively inhibited the growth of HUVECs at all
the treated concentrations (1–25 µM). These results indicated that
ahpatinins C, E, and G suppressed the growth of HUVECs induced by
VEGF without cytotoxicity.
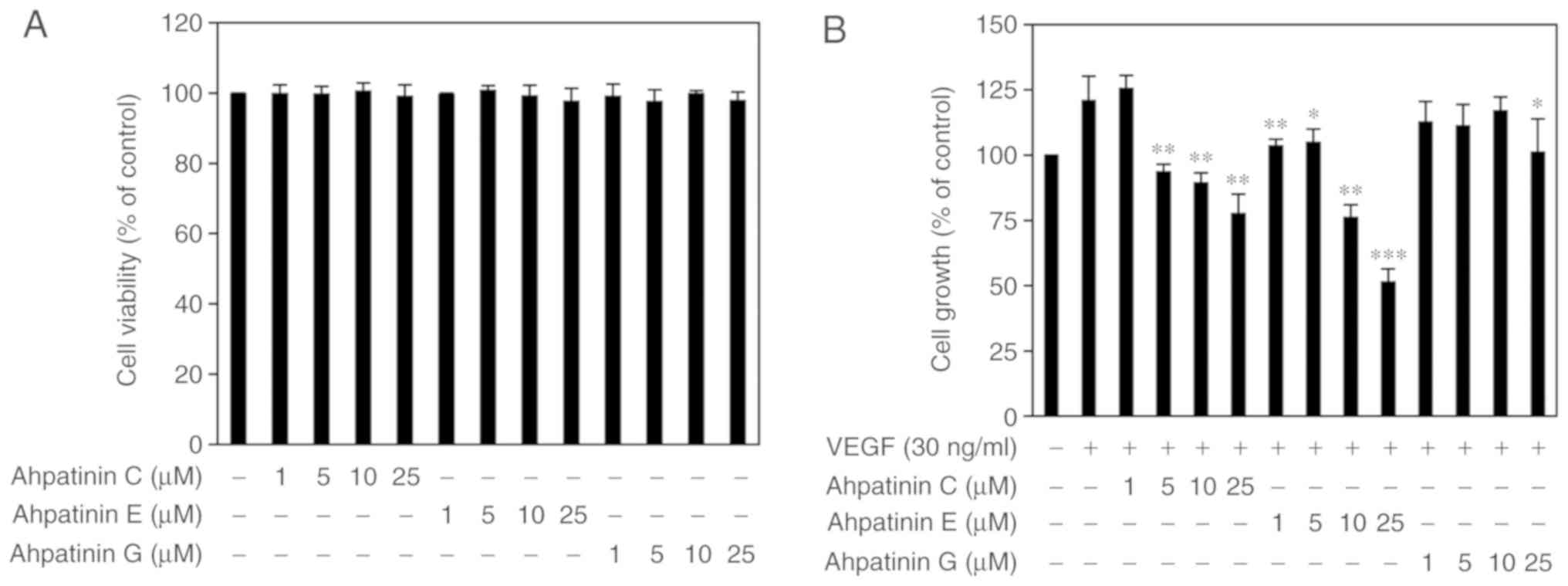 | Figure 2.Antiproliferative activities of
ahpatinins C, E, and G in HUVECs. (A) The effects of ahpatinins C,
E, and G on the cytotoxicity of HUVECs. Cells were treated with the
various concentrations (1–25 µM) of ahpatinins C, E, and G and
incubated for 72 h. Cell viability was measured by the trypan blue
assay. (B) The effects of ahpatinins C, E, and G on the growth of
HUVECs. Cells were treated with ahpatinins C, E, and G (1–25 µM) in
the presence of VEGF (30 ng/ml) and incubated for 72 h. *P<0.05,
**P<0.01 and ***P<0.001 vs. the VEGF control. Each value
represents the mean ± SE from three independent experiments.
HUVECs, human umbilical vein endothelial cells; VEGF, vascular
endothelial growth factor. |
Effects of ahpatinins C, E, and G on
in vitro angiogenesis
The effects of ahpatinins C, E, and G on the key
angiogenic phenotypes such as endothelial cell invasion, adhesion,
and tube formation, were next determined (20). Serum-starved HUVECs were stimulated
by VEGF with or without ahpatinins C, E, and G (10 and 20 µM), and
their inhibitory activities on VEGF-induced angiogenesis were
observed through chemoinvasion, adhesion, and tube formation
assays. As revealed in Fig. 3A-C,
ahpatinins C, E, and G dose-dependently decreased the invasiveness,
adhesion, and tube forming ability of HUVECs stimulated by VEGF.
Among them, ahpatinin E exhibited the greatest antiangiogenic
activity. These results demonstrated that ahpatinins C, E, and G
significantly inhibited angiogenesis without exhibiting
cytotoxicity on endothelial cells in vitro.
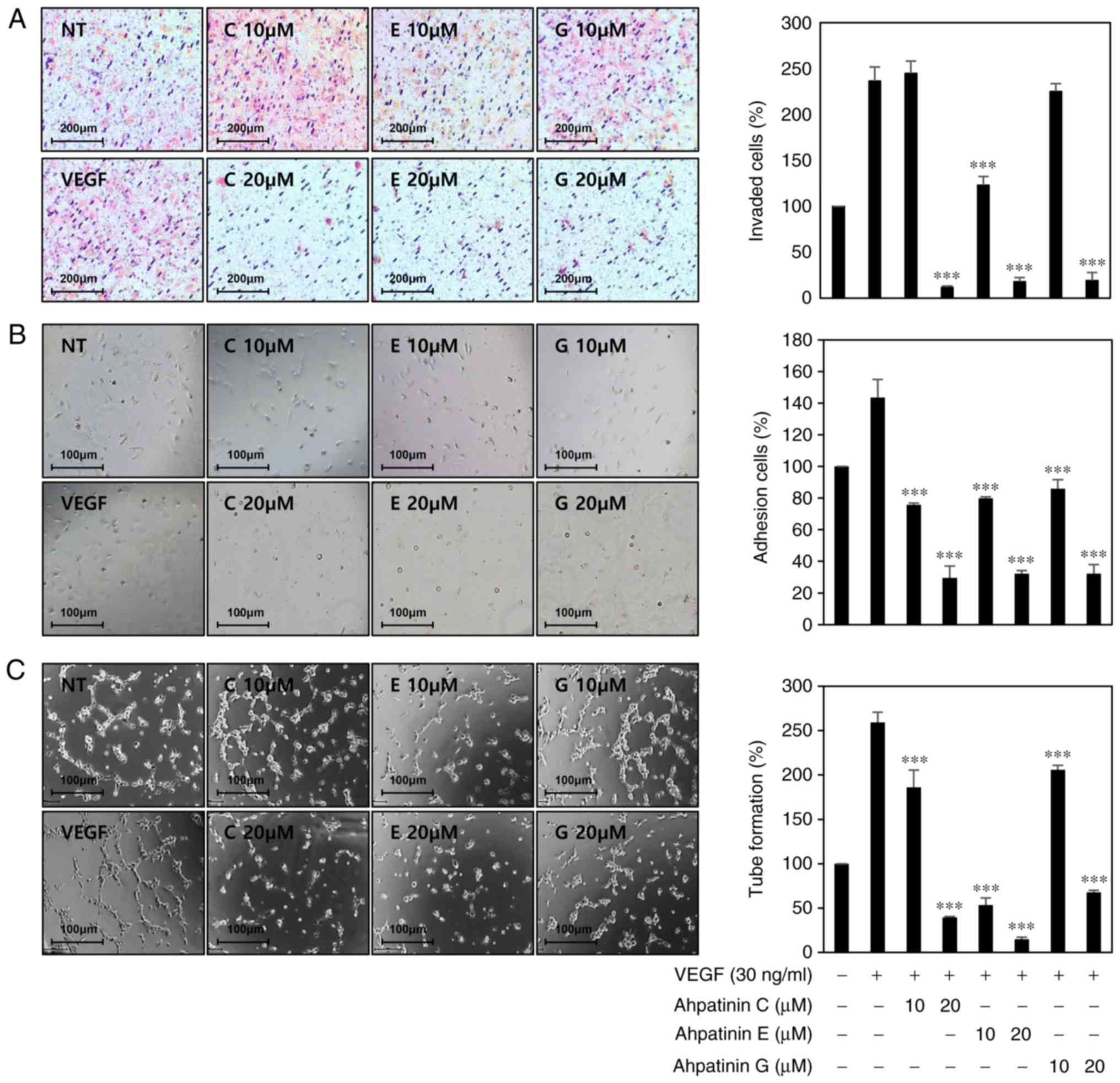 | Figure 3.In vitro antiangiogenic
activities of ahpatinins C, E, and G in HUVECs. (A-C) The
inhibitory effects of ahpatinins C, E, and G on the (A) invasion,
(B) adhesion, and (C) tube formation of HUVECs induced by VEGF.
Serum-starved HUVECs were stimulated with VEGF (30 ng/ml) in the
presence or absence of ahpatinins C, E, and G (10 and 20 µM). The
basal levels of invasion, adhesion, and tube formation of HUVECs
that were incubated in serum-free medium without VEGF were
normalized to 100%. ***P<0.001 vs. the VEGF control. Each value
represents the mean ± SE from three independent experiments.
HUVECs, human umbilical vein endothelial cells; VEGF, vascular
endothelial growth factor. |
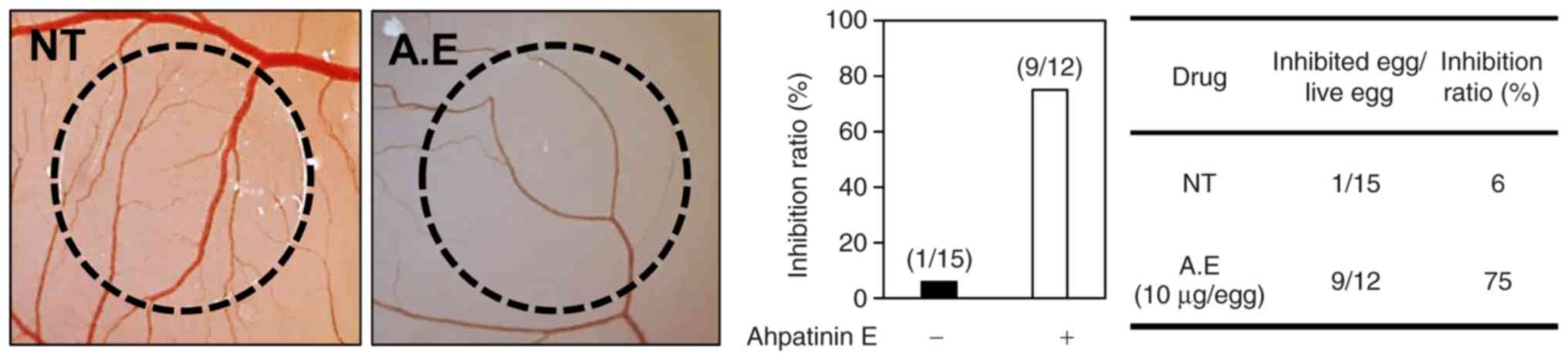
Effects of ahpatinin E on in vivo
angiogenesis
To further assess the maximum inhibitory effects of
ahpatinin E on in vitro and in vivo angiogenesis, a
CAM assay was employed. Coverslips containing vehicle alone or
ahpatinin E were located on the CAM surface, and the
neovascularized zones were observed under a microscope. The
ahpatinin E-treated CAMs had avascular zones as compared to the
vehicle-treated ones (Fig. 4). The
antiangiogenic response was calculated as the percentage of
inhibited eggs relative to the total number of live eggs tested.
The inhibition of neovascularization on the control coverslips was
6% (n=15), whereas ahpatinin E potently suppressed the angiogenesis
of the CAMs (75% at 10 µg/egg, n=12), without any rupture of the
preexisting vascular tubes. In conclusion, these results
demonstrated that ahpatinin E significantly inhibited angiogenesis
both in vitro and in vivo without exhibiting
cytotoxicity against endothelial cells.
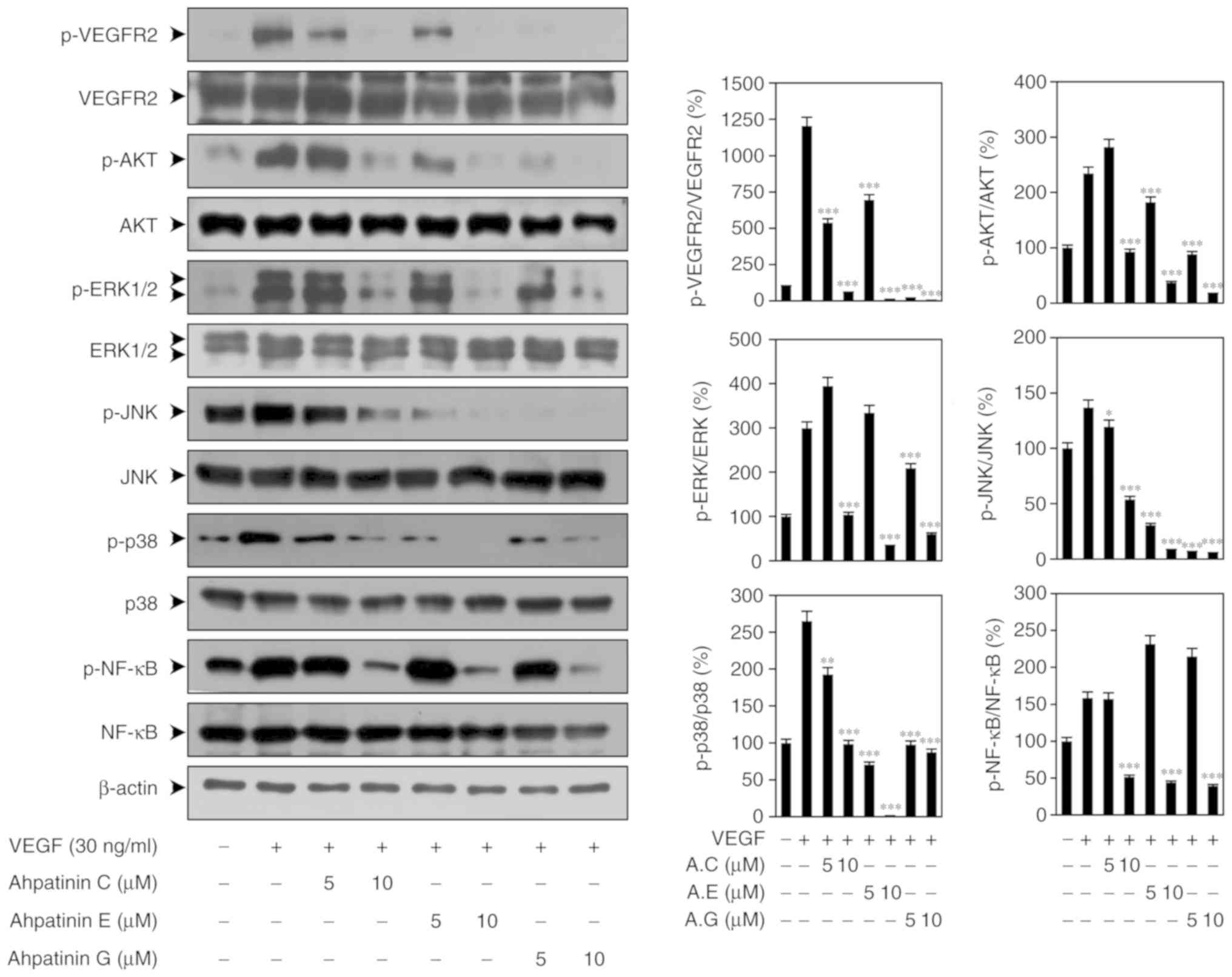
Effects of ahpatinins C, E, and G on
VEGFR2-mediated signal transduction
VEGF binding to VEGFR2 at the surface of endothelial
cells leads to dimerization and autophosphorylation of specific
tyrosine residues in the cytoplasmic domain of VEGFR2 resulting in
the subsequent activation of multiple downstream signaling
mediators, such as AKT, MAPKs, and NF-κB that promote angiogenesis
(13–20). Thus, it was investigated whether the
antiangiogenic activities of ahpatinins C, E, and G are associated
with the suppression of VEGFR2-mediated signaling pathways. As
revealed in Fig. 5, ahpatinins C,
E, and G effectively suppressed the phosphorylation of VEGFR2, AKT,
ERK1/2, JNK, p38, and NF-κB induced by VEGF, without affecting the
total protein levels. Collectively, these data indicated that
ahpatinins C, E, and G may exert antiangiogenic activities by
inhibiting VEGFR2-mediated downstream signaling cascades in
HUVECs.
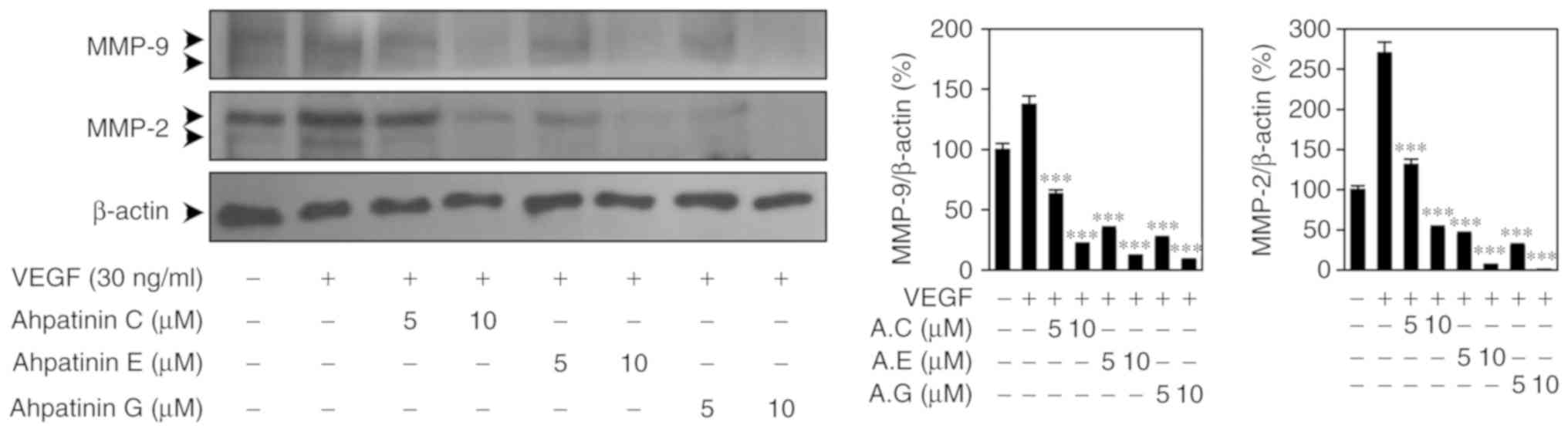
Effects of ahpatinins C, E, and G on
MMP-2/-9 expression
MMPs degrade various ECM proteins present in the
blood vessel basement membrane during angiogenesis (33). Therefore, the effects of ahpatinins
C, E, and G on the expression of MMP-2/-9 stimulated by VEGF were
examined. As revealed in Fig. 6,
ahpatinins C, E, and G significantly decreased the expression
levels of MMP-2 and MMP-9 in HUVECs, indicating that the
antiangiogenic effects of ahpatinins C, E, and G may be associated
with the downregulation of MMP-2/-9 expression.
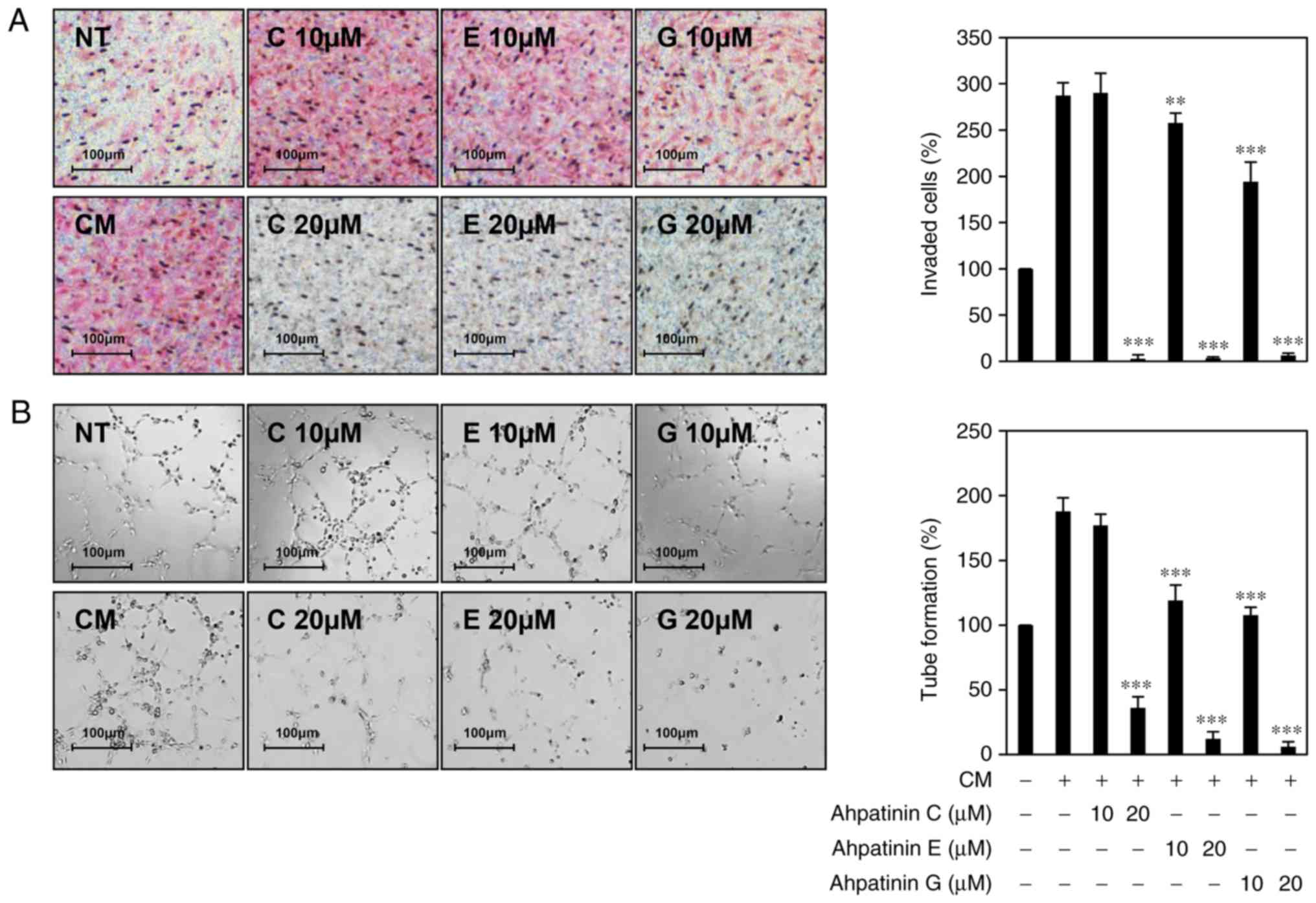
Effects of ahpatinins C, E, and G on
tumor cell-induced angiogenesis
Since tumor angiogenesis is a critical hallmark of
tumor growth and metastasis, blocking tumor-induced angiogenesis is
a promising strategy in limiting cancer progression (34,35).
To evaluate whether ahpatinins C, E, and G inhibit tumor
cell-induced angiogenesis, the effects of ahpatinins C, E, and G on
the invasion and tube formation of HUVECs stimulated by U87MG
glioblastoma cells were examined. As revealed in Fig. 7A, the HUVECs co-cultured with U87MG
cells exhibited significant invasion compared to HUVECs alone. In
contrast, when U87MG cells were treated with 20 µM of ahpatinins C,
E, and G, the induced invasion of HUVECs was completely inhibited.
In addition, the tube formation of HUVECs was increased by the
conditioned medium obtained from U87MG cells compared to the
control (medium only), whereas conditioned medium obtained from the
U87MG cells treated with ahpatinins C, E, and G, effectively
prevented the induced tube formation of HUVECs (Fig. 7B). These data revealed that
ahpatinins C, E, and G could inhibit tumor cell-induced
angiogenesis.
Effects of ahpatinins C, E, and G on
hypoxia-induced HIF-1a protein expression
Activation of HIF-1α in the hypoxic tumor cells
results in an increased expression of proangiogenic growth factors
such as VEGF (36,37). To confirm the role of HIF-1α in
mediating the antiangiogenic effects of ahpatinins C, E, and G, the
inhibitory activities of ahpatinins C, E, and G on the HIF-1α
expression of U87MG glioblastoma cells were determined. As revealed
in Fig. 8A, treatment with 10 and
20 µM of ahpatinins C, E, and G decreased the hypoxia-induced
accumulation of HIF-1α protein in U87MG cells. Furthermore, the
effects of ahpatinins C, E, and G on the expression of VEGF induced
by hypoxia were investigated. Ahpatinins C, E, and G (10 µM) also
reduced VEGF production by U87MG cells under hypoxic conditions
(Fig. 8B). These results indicated
that ahpatinins C, E, and G could suppress tumor angiogenesis
through the downregulation of HIF-1α and its downstream target
gene, VEGF.
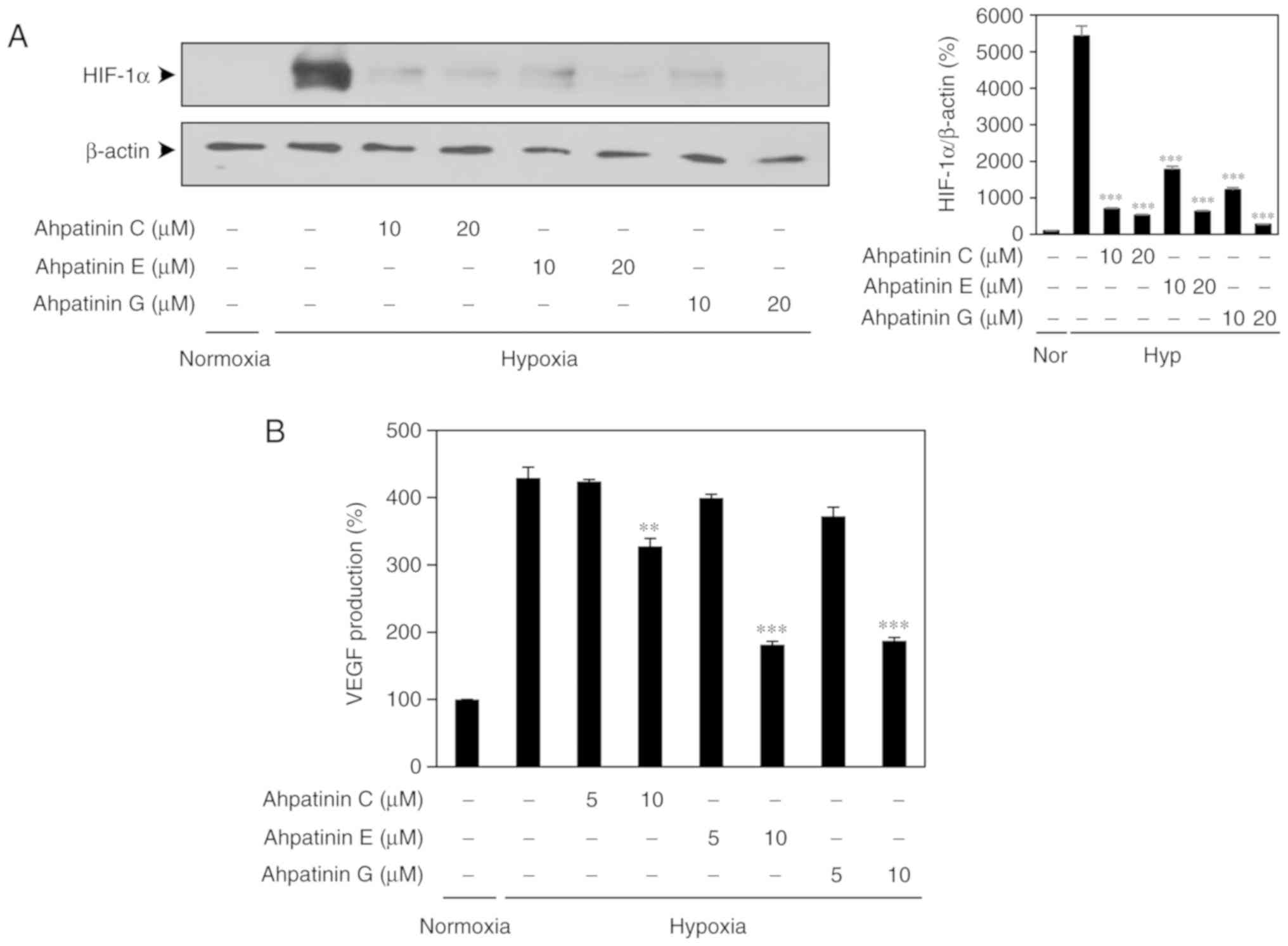 | Figure 8.HIF-1α inhibitory effects of
ahpatinins C, E, and G. (A) The effects of ahpatinins C, E, and G
on HIF-1α protein accumulation. U87MG cells were pretreated with
ahpatinins C, E, and G (10 and 20 µM) for 1 h and then exposed to
1% O2 for 8 h. Protein levels were detected by western
blot analysis. The level of β-actin was used as an internal
control. (B) The effects of ahpatinins C, E, and G on VEGF
expression. U87MG cells were pretreated with ahpatinins C, E, and G
(5 and 10 µM) for 1 h and then exposed to 1% O2 for 8 h.
The concentration of VEGF protein in the culture supernatant was
determined by a VEGF specific ELISA. **P<0.01 and ***P<0.001
vs. the hypoxic control. Each value represents the mean ± SE from
three independent experiments. HIF-1α, hypoxia-inducible factor
VEGF, vascular endothelial growth factor. |
Discussion
The screening of new antiangiogenic drug candidates
from microbial products has been successfully implemented for over
a decade (25,26,38).
Recently, octaminomycins A and B, isolated from Streptomyces
sp. RK85-270 were revealed to inhibit angiogenesis by reducing
the protein stabilities of VEGFR2, AKT, and ERK1/2 and the
activities of MMP-2 and MMP-9 in HUVECs (25). Two cyclic hexapeptides from the soil
fungus Penicillium sp. FN070315, PF1171A, and PF1171C
exerted their antiangiogenic activities by downregulating both
VEGFR2 and HIF-1α (26).
Elaiophylin, which was originally isolated from Streptomyces
melanosporus, exhibited a potent antiangiogenic activity
through suppression of the activation of VEGFR2 and its downstream
signaling mediators, AKT, ERK1/2, JNK, p38, and NF-κB (38). Through our continuing efforts in the
search for potential angiogenesis inhibitors from microbial-derived
natural products, the antiangiogenic effects and underlying
molecular mechanisms of ahpatinins C, E, and G from a soil-derived
Streptomyces sp. 15JA150 were newly revealed in the present
study.
In previous studies, ahpatinins were reported to
have inhibitory activities on renin and pepsin (27,28).
Ahpatinin C, an aspartic proteinase specific inhibitor, also
suppressed the receptor activator of NF-κB ligand (RANKL)-induced
osteoclast differentiation through the downregulation of ERK
signaling and expression of the nuclear factor of the activated T
cells 1 (NFATc1) (39). Notably,
ahpatinin C inhibited the invasion, tube formation, and MMP-9
activity of HUVECs and the angiogenesis of chick CAM induced by
cathepsin D, which was identified to enhance tumor angiogenesis,
growth, and metastasis (29,40,41).
However, the inhibitory activities of ahpatinins C, E, and G
against VEGF-induced angiogenesis and their underlying molecular
mechanisms have not yet been reported.
In the present study, it was demonstrated that
ahpatinins C, E, and G effectively disrupted the key processes of
angiogenesis stimulated by VEGF including the proliferation,
invasion, adhesion, and tube formation of HUVECs, at sub-toxic
concentrations. Moreover, ahpatinin E, which possessed a more
potent antiangiogenic activity compared to that of ahpatinins C and
G in vitro, significantly suppressed the in vivo
angiogenesis of CAM in chick embryos without any rupture of the
preexisting blood vessels. Although ahpatinins C, E, and G
exhibited inhibitory activities against VEGF-induced angiogenesis,
further investigations identifying the relationship between their
structures and activities could provide new insights into how they
regulate angiogenesis.
Angiogenesis is promoted by the interactions between
various proangiogenic factors and their receptors. VEGF, which is
recognized as the major mediator of angiogenesis in cancer, can
bind to and activate VEGFR1 (Flt-1) and VEGFR2 (KDR/Flk-1)
(42,43). However, the tyrosine kinase activity
of VEGFR2 is approximately 10-fold stronger than that of VEGFR1.
Accordingly, the major proangiogenic signal generally originates
from VEGFR2 activated by VEGF. The binding of VEGF to VEGFR2 on the
endothelial cell surface promotes receptor dimerization, enabling
autophosphorylation of the intracellular tyrosine residues. The
activation of VEGFR2 contributes to the phosphorylation of multiple
downstream signaling molecules such as AKT, ERK1/2, JNK, p38, and
NF-κB, and these molecules promote the proangiogenic cellular
responses, including endothelial cell survival, proliferation,
invasion, and migration, as well as vascular permeability and
inflammation. Therefore, the blockade of VEGFR2-mediated signal
transduction has been considered as a powerful therapeutic strategy
for inhibiting angiogenesis. In the present study, the
phosphorylation of VEGFR2 by VEGF was significantly inhibited by
ahpatinins C, E, and G, resulting in the suppression of the
VEGF-induced activation of downstream signaling effectors,
including AKT, ERK1/2, JNK, p38, and NF-κB. These results indicated
that ahpatinins C, E, and G exhibited antiangiogenic activities by
blocking VEGFR2-dependent downstream signaling cascades in
HUVECs.
HIF-1α plays a key role in the regulation of the
expression of numerous genes involved in the metabolic adaptation
to low oxygen, survival, and angiogenesis (21–23).
The reduced HIF-1α activity has been closely associated with
promoted tumor angiogenesis and aggressive tumor growth in numerous
cancers, resulting in the overexpression of VEGF and other genes
that are involved in the induction of angiogenesis. HIF-1α can be
activated by tumor hypoxia, a wide range of tumor growth-promoting
stimuli, and oncogenic pathways. HIF-1α has, therefore, been
suggested as a therapeutic target for impeding tumor angiogenesis.
The present data revealed that ahpatinins C, E, and G suppressed
the angiogenesis-promoting ability of U87MG glioblastoma cells
present in a hypervascular tumor with a high level of HIF-1α
expression. In addition, ahpatinins C, E, and G reduced the
hypoxia-induced accumulation of HIF-1α protein in U87MG cells. They
also decreased the VEGF production of U87MG cells under hypoxic
conditions, indicating that ahpatinins C, E, and G could inhibit
tumor cell-induced angiogenesis by downregulating HIF-1α and its
target gene, VEGF.
Collectively, the present findings provide a new
outlook on the therapeutic aspect for ahpatinins C, E, and G as
potent inhibitors of tumor angiogenesis through the dual blockade
of the proangiogenic signaling mediated by VEGFR2 and the
expression of HIF-1α. Further studies on how ahpatinins C, E, and G
regulate both the VEGFR2 and HIF-1α pathways will help to fully
understand the antiangiogenic mechanisms of action of these natural
microbial products.
Acknowledgements
The authors appreciate the Korea Basic Science
Institute, Ochang, Korea, for providing the NMR and HRESIMS
data.
Funding
The present research was supported by Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (NRF-2016R1D1A1B03932956)
and the NRF grant funded by the Ministry of Science and ICT (no.
2019R1A2C1009033). This work was also supported by an International
Joint Research Project (ASIA-16-011) of the NST (National Research
Council of Science & Technology) and the KRIBB Research
Initiative Program funded by the Ministry of Science ICT (MSIT) of
the Republic of Korea.
Availability of data and materials
The datasets generated and analyzed during the
current study are available from the corresponding author upon
reasonable request.
Authors' contributions
HJJ conceived and designed the experiments. JMH and
JPJ performed the experiments and analyzed the data. HJJ, JHJ and
JSA contributed reagents/materials/analysis tools. JMH and HJJ
wrote the manuscript. JPJ, JHJ and JSA reviewed and edited the
manuscript. All authors read and approved the final manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ellis LM, Liu W, Ahmad SA, Fan F, Jung YD,
Shaheen RM and Reinmuth N: Overview of angiogenesis: Biologic
implications for antiangiogenic therapy. Semin Oncol. 28 (5 Suppl
16):S94–S104. 2001. View Article : Google Scholar
|
|
2
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weis SM and Cheresh DA: Tumor
angiogenesis: Molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Li JX, Wang YQ and Miao ZH:
Tanshinone I inhibits tumor angiogenesis by reducing STAT3
phosphorylation at TYR705 and hypoxia-induced HIF-1α accumulation
in both endothelial and tumor cells. Oncotarget. 6:16031–16042.
2015.PubMed/NCBI
|
|
5
|
Jain RK: Antiangiogenesis strategies
revisited: From starving tumors to alleviating hypoxia. Cancer
Cell. 26:605–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Loges S, Schmidt T and Carmeliet P:
Mechanisms of resistance to anti-angiogenic therapy and development
of third-generation anti-angiogenic drug candidates. Genes Cancer.
1:12–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferrara N, Gerber HP and LeCounter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karkkainen MJ and Petrova TV: Vascular
endothelial growth factor receptors in the regulation of
angiogenesis and lymphangiogenesis. Oncogene. 19:5598–5605. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kowanetz M and Ferrara N: Vascular
endothelial growth factor signaling pathways: Therapeutic
perspective. Clin Cancer Res. 12:5018–5022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sachdev JC and Jahanzeb M: Evolution of
bevacizumab-based therapy in the management of breast cancer. Clin
Breast Cancer. 8:402–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salter JT and Miller KD: Antiangiogenic
agents in breast cancer. Cancer Invest. 25:518–526. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abhinand CS, Raju R, Soumya SJ, Arya PS
and Sudhakaran PR: VEGF-A/VEGFR2 signaling network in endothelial
cells relevant to angiogenesis. J Cell Commun Signal. 10:347–354.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gomes E and Rockwell P: p38 MAPK as a
negative regulator of VEGF/VEGFR2 signaling pathway in serum
deprived human SK-N-SH neuroblastoma cells. Neurosci Lett.
431:95–100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling-in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kyosseva SV: Mitogen-activated protein
kinase signaling. Int Rev Neurobiol. 59:201–220. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noort AR, van Zoest KP, Weijers EM,
Koolwijk P, Maracle CX, Novack DV, Siemerink MJ, Schlingemann RO,
Tak PP and Tas SW: NF-κB-inducing kinase is a key regulator of
inflammation-induced and tumour-associated angiogenesis. J Pathol.
234:375–385. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moulder JE and Rockwell S: Tumor hypoxia:
Its impact on cancer therapy. Cancer Metastasis Rev. 5:313–341.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jubb AM, Buffa FM and Harris AL:
Assessment of tumour hypoxia for prediction of response to therapy
and cancer prognosis. J Cell Mol Med. 14:18–29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Avni R, Cohen B and Neeman M: Hypoxic
stress and cancer: Imaging the axis of evil in tumor metastasis.
NMR Biomed. 24:569–581. 2011.PubMed/NCBI
|
|
24
|
Bernardes N, Seruca R, Chakrabarty AM and
Fialho AM: Microbial-based therapy of cancer: Current progress and
future prospects. Bioeng Bugs. 1:178–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jang JP, Han JM, Jung HJ, Osada H, Jang JH
and Ahn JS: Anti-angiogenesis effects induced by octaminomycins A
and B against HUVECs. J Microbiol Biotechnol. 28:1332–1338. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jang JP, Jung HJ, Han JM, Jung N, Kim Y,
Kwon HJ, Ko SK, Soung NK, Jang JH and Ahn JS: Two cyclic
hexapeptides from penicillium sp. FN070315 with antiangiogenic
activities. PLoS One. 12:e01843392017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Omura S, Imamura N, Kawakita K, Mori Y,
Yamazaki Y, Masuma R, Takahashi Y, Tanaka H, Huang LY and Woodruff
HB: Ahpatinins, new acid protease inhibitors containing
4-amino-3-hydroxy-5-phenylpentanoic acid. J Antibiot (Tokyo).
39:1079–1085. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Y, Takada K, Nogi Y, Okada S and
Matsunaga S: Lower homologues of ahpatinin, aspartic protease
inhibitors, from a marine Streptomyces sp. J Nat Prod.
77:1749–1752. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu L, Roth JM, Brooks P, Luty J and
Karpatkin S: Thrombin up-regulates cathepsin D which enhances
angiogenesis, growth, and metastasis. Cancer Res. 68:4666–4673.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morjen M, Honoré S, Bazaa A,
Abdelkafi-Koubaa Z, Ellafi A, Mabrouk K, Kovacic H, El Ayeb M,
Marrakchi N and Luis J: PIVL, a snake venom kunitz-type serine
protease inhibitor, inhibits in vitro and in vivo angiogenesis.
Microvasc Res. 95:149–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Drewes CC, Dias RY, Hebeda CB, Simons SM,
Barreto SA, Ferreira JM Jr, Chudzinski-Tavassi AM and Farsky SH:
Actions of the kunitz-type serine protease inhibitor amblyomin-X on
VEGF-A-induced angiogenesis. Toxicon. 60:333–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu M, Li B, Wang J and Xiao R: The
anti-angiogenic activity of a cystatin F homologue from the buccal
glands of lampetra morii. Mar Drugs. 16:E4772018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hass TL: Endothelial cell regulation of
matrix metalloproteinases. Can J Physiol Pharmacol. 83:1–7. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bergers G and Benjamin LE: Tumorigenesis
and the angiogenic switch. Nat Rev Cancer. 3:401–410. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
André T, Chastre E, Kotelevets L, Vaillant
JC, Louvet C, Balosso J, Le Gall E, Prévot S and Gespach C: Tumoral
angiogenesis: Physiopathology, prognostic value and therapeutic
perspectives. Rev Med Interne. 19:904–913. 1998.(In French).
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Badewiijns MM, van Vlodrop IJ, Vermeulen
PB, Soetekouw PM, van Engeland M and de Bruïne AP: VHL and HIF
signalling in renal cell carcinogenesis. J Pathol. 221:125–138.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lim HN, Jang JP, Han JM, Jang JH, Ahn JS
and Jung HJ: Antiangiogenic potential of microbial metabolite
elaiophylin for targeting tumor angiogenesis. Molecules.
23:E5632018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoshida H, Okamoto K, Iwamoto T, Sakai E,
Kanaoka K, Hu JP, Shibata M, Hotokezaka H, Nishishita K, Mizuno A
and Kato Y: Pepstatin A, an aspartic proteinase inhibitor,
suppresses RANKL-induced osteoclast differentiation. J Biochem.
139:583–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liaudet-Coopman E, Beaujouin M, Derocq D,
Garcia M, Glondu-Lassis M, Laurent-Matha V, Prébois C, Rochefort H
and Vignon F: Cathepsin D: Newly discovered functions of a
long-standing aspartic protease in cancer and apoptosis. Cancer
Lett. 237:167–179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Trincheri NF, Nicotra G, Follo C, Castino
R and Isidoro C: Resveratrol induces cell death in colorectal
cancer cells by a novel pathway involving lysosomal cathepsin D.
Carcinogenesis. 28:922–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang J, Zhao P, Fu Z, Chen X, Liu N, Lu
A, Li R, Shi L, Pu P, Kang C and You Y: Glioma cells enhance
endothelial progenitor cell angiogenesis via VEGFR-2, not VEGFR-1.
Oncol Rep. 20:1457–1463. 2008.PubMed/NCBI
|
|
43
|
Liang X, Xu F, Li X, Ma C, Zhang Y and Xu
W: VEGF signal system: The application of antiangiogenesis. Curr
Med Chem. 21:894–910. 2014. View Article : Google Scholar : PubMed/NCBI
|