Introduction
In 2018, esophageal cancer ranked seventh for cancer
incidence and sixth as the leading cause of cancer-related
mortality in the world, and ~70% of cases occurred in men. In men,
the rates were also 2-fold higher in Human Development Index (HDI)
countries, with mortality rates ranking fifth in these countries
(1). Esophageal squamous cell
carcinomas (ESCC) and esophageal adenocarcinomas (EAC) account for
>95 % of the various esophageal malignancies. Moreover, the
incidence of EAC is rapidly increasing in the United States and
Western Europe (2). Esophagectomy
alone is associated with a high rate of recurrence and low 5-year
survival rates. A recent clinical trial revealed that neoadjuvant
chemotherapy significantly improved survival in patients with
resectable tumors (3). Conventional
chemotherapeutic agents can effectively kill actively proliferating
cells, however, the non-selective targeting can cause the
destruction of both healthy and cancerous cells. Ultimately,
chemotherapy leads to multiple side effects and the occurrence of
drug resistance in patients. Therefore, an important approach is to
identify low-toxicity and non-cancer agents for combination therapy
in order to reduce the dose requirement of chemotherapeutic agents
(4). Drug repurposing is an
efficient approach, using clinically-approved drugs for different
disease treatments bypassing the long streamline of the drug
discovery process (5). In order to
enhance the killing efficacy of cancer cells and ameliorate the
side effects caused by cancer treatments, it is significant to
study and evaluate both the feasibility and efficacy of combination
therapy with chemotherapeutic agents plus repurposed non-cancer
drugs targeting similar pathways found in cancer (6). Based on this concept, searching for
non-cancer clinical medications and evaluating the feasibility of
combination therapy of these non-cancer clinical medications and
chemotherapeutic agents for esophageal cancers is the focus of this
research.
Niclosamide is an FDA-approved chewable tablet
consumed orally against tapeworms and schistosomiasis. The
mechanism of niclosamide action is through the inhibition of
glucose uptake, oxidative phosphorylation, and anaerobic metabolism
in the target worms (7). Except for
antiprotozoal function, the antineoplastic effects of niclosamide
has been revealed in many human cancer cells, including
adrenocortical carcinoma (8),
breast (9–11), colorectal (12,13)
and cervical cancer (14),
glioblastoma (15), hepatocellular
carcinoma (16,17), head and neck (18,19)
and lung cancer (20,21), leukemia (22,23),
nasopharyngeal cancer (24),
osteosarcoma (25), oral squamous
cell carcinoma (26,27), ovarian (28–30),
prostate (31–33), renal (34) and thyroid cancer (35). Niclosamide treatment was reported to
induce apoptosis of cancer cells in vitro (7,10,23)
and suppress tumor size in animal studies (11,29).
Moreover, the combination of anticancer agents with niclosamide
synergistically suppressed cell proliferation of acute myelogenous
leukemia, head and neck, ovarian, prostate and non-small lung
cancer (19,21,23,30,31).
However, whether niclosamide is effective against esophageal cancer
has not been investigated yet.
The molecular mechanisms underlying the
antineoplastic effect of niclosamide have been explored in many
human malignant cancers, indicating that niclosamide exhibits
anticancer activity by suppressing many oncogenic signaling
pathways concurrently (7,13,17,23,27,28,30,36).
For instance, niclosamide has been identified as a direct inhibitor
of signal transducer and activator of transcription 3 (STAT3)
through interaction with the DNA-binding domain (37). In ovarian cancer, niclosamide
significantly decreased the expression of proteins in the
wingless/integrated (Wnt), mammalian target of rapamycin (mTOR) and
STAT3 pathways and caused significant inhibition of proliferation
of cells (28). In acute myeloid
leukemia, niclosamide could induce apoptosis of AML blast cells
through inhibition of the nuclear factor-κB (NF-κB) pathway and
increasing the production of reactive oxygen species (23). In lung and head and neck cancers,
niclosamide suppressed erlotinib-induced STAT3 phosphorylation, and
a combination of erlotinib and niclosamide decreased tumor size in
animal model experiments (19,21).
In advanced prostate cancer, niclosamide blocked the interleukin 6
(IL6)/STAT3/androgen receptor (AR) pathway to overcome enzalutamide
resistance and inhibit migration and invasion (31).
In the present study, the antineoplastic effects of
niclosamide on esophageal cancer cells were investigated and it was
revealed that niclosamide suppressed the STAT3 signaling pathway
and inhibited cell proliferation in esophageal cancer cells.
Niclosamide also induced cell apoptosis and G1-phase arrest of the
cell cycle. Furthermore, the combination treatment of niclosamide
and chemotherapeutic drugs selectively reduced the dose requirement
of the chemotherapeutic drugs in order to obtain the
IC50 efficacy. These findings indicated that niclosamide
may be used as a single or combined drug treatment for esophageal
cancer.
Materials and methods
Reagents
Niclosamide (product no. N3510), 5-fluorouracil
(5-FU) (product no. F6627), cisplatin (P4394), and paclitaxel
(T7402) were purchased from Sigma-Aldrich (Merk KGaA). Cisplatin
was dissolved in ddH2O, whereas, niclosamide, 5-FU and
paclitaxel were dissolved in dimethyl sulfoxide (DMSO). The solvent
was routinely used in the control group of the experiment.
Cell culture
Esophageal cancer cell lines, BE3 (adenocarcinoma),
CE48T/VGH and CE81T/VGH (squamous cell carcinoma) were courtesy of
Dr Yen (38) and Dr Lee (39), respectively. BE3 cells were cultured
in Dulbecco's Modified Eagle's Medium (DMEM) and CE48T and CE81T
were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco
BRL; Thermo Fisher Scientific, Inc.), supplemented with 10%
heat-inactivated fetal bovine serum, 1% penicillin/streptomycin
solution (Gibco-BRL; Thermo Fisher Scientific, Inc.). The cells
were grown in a humidified incubator containing 5% CO2
at 37°C.
MTS assay
To determine the cytotoxicity of niclosamide and the
combined effect of niclosamide and chemotherapeutic agents, cells
were seeded in 96-well plates overnight and treated for 72 h with
different concentrations of niclosamide (1.25–20 µM) or co-treated
with 2.5 µM niclosamide and serial doses of chemotherapeutic drugs,
or DMSO as a vehicle control. The cell viability was then evaluated
by MTS assay. Twenty microliters of the MTS CellTiter 96 Cell
Proliferation Assay reagent (Promega Corporation) were added to
each well. After 2 h of incubation at 37°C, the absorbance of the
colored product was measured on an EPOCH2 microplate reader (BioTek
Instruments, Inc.).
Colony-forming assay
Esophageal cancer cell lines to be tested were
trypsinized and dissociated into single-cell suspensions for
plating in 6-well plates. An agar suspension (0.3% agar) containing
colony-forming cells (2×104 cells/well) is plated over
an agar underlay (0.6% agar). Niclosamide (2.5 µM) was delivered to
the cells. Tissue culture medium was replenished every 3 days.
After 21 days of culture, the cells were washed twice with
phosphate-buffered saline (PBS) and incubated in fresh medium with
MTS reagent for 2 h at 37°C. The living colony formation efficiency
was determined by formation of dark-colored formazan dye by
reduction of the tetrazolium salt MTS by metabolically active
cells. Digital images of the plates were captured by Nikon
camera.
Western blot analysis
To evaluate the protein expression levels after
niclosamide treatment, western blot analysis was performed. The
cells were dissolved in a lysis buffer containing 50 mM Tris-HCl,
pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA, 5
mM sodium orthovanadate, 25 mM sodium fluoride, 5 mM sodium
pyrophosphate decahydrate and 5 mM β-glycerophosphate. Protein
concentration was quantified by Bio-Rad protein assay. Fifty
micrograms of protein was fractionated via 10 or 12% SDS-PAGE and
transferred to Immobilon Transfer Membranes (EMD Millipore), and
then the membranes were blocked in 5% skim milk at room temperature
for 1 h. Primary antibodies used in the present study included
those against β-actin (GTX109639) (dilution 1:10,000; GeneTex,
Inc.), poly(ADP-ribose) polymerase (PARP) (9542S), β-catenin
(8480S), STAT3 (4904S), phosphor-STAT3 (9145S) (dilution 1:1,000;
Cell Signaling Technology, Inc.), cyclin D1 (H295), cyclin E1
(HE-12), cyclin A (H432), and cyclin B (GNS1) (dilution 1:1,000;
Santa Cruz Biotechnology, Inc.). The HRP-conjugated secondary
antibodies recognizing mouse-IgG (7076) or rabbit-IgG (7074) were
purchased from Cell Signaling Technology, Inc. and those
recognizing goat-IgG (SC-2020) were purchased from Santa Cruz
Biotechnology, Inc. (dilution, ~1:5,000–10,000). The
chemiluminescence signal was developed with Immobilon Western
Chemiluminescent HRP Substrate (WBKLS0500; Millipore) and
visualized and recorded using the BioSpectrum UVP810 Imaging System
with VisionWorks software (GE Healthcare Life Sciences).
Real-time quantitative RT-PCR
(RT-qPCR) analysis
To quantify the endogenous mRNA expression levels,
qRT-PCR was performed. Total RNA of cells was isolated using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. One microgram of total RNA was
used to synthesize the cDNA using SuperScript III reverse
transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) with
Oligo-dT primer. Quantitative PCR was performed using Kapa SYBR
fast qPCR master mix (Kapa Biosystems, Inc.). The primer sequences
for qPCR were as follows: GAPDH forward, 5′-GAAGGTGAAGGTCGGAGT-3′
and reverse, 5′-GAAGATGGTGATGGGATTTC-3′; p21 forward,
5′-GAAGACCATGTGGACCTGTC-3′ and reverse, 5′-TCCTCTTGGAGAAGATCAGC-3′;
p27 forward, 5′-GTTTCAGACGGTTCCCCAAA-3′ and reverse,
5′-CCTTGCTTCATCAAGCAGTG-3′. The qPCR reaction was run in a final
volume of 20 µl containing 1 µl of reverse transcriptase product,
10 µl of 2X Kapa SYBR fast qPCR master mix, and 0.6 µl of each
primer (10 µM). The qPCR mixtures were pre-incubated at 95°C for 3
min, followed by 40 cycles at 95°C for 3 sec and 60°C for 30 sec.
All qPCR was performed in triplicate using an iQ5 Real-Time PCR
detection system (Bio-Rad Laboratories, Inc.). Relative expression
was calculated using the 2−ΔΔCq method (40).
Annexin V and propidium iodide (PI)
double staining
Following drug treatment, the cells were collected
and washed twice with cold PBS, and stained with 5 µl of Annexin
V-FITC and 10 µl of PI (5 g/ml) in 0.5 ml of binding buffer (10 mM
HEPES, pH 7.4; 140 mM NaOH; 2.5 mM CaCl2) for 15 min at
room temperature in the dark. The apoptotic cells were determined
using a BD FACSVerse Flow Cytometer (BD Biosciences).
Cell cycle analysis
After niclosamide treatments, the cells were
collected and fixed with cold 80% ethanol at −20°C for 1 h. The
cells were then washed twice with PBS and stained with 0.5 ml
PI/RNase at RT for 30 min. The samples were analyzed by BD
FACSVerse Flow Cytometer.
Statistical analysis
All values were expressed as the mean ± standard
error. The Student's t-test was used to analyze the results, and a
P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
Niclosamide suppresses the STAT3
signaling pathway and inhibits the cell growth of esophageal cancer
cell lines
Niclosamide, a direct STAT3 inhibitor, had been
reported to exhibit the ability to suppress multiple oncogenic
signaling transduction pathways. In order to explore the effect of
niclosamide on STAT3 and β-catenin signaling pathways of cultured
esophageal cancer cells, CE48T, CE81T and BE3 cells were treated
with 10 µM niclosamide for 24 h, and the total forms of STAT3 and
β-catenin and phosphorylated (p)-STAT3 (Y705) were investigated by
western blot analysis. As revealed in Fig. 1A, there were different expression
levels of β-catenin in these cell lines. The protein levels of
endogenous β-catenin could only be detected in CE48T and CE81T
cells, but not in BE3 cells. Moreover, niclosamide treatment did
not markedly affect the protein level of β-catenin. Conversely, the
STAT3 signaling pathway was constitutively active in these cancer
cells, and niclosamide treatment markedly suppressed STAT3 (Y705)
phosphorylation. Furthermore, to explore the effect of niclosamide
on the proliferation of esophageal cancer cells, these cells were
treated with niclosamide at different concentrations for 72 h and
then cell viability was assessed by MTS assay. As revealed in
Fig. 1B, the cell viabilities of
these three cancer cell lines were dose-dependently decreased by
niclosamide treatment. The IC50 concentrations of
niclosamide were 2.8, 11.3, and 5.1 µM for CE48T, CE81T and BE3,
respectively. Moreover, a clonogenic assay was conducted to
investigate the long-term growth inhibitory effect of niclosamide
on esophageal cancer cells. Since CE81T cells did not grow on the
soft agar to form a colony, only BE3 and CE48T cells were repeated
and long-term treated with a physiological concentration of
niclosamide (2.5 µM) for 21 days and living colony formation
efficiency was determined by formation of dark-colored formazan dye
by reduction of the tetrazolium salt MTS by metabolically active
cells. As revealed in Fig. 1C,
niclosamide completely inhibited colony formation in BE3 and CE48T
cells, indicating that niclosamide may be considered as a potential
inhibitor for esophageal cancer cell growth.
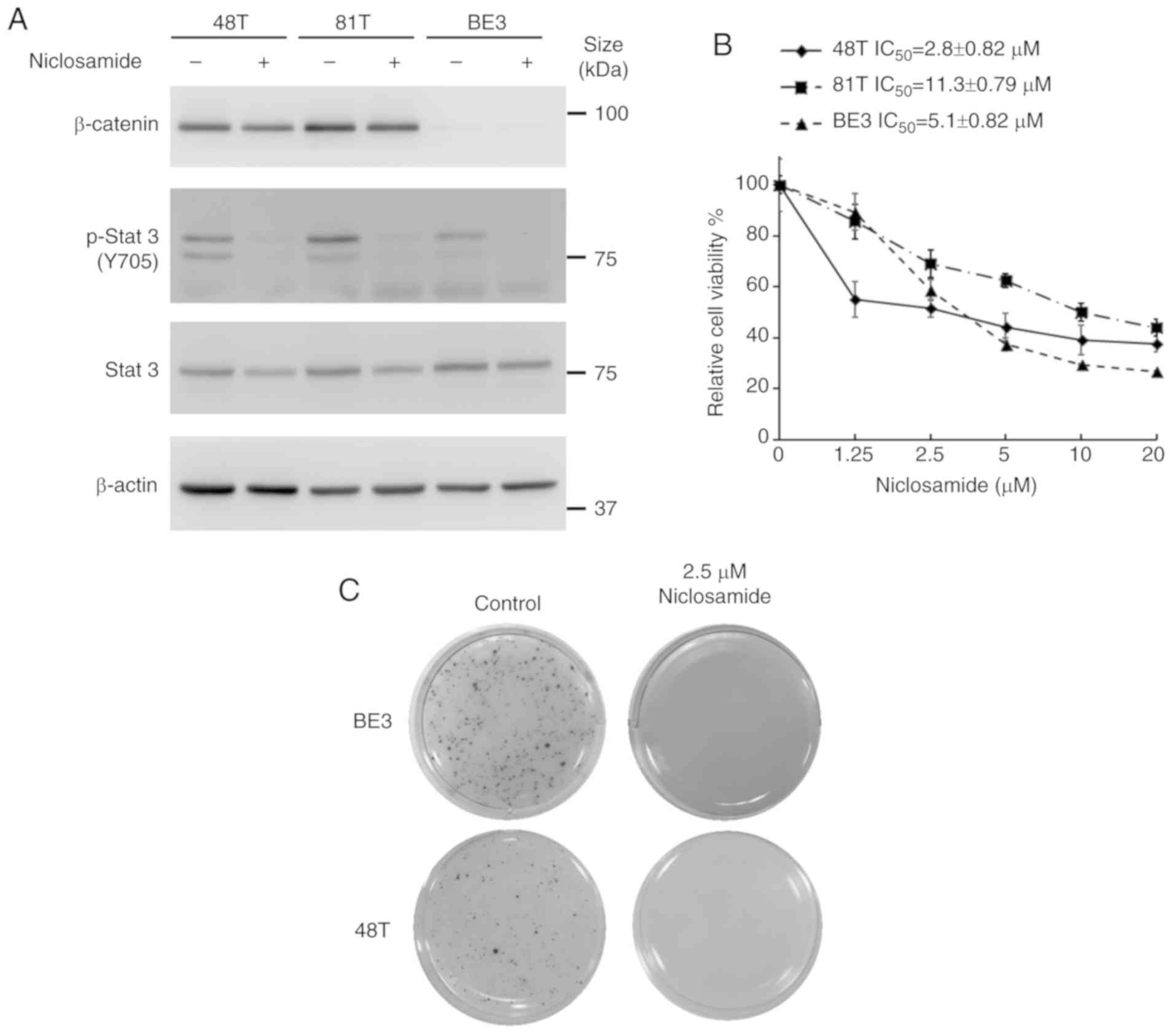 | Figure 1.Niclosamide treatment blocks the
STAT3 signaling pathway and inhibits the cell growth of human
esophageal cancer cell lines. (A) CE48T, CE81T and BE3 cells were
treated with 10 µM niclosamide for 24 h, and the phosphorylation of
Y705 and the total form of STAT3, as well as β-catenin, were
investigated by western blot assay using β-actin as a loading
control. (B) Cancer cells were seeded in 96-well plates and treated
with a concentration series of niclosamide (0, 1.25, 2.5, 5, 10 or
20 µM) for 72 h, and then the relative cell viabilities were
determined by MTS assay. Error bars represent the standard
deviation. The viability of control cells was designated as 100%,
and that of the other groups were expressed as the percent of the
control. (C) Colony-forming ability of CE48T and BE3 cells analyzed
by clone formation assay. Images of living cell clones indicated by
the formation of dark-colored formazan dye by reduction of the
tetrazolium salt MTS by metabolically active cells. STAT3, signal
transducer and activator of transcription 3; MTS,
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium. |
Niclosamide treatment induces
apoptosis of esophageal cancer cell lines
To further explore if the decrease in esophageal
cancer cell viability was a consequence of niclosamide-induced
apoptosis, BE3 cells treated with niclosamide were subjected to
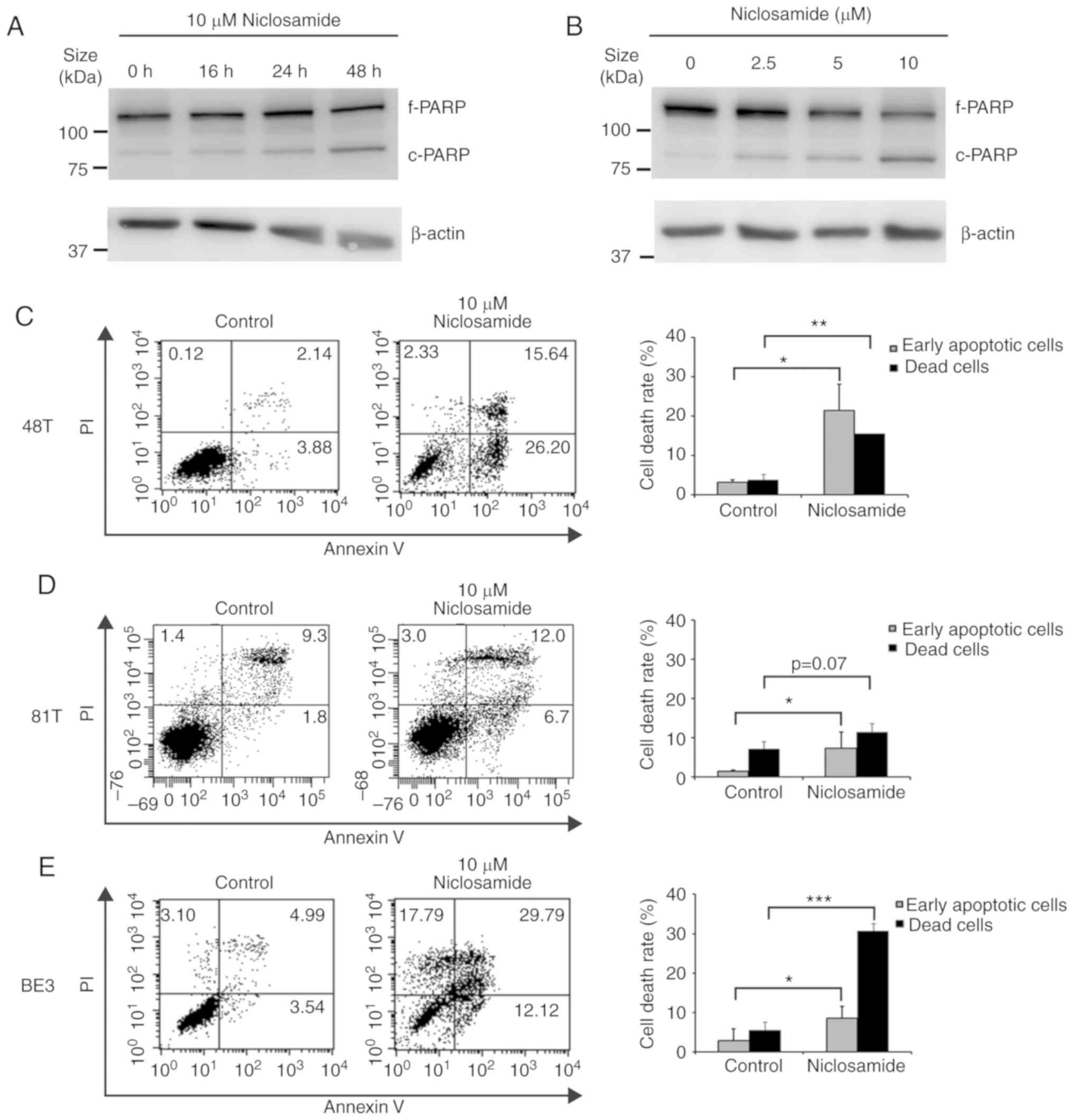
western blot analysis of cleaved PARP. As revealed in Fig. 2A and B, the highest level of cleaved
PARP product was detected at 10 µM niclosamide and 48 h of
treatment, indicating that the apoptotic level was positively
associated with the niclosamide dose and exposure time in BE3
cells. To further demonstrate the induction of apoptosis by
niclosamide, CE48T, CE81T and BE3 cells were treated with
niclosamide for 48 h, and subjected to Annexin V and PI
double-staining assay. The early apoptotic and dead cells were
quantified by flow cytometric analysis. As revealed in Fig. 2C-E, niclosamide significantly
(P<0.05) increased the proportion of early apoptotic cells in
the three cell lines, but only significantly increased the
proportion of dead cells in CE48T (P<0.005) and BE3
(P<0.0005) cells. The results indicated that the reduction of
cell viabilities of CE48T and BE3 cells was mainly caused by
niclosamide-induced cell apoptosis, but was partial to that of
CE81T cells.
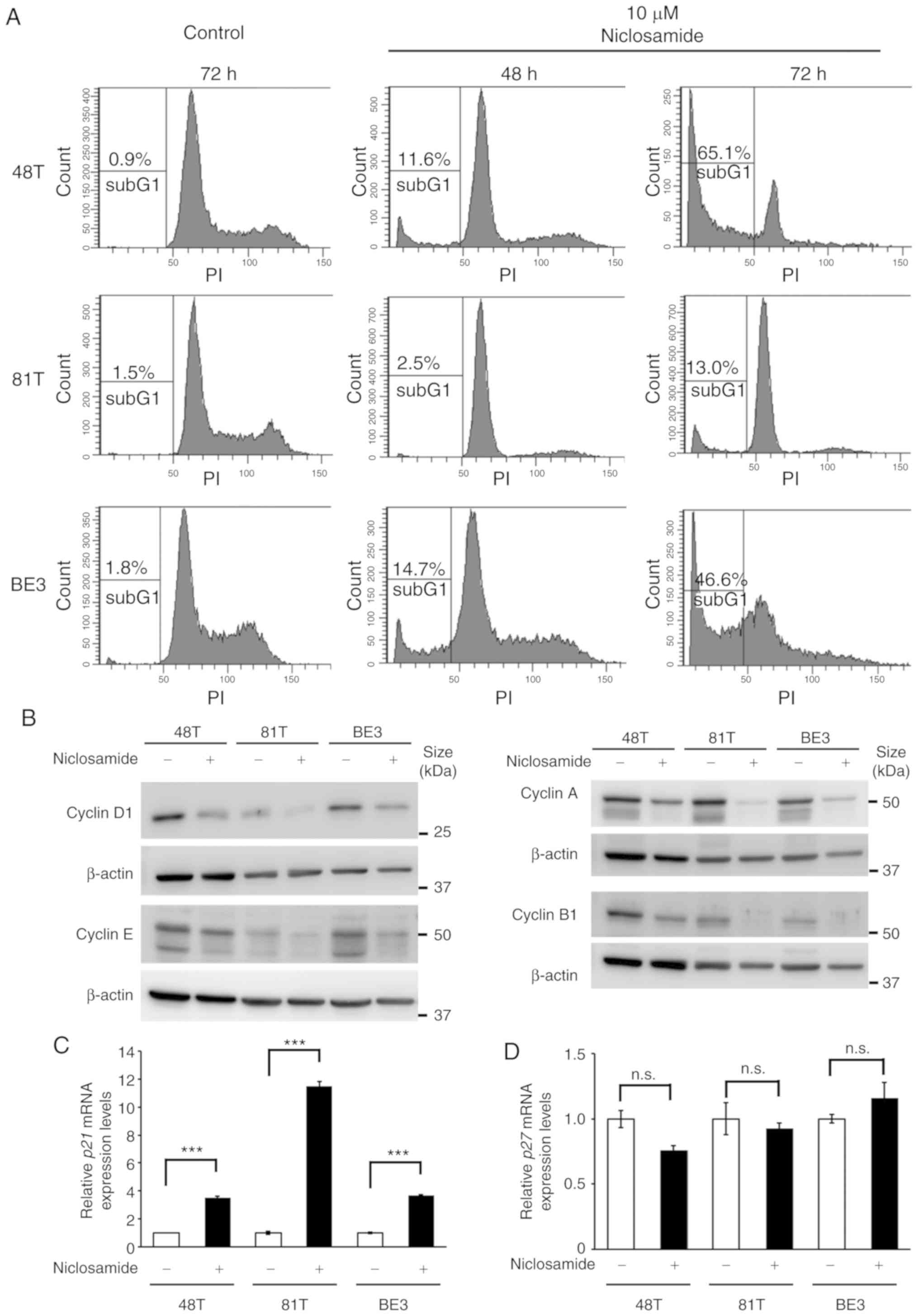
Niclosamide treatment causes G1-phase
arrest in CE81T cells
Given that only a small proportion of apoptotic
cells were detected in CE81T cells after treatment with 10 µM
niclosamide for 48 h (Fig. 2D),
cell cycle dynamics were analyzed by flow cytometric analysis to
investigate why niclosamide suppressed cell growth of CE81T cells.
The cell cycle analysis revealed that after treatment with 10 µM
niclosamide for 72 h a high proportion of sub-G1 (cell debris) was
only detected in CE48T and BE3 cells, while a low proportion of
sub-G1 was revealed in CE81T cells. Moreover, an apparent G1 peak
was only revealed in CE81T cells, as well as absence of S and G2/M
phases (Fig. 3A). In addition, the
physiological concentration of niclosamide (2.5 µM) was also used
to conduct cell cycle analysis in CE48T, CE81T and BE3 cells. The
proportion of sub-G1 cells was increased in a time-dependent manner
in CE48T and BE3 cells after 48 and 72 h treatment. In addition, at
72 h of treatment, the proportion of G1-phase cells was decreased
and the cells of the S and G2/M phases were still detectable. In
contrast, in CE81T cells, the proportion of sub-G1 cells did not
significantly increase, the proportion of G1-phase cells was
increased and the proportion of S-phase cells was decreased after
72 h of treatment (Fig. S1). This
finding indicated that niclosamide induced G1-phase arrest in CE81T
cells. Cyclin is a family of proteins that control the progression
of cells through the cell cycle. The protein levels of various
cyclins in CE48T, CE81T and BE3 cells were assessed by western blot
assay, and it was revealed that all of the expression levels of
cyclin D1, E, A and B1 were almost completely lost in CE81T cells
treated with niclosamide for 48 h (Fig.
3B). In addition, the expression of cell cycle inhibitor genes,
p21 and p27, were also examined in these cells
treated with or without 10 µM niclosamide for 24 h. As revealed in
Fig. 3C and D, an ~11-fold increase
in p21 mRNA expression was detected in niclosamide-treated
CE81T cells, as well as an ~3.5-fold increase in
niclosamide-treated CE48T and BE3 cells. In contrast, niclosamide
treatment did not affect p27 mRNA expression in these
cells.
Niclosamide co-treatment selectively
reduces the dosage of anticancer drugs for achieving
IC50 in esophageal cancer cells
A previous study reported that the combination of
niclosamide and anticancer drugs resulted in a synergistic
antitumor efficacy against xenografted AML tumor cells (23). 5-FU, cisplatin, and paclitaxel are
three frontline chemotherapeutic drugs currently used in clinical
esophageal therapy (41–43). Therefore, they were used in the
present experiment to explore whether the treatment of these
anticancer drugs combined with niclosamide could have an additive
benefit of antitumor efficacy for esophageal cancer cells.
Niclosamide appears to be minimally absorbed from the
gastrointestinal tract and a pharmacokinetics study revealed that
the oral dosage of niclosamide for adults in cytocidal treatment
was 2 g as a single dose, leading to maximal serum concentration of
0.76 to 18.35 µM (44). In the
present study, the esophageal cancer cell lines were treated with
the combination of 2.5 µM niclosamide plus different
chemotherapeutic drugs for 72 h, and cell viability was assessed by
MTS assay. In CE48T and BE3 cells, the IC50 values of
5-FU, cisplatin, and paclitaxel were reduced in the combination
groups of niclosamide, when compared to the anticancer drug
administrated alone (Fig. 4A and
C). In CE81T cells, the niclosamide combination only reduced
the IC50 values of 5-FU and paclitaxel as compared with
anticancer drug treatment alone (Fig.
4B). To further validate the benefit of combination treatment
with niclosamide, the proportion of apoptotic cells (Annexin
V+ cells) induced by the anticancer drug alone, or in
combination with niclosamide was investigated by flow cytometric
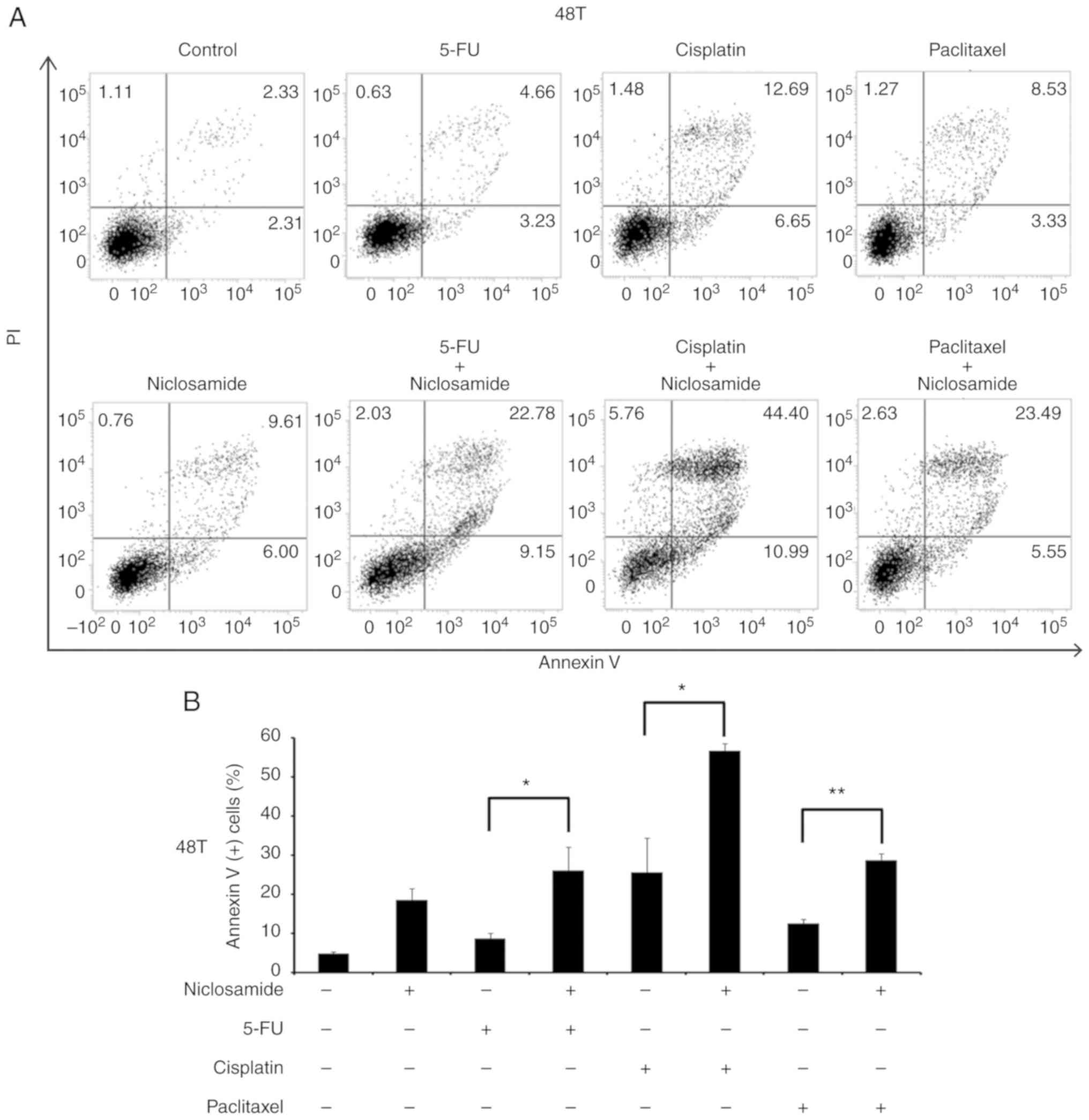
analysis. As revealed in Fig. 5,
the treatment of 2.5 µM niclosamide for 48 h induced 18.46±3.06% of
apoptotic cells in CE48T cells, and the combination of niclosamide
and 5-FU (50 µM) caused a higher proportion of apoptotic cells than
5-FU treatment alone (26.14±5.89% vs. 8.62±1.47%; P<0.05).
Moreover, the combination of niclosamide and cisplatin (10 µM)
resulted in a higher proportion of apoptotic cells than cisplatin
treatment alone (56.68±1.82% vs. 25.57±8.81%; P<0.05), as well
as the combination of niclosamide and paclitaxel (10 nM) which
induced a higher proportion of apoptotic cells than paclitaxel
treatment alone (28.69±1.75% vs. 12.54±1.12%; P<0.005). In CE81T
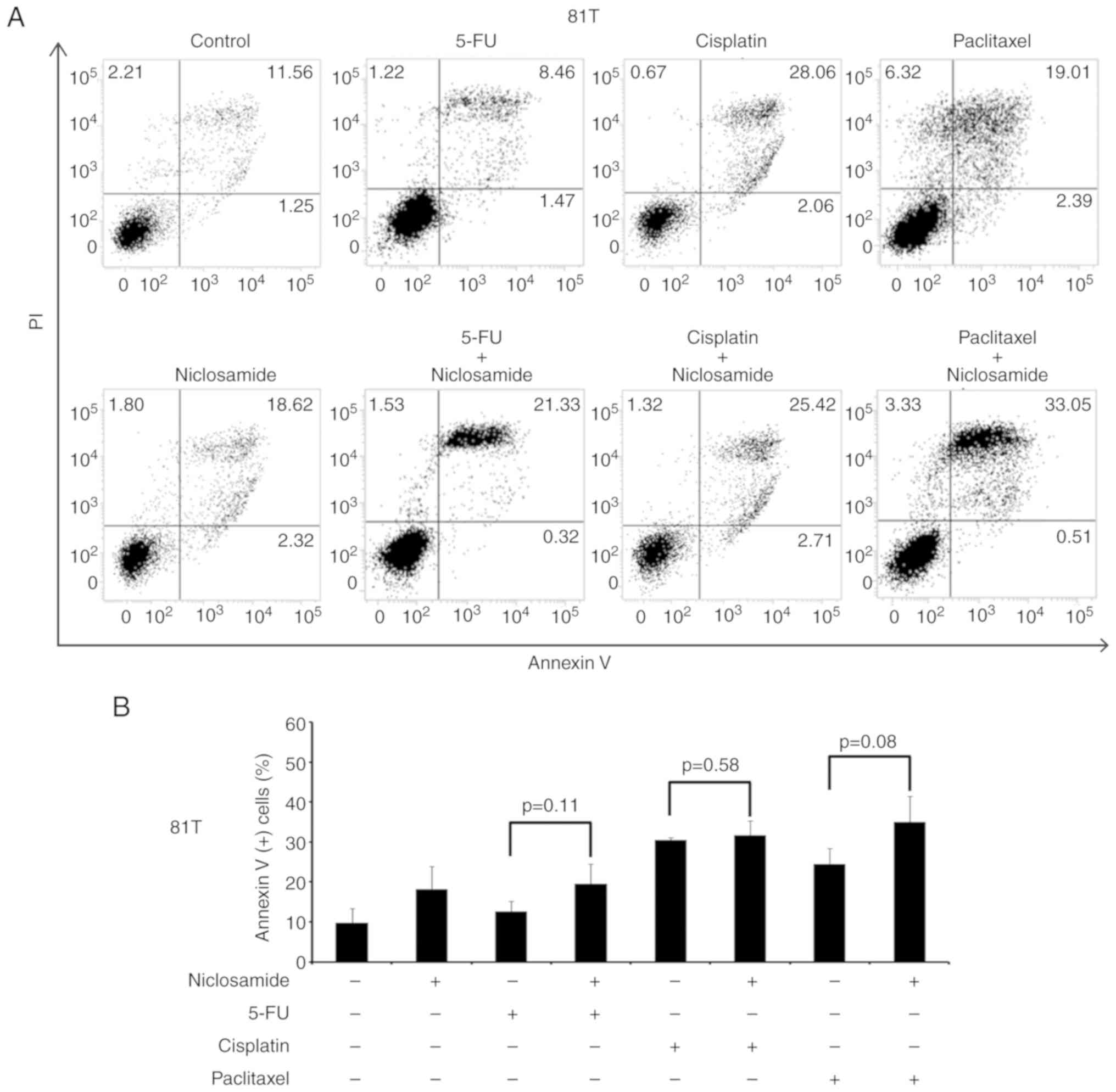
cells, there was no statistically significant difference detected
in the proportion of apoptotic cells between treatment with these
three chemotherapeutic agents alone and the combination of
niclosamide with 5-FU, cisplatin, or paclitaxel (as revealed in
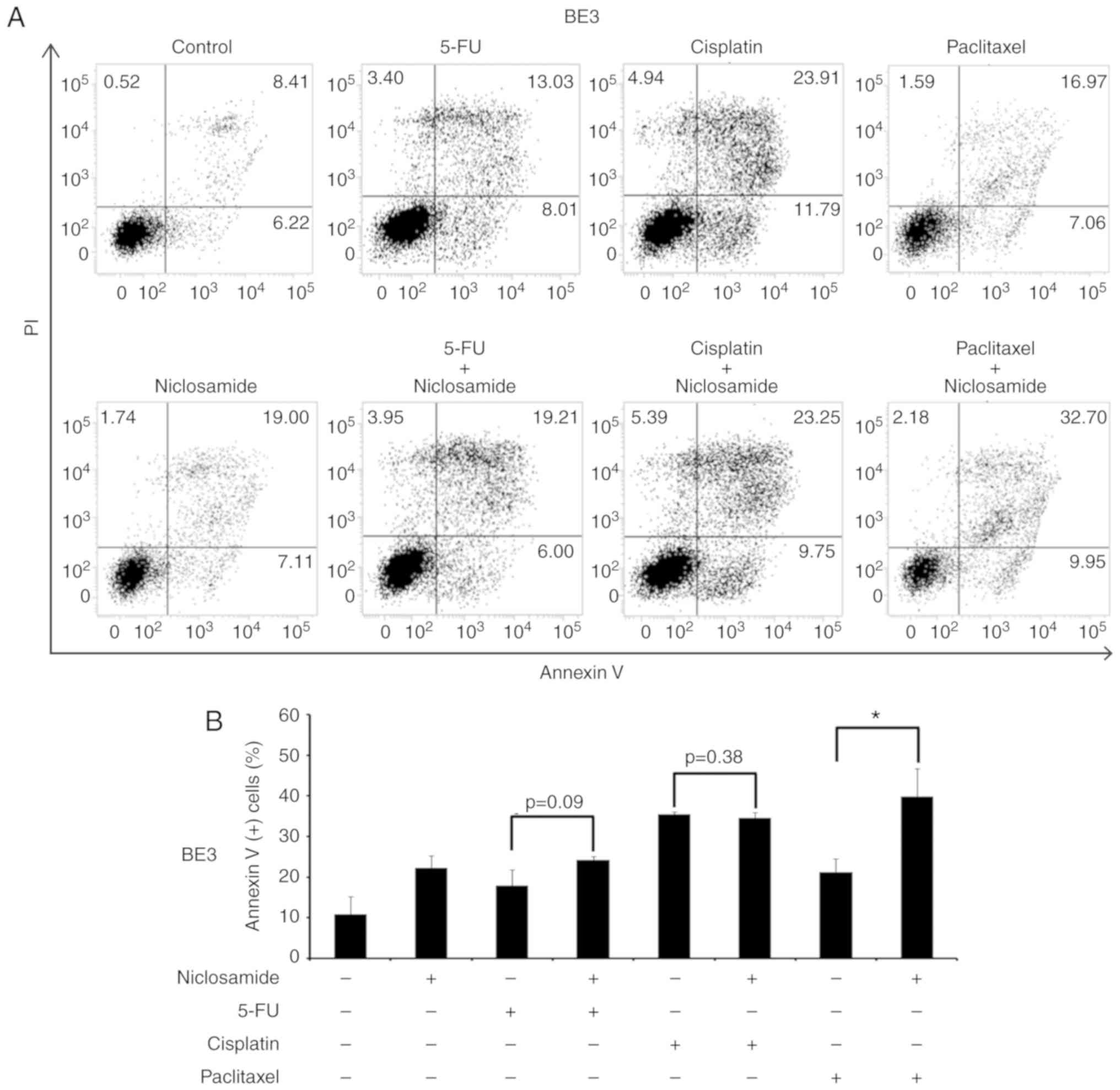
Fig. 6). In BE3 cells, the
combination of niclosamide and paclitaxel induced a higher
proportion of apoptotic cells than paclitaxel treatment alone
(39.88±6.83% vs. 21.26±3.26%; P<0.05) (as revealed in Fig. 7). The results of the cell apoptosis
assays were similar with the findings of the cell viability assays,
indicating that niclosamide selectively reduced the dosage
requirement of anticancer drug for obtaining the IC50 in
esophageal cancer cells.
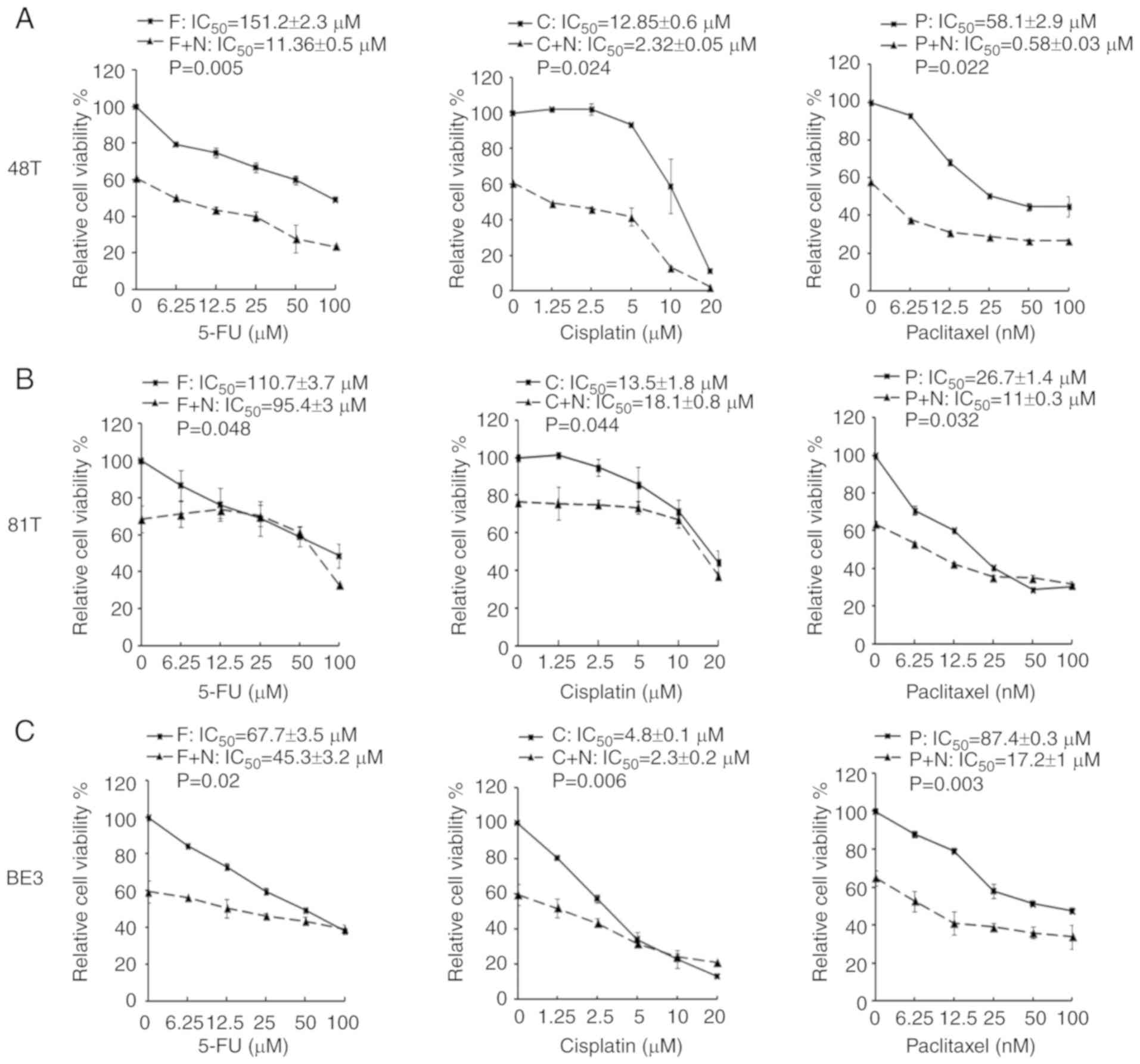 | Figure 4.MTS assays of esophageal cancer cells
treated with a chemotherapeutic drug alone or in combination with
niclosamide. Esophageal cancer cells (A) CE48T, (B) CE81T and (C)
BE3 cells were seeded in 96-well plates and treated for 72 h with
serial doses of 5-FU, cisplatin, paclitaxel, or in combination with
2.5 µM of niclosamide. Cell viability was then analyzed by MTS
assay. F, C, and P indicate the treatment of 5-FU, cisplatin, or
paclitaxel, respectively. F+N, C+N, and P+N indicate the
combination treatment of niclosamide with 5-FU, cisplatin, or
paclitaxel, respectively. 5-FU, 5-fluorouracil. |
Discussion
The current chemotherapeutic strategy is inevitably
accompanied by a variety of dose-dependent and sometimes lethal
adverse effects. Thus, it is worth exploring cost-effective drug
repurposing and combinations to ameliorate unpleasant adverse
effects following traditional chemotherapy. Niclosamide is an
FDA-approved anthelmintic drug which exerts antineoplastic
activity. The antineoplastic efficacy of niclosamide in numerous
human cancer studies (8–35) has been demonstrated. However,
whether niclosamide is active against esophageal cancer is unknown.
Herein, the present study for the first time demonstrated that
niclosamide exhibited antineoplastic effects in esophageal cancer
cell lines.
Niclosamide has been identified as a direct STAT3
inhibitor through interaction with the DNA-binding domain (37). In addition to its role in STAT3
inhibition, niclosamide has also been revealed to concurrently
inhibit multiple intracellular signaling pathways, including the
Wnt, Notch 1, mTOR and NF-κB signaling cascades (10,45).
In many solid and hematological cancers, STAT3 is constitutively
active, promoting tumor survival and progression. For example, a
previous study by Huang et al reported that positive p-STAT3
expression was observed in 47.7% of patients with esophageal
squamous cell carcinoma. Patients with p-STAT3 positive disease had
significantly worse clinical outcomes in terms of median
progression-free survival (5.0 vs. 6.9 months, P<0.001) and
median overall survival (8.9 vs. 9.9 months, P=0.026) (46). In addition, Wang et al
reported that constitutively activated STAT3 in normal human
esophageal epithelium cells (HET-1A) promoted proliferation and
decreased apoptosis. Moreover, STAT3 activated HET-1A cells to form
tumors in vivo (47),
suggesting that aberrant STAT3 expression plays a crucial role in
esophageal carcinogenesis. Conversely, STAT3 knockdown
significantly reduced cell proliferation of OE21 (ESCC) and OE33
(EAC) cells, and a reduced STAT3 level was revealed to be
associated with significant downregulation of cell cycle genes in
both cell lines (48), indicating
that STAT3 plays a pivotal role in cell proliferation of esophageal
cancer cells. Through western blot analysis, it was revealed that
the STAT3 signaling pathway, but not the Wnt pathway, was
constitutively active in the three esophageal cancer cell lines. In
addition, niclosamide treatment did not only markedly suppress the
phosphorylation of Y705 of STAT3, but also slightly decreased the
total protein levels of STAT3. Similar results were also obtained
by Yoshikawa et al in a niclosamide-treated ovarian clear
cell line (KK) (49). However, the
mechanism resulting in the decrease of STAT3 total protein form by
niclosamide treatment is still unclear. The IC50 values
of niclosamide were 2.8, 11.3, and 5.1 µM for CE48T, CE81T and BE3
cells, respectively. Notably, ESCC CE81T cells were more resistant
than ESCC CE48T cells to niclosamide. Similar resistant responses
were revealed in a previous study in which CE81T cells were
resistant to photofrin-induced inhibition of EGFR as well as to
photofrin-mediated dark toxicity, compared with CE48T cells
(39). Since niclosamide is
minimally absorbed from the gastrointestinal tract, 2.5 µM
niclosamide was used, a concentration less than the IC50
value, to investigate the long-term growth inhibitory effect of
niclosamide in esophageal cancer cells by colony-forming assay.
Niclosamide completely inhibited colony formation in BE3 and CE48T
cells after 21 days of treatment, indicating that niclosamide may
be considered as a potential inhibitor of esophageal cancer cell
growth, as demonstrated in other types of cancer cells.
According to the results of western blot analysis of
PARP and the Annexin V/PI staining assay, it was demonstrated that
the inhibitory effect of niclosamide on cell growth in esophageal
adenocarcinoma cell line BE3 was caused by niclosamide-induced
apoptosis. By contrast, in esophageal squamous cell carcinoma cell
lines, niclosamide significantly (P<0.05) increased the
proportion of early apoptotic cells in both CE48T and CE81T cells,
however only significantly increased the proportion of dead cells
in CE48T (P<0.005), but not in CE81T cells. After 48 h of
treatment of niclosamide, only a small proportion of Annexin
V-positive cells were detected in CE81T, compared with CE48T and
BE3 cells. The detailed mechanism of the different response of
CE81T cells to niclosamide compared to CE48T and BE3 cells remains
unclear and further investigation is required.
Previous studies in oral squamous cell carcinoma
(26), adrenocortical carcinoma
(8) and head and neck cancer
(18) and osteosarcoma (25) revealed that niclosamide could induce
G1-phase arrest of the cell cycle. Through flow cytometric analysis
using DNA dye PI staining, it was revealed that a high proportion
of sub-G1 cells were detected in CE48T and BE3 cells after 10 µM
niclosamide treatment for 72 h, while only a small proportion of
sub-G1 was detected in CE81T cells. Moreover, an evident G1 peak
was revealed in CE81T cells. Similar experiments were also
performed using the physiological concentration of niclosamide (2.5
µM) and the results were consistent with the high dose treatment.
In addition, the protein levels of the various cyclins were
explored by western blot assay, and it was revealed that the
protein levels of cyclin D1, E, A and B1 almost completely
disappeared in CE81T cells after niclosamide treatment for 48 h. As
G1-phase arrest, cell cycle checkpoint proteins, such as p21 and
p27, may mediate assembly and activation of Cdk4(6)-cyclin D in the
cytoplasm, as well as inhibiting their activity in the nucleus.
Ectopic induction of p21 or p27 expression completely arrested
cells at the G1 phase of the cell cycle by p27 and at G1 and G2
phases by p21 (50). Therefore, in
the present study, it was explored whether p21, p27 or both were
involved in niclosamide-induced G1-phase arrest in CE81T cells. As
revealed, niclosamide after 24 h of treatment significantly
increased p21 mRNA expression in CE81T cells, but not
p27 mRNA. These findings demonstrated that niclosamide
induced G1-phase arrest in ESCC CE81T cells, however, the detailed
mechanism is not clear yet.
Niclosamide may be beneficial not only as a
monotherapy but also in combination therapy. Niclosamide has been
demonstrated to increase the sensitivity of cancer cells for
radiation therapy in lung cancer (51), nasopharyngeal cancer (24) and breast cancer (9). Furthermore, niclosamide was also
revealed to exert a synergistic effect with numerous cancer drugs
in human cancer cells. Compared with cancer drug treatment alone,
the combination treatment of niclosamide with bicalutamide,
cisplatin, paclitaxel, carboplatin, erlotinib, dasatinib, or
sorafenib exhibited an improved antineoplastic efficacy in prostate
(32), lung (52), cervical (14), ovarian (30), colon (53), chronic myeloid leukemia (22) and renal cancer (34), respectively. To date, the frontline
chemotherapeutic agents for advanced esophageal cancer cells are
5-FU, cisplatin, and paclitaxel. In the present study, the
antineoplastic efficacy of combination treatment of niclosamide
(2.5 µM) plus 5-FU, cisplatin, or paclitaxel was assessed in
esophageal cancer cells, and it was revealed that niclosamide
co-treatment selectively reduced the dosage of anticancer drugs for
obtaining the IC50. In CE48T and BE3 cells, the
IC50 values of 5-FU, cisplatin, or paclitaxel were
reduced in the combination groups with niclosamide, when compared
to the anticancer drug administered alone. In CE81T cells,
niclosamide combination treatment only reduced the IC50
values of 5-FU and paclitaxel as compared with the anticancer drug
treatments alone. The beneficial effects of niclosamide combination
on inhibition of cancer cell proliferation were further validated
in the cell apoptosis assays. These findings indicated that
niclosamide may be used as a repurposing drug to reduce the dosage
requirement of anticancer drug for achieving the IC50 in
esophageal cancer cells.
Sustained inhibition of tumor colony formation by
long-term niclosamide treatment with potentially achievable serum
concentration is indicative of a novel maintenance strategy for
esophageal cancers adjunctive to current curatively local
treatments or palliative chemotherapy with limited choices and
survival benefits but well-known side effects. Due to approved
indication of niclosamide as an antiparasitic, there is little
clinical data with regard to the adverse effects of prolonged
niclosamide treatment. Andrews et al reviewed associated
results in laboratory animals (including mice, rats and dogs) and
reported few cumulative or long-term toxicities across species.
They mentioned that there was no indication that a higher incidence
of tumors occurred. From all long-term experiments it is evident
that the closely-related substances niclosamide ethanolamine salt
and niclosamide are not high-risk substances (44). To date, at least three clinical
trials for niclosamide in prostate and colon cancers are
registered, due to cumulative encouraging laboratory results. There
is no reported early termination due to unexpected/unacceptable
adverse effects. It is anticipated that if the combination of
niclosamide and chemotherapeutic drugs was used clinically it would
offer two important advantages: A reduction in the dose requirement
of chemotherapeutic drugs, and amelioration of the side effects
caused by aggressive treatment with high doses of chemotherapy.
Supplementary Material
Supporting Data
Acknowledgements
Esophageal adenocarcinoma cell line (BE3) was
generously provided by Dr Chia-Jui Yen (Division of Hematology and
Oncology, Department of Internal Medicine, National Cheng Kung
University Hospital, College of Medicine, National Cheng Kung
University, Tainan, Taiwan) and esophageal squamous cell carcinoma
cell lines (CE48T and CE81T) were kindly provided from Dr Jang-Ming
Lee (Department of Surgery, National Taiwan University Hospital and
National Taiwan University College of Medicine, Taipei, Taiwan). We
thank the staff of the Second Core Lab, Department of Medical
Research, National Taiwan University Hospital for technical support
during the study.
Funding
The present study was financially supported partly
from NTUH grants i) NTUH 99S-1309, ii) NTUH 101-S1867, iii) 107-14,
and iv) 108-13, and partly from the National Science Council of the
Republic of China grants i) NSC 98-2314-B-002-091-MY3 and ii) NSC
101-2320-B-002-010-MY3). We also thank Dr Jau-Min Wong, Dr Mei-Lin
Wu and ‘Liver Disease Prevention & Treatment Research
Foundation, Taiwan’ for partial funding support.
Availability of data and materials
All data used during the current study are available
from the corresponding author on reasonable request.
Authors' contributions
MCL and BRL conceived and designed the experiments.
MCL and YJH performed the experiments. MCL, BRL and YKC collected
and analyzed the data, and interpreted the findings. MCL wrote the
manuscript. BRL and YKC edited the manuscript. All authors have
read and approved the final manuscript and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
There authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AML
|
acute myelogenous leukemia
|
|
AR
|
androgen receptor
|
|
EAC
|
esophageal adenocarcinoma
|
|
EGFR
|
epidermal growth factor receptor
|
|
ER
|
estrogen receptor
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
5-FU
|
5-fluorouracil
|
|
FDA
|
Food and Drug Administration
|
|
IC50
|
half maximal inhibitory
concentration
|
|
IL6
|
interleukin 6
|
|
LRP6
|
lipoprotein receptor-related protein
6
|
|
mTOR
|
mammalian target of rapamycin
|
|
MTS
|
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
|
|
NF-κB
|
nuclear factor-κB
|
|
PI
|
propidium iodide
|
|
PI3K
|
phosphoinositide 3 kinase
|
|
ROS
|
reactive oxygen species
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
WNT
|
wingless and int-1
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang K, Johnson A, Ali SM, Klempner SJ,
Bekaii-Saab T, Vacirca JL, Khaira D, Yelensky R, Chmielecki J,
Elvin JA, et al: Comprehensive genomic profiling of advanced
esophageal squamous cell carcinomas and esophageal adenocarcinomas
reveals similarities and differences. Oncologist. 20:1132–1139.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu B, Bo Y, Wang K, Liu Y, Tang X, Zhao
Y, Zhao E and Yuan L: Concurrent neoadjuvant chemoradiotherapy
could improve survival outcomes for patients with esophageal
cancer: A meta-analysis based on random clinical trials.
Oncotarget. 8:20410–20417. 2017.PubMed/NCBI
|
|
4
|
Bayat Mokhtari R, Homayouni TS, Baluch N,
Morgatskaya E, Kumar S, Das B and Yeger H: Combination therapy in
combating cancer. Oncotarget. 8:38022–38043. 2017.PubMed/NCBI
|
|
5
|
Yang X, Huang WT, Wu HY, He RQ, Ma J, Liu
AG and Chen G: Novel drug candidate for the treatment of several
softtissue sarcoma histologic subtypes: A computational method
using survivalassociated gene signatures for drug repurposing.
Oncol Rep. 41:2241–2253. 2019.PubMed/NCBI
|
|
6
|
Sun W, Sanderson PE and Zheng W: Drug
combination therapy increases successful drug repositioning. Drug
Discov Today. 21:1189–1195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan JX, Ding K and Wang CY: Niclosamide,
an old antihelminthic agent, demonstrates antitumor activity by
blocking multiple signaling pathways of cancer stem cells. Chin J
Cancer. 31:178–184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Satoh K, Zhang L, Zhang Y, Chelluri R,
Boufraqech M, Nilubol N, Patel D, Shen M and Kebebew E:
Identification of niclosamide as a novel anticancer agent for
adrenocortical carcinoma. Clin Cancer Res. 22:3458–3466. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim
H, Yin T, Fan S and Shen Q: Activation of STAT3 and Bcl-2 and
reduction of reactive oxygen species (ROS) promote radioresistance
in breast cancer and overcome of radioresistance with niclosamide.
Oncogene. 37:5292–5304. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ye T, Xiong Y, Yan Y, Xia Y, Song X, Liu
L, Li D, Wang N, Zhang L, Zhu Y, et al: The anthelmintic drug
niclosamide induces apoptosis, impairs metastasis and reduces
immunosuppressive cells in breast cancer model. PLoS One.
9:e858872014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang YC, Chao TK, Chang CC, Yo YT, Yu MH
and Lai HC: Drug screening identifies niclosamide as an inhibitor
of breast cancer stem-like cells. PLoS One. 8:e745382013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suliman MA, Zhang Z, Na H, Ribeiro AL,
Zhang Y, Niang B, Hamid AS, Zhang H, Xu L and Zuo Y: Niclosamide
inhibits colon cancer progression through downregulation of the
Notch pathway and upregulation of the tumor suppressor miR-200
family. Int J Mol Med. 38:776–784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Ren XR, Piao H, Zhao S, Osada T,
Premont RT, Mook RA Jr, Morse MA, Lyerly HK and Chen W:
Niclosamide-induced Wnt signaling inhibition in colorectal cancer
is mediated by autophagy. Biochem J. 476:535–546. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen L, Wang L, Shen H, Lin H and Li D:
Anthelminthic drug niclosamide sensitizes the responsiveness of
cervical cancer cells to paclitaxel via oxidative stress-mediated
mTOR inhibition. Biochem Biophys Res Commun. 484:416–421. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng B, Morales LD, Zhang Y, Mito S and
Tsin A: Niclosamide induces protein ubiquitination and inhibits
multiple pro-survival signaling pathways in the human glioblastoma
U-87 MG cell line. PLoS One. 12:e01843242017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang C, Zhou X, Xu H, Shi X, Zhao J, Yang
M, Zhang L, Jin X, Hu Y, Li X, et al: Niclosamide inhibits cell
growth and enhances drug sensitivity of hepatocellular carcinoma
cells via STAT3 signaling pathway. J Cancer. 9:4150–4155. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tomizawa M, Shinozaki F, Motoyoshi Y,
Sugiyama T, Yamamoto S, Sueishi M and Yoshida T: Niclosamide
suppresses Hepatoma cell proliferation via the Wnt pathway. Onco
Targets Ther. 6:1685–1693. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han Z, Li Q, Wang Y, Wang L, Li X, Ge N,
Wang Y and Guo C: Niclosamide induces cell cycle arrest in G1 phase
in head and neck squamous cell carcinoma through Let-7d/CDC34 axis.
Front Pharmacol. 9:15442019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li R, You S, Hu Z, Chen ZG, Sica GL, Khuri
FR, Curran WJ, Shin DM and Deng X: Inhibition of STAT3 by
niclosamide synergizes with erlotinib against head and neck cancer.
PLoS One. 8:e746702013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim MO, Choe MH, Yoon YN, Ahn J, Yoo M,
Jung KY, An S, Hwang SG, Oh JS and Kim JS: Antihelminthic drug
niclosamide inhibits CIP2A and reactivates tumor suppressor protein
phosphatase 2A in non-small cell lung cancer cells. Biochem
Pharmacol. 144:78–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK,
Sica GL, Ramalingam SS, Curran WJ, Khuri FR and Deng X: Niclosamide
overcomes acquired resistance to erlotinib through suppression of
STAT3 in non-small cell lung cancer. Mol Cancer Ther. 12:2200–2212.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Z, Li Y, Lv C, Wang L and Song H:
Anthelmintic drug niclosamide enhances the sensitivity of chronic
myeloid leukemia cells to dasatinib through inhibiting
Erk/Mnk1/eIF4E pathway. Biochem Biophys Res Commun. 478:893–899.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin Y, Lu Z, Ding K, Li J, Du X, Chen C,
Sun X, Wu Y, Zhou J and Pan J: Antineoplastic mechanisms of
niclosamide in acute myelogenous leukemia stem cells: Inactivation
of the NF-kappaB pathway and generation of reactive oxygen species.
Cancer Res. 70:2516–2527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Li H, Zhan D, Xiang M, Yang J, Zuo
Y, Yu Y, Zhou H, Jiang D, Luo H, et al: Niclosamide sensitizes
nasopharyngeal carcinoma to radiation by downregulating Ku70/80
expression. J Cancer. 9:736–744. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liao Z, Nan G, Yan Z, Zeng L, Deng Y, Ye
J, Zhang Z, Qiao M, Li R, Denduluri S, et al: The anthelmintic drug
niclosamide inhibits the proliferative activity of human
osteosarcoma cells by targeting multiple signal pathways. Curr
Cancer Drug Targets. 15:726–738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Ding R, Han Z, Ma Z and Wang Y:
Targeting of cell cycle and let-7a/STAT3 pathway by niclosamide
inhibits proliferation, migration and invasion in oral squamous
cell carcinoma cells. Biomed Pharmacother. 96:434–442. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li X, Yang Z, Han Z, Wen Y, Ma Z and Wang
Y: Niclosamide acts as a new inhibitor of vasculogenic mimicry in
oral cancer through upregulation of miR-124 and downregulation of
STAT3. Oncol Rep. 39:827–833. 2018.PubMed/NCBI
|
|
28
|
Arend RC, Londono-Joshi AI, Gangrade A,
Katre AA, Kurpad C, Li Y, Samant RS, Li PK, Landen CN, Yang ES, et
al: Niclosamide and its analogs are potent inhibitors of
Wnt/β-catenin, mTOR and STAT3 signaling in ovarian cancer.
Oncotarget. 7:86803–86815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yo YT, Lin YW, Wang YC, Balch C, Huang RL,
Chan MW, Sytwu HK, Chen CK, Chang CC, Nephew KP, et al: Growth
inhibition of ovarian tumor-initiating cells by niclosamide. Mol
Cancer Ther. 11:1703–1712. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arend RC, Londoño-Joshi AI, Samant RS, Li
Y, Conner M, Hidalgo B, Alvarez RD, Landen CN, Straughn JM and
Buchsbaum DJ: Inhibition of Wnt/β-catenin pathway by niclosamide: A
therapeutic target for ovarian cancer. Gynecol Oncol. 134:112–120.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu C, Lou W, Armstrong C, Zhu Y, Evans CP
and Gao AC: Niclosamide suppresses cell migration and invasion in
enzalutamide resistant prostate cancer cells via Stat3-AR axis
inhibition. Prostate. 75:1341–1353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu C, Armstrong CM, Lou W, Lombard AP,
Cucchiara V, Gu X, Yang JC, Nadiminty N, Pan CX, Evans CP and Gao
AC: Niclosamide and bicalutamide combination treatment overcomes
enzalutamide- and bicalutamide-resistant prostate cancer. Mol
Cancer Ther. 16:1521–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schweizer MT, Haugk K, McKiernan JS,
Gulati R, Cheng HH, Maes JL, Dumpit RF, Nelson PS, Montgomery B,
McCune JS, et al: A phase I study of niclosamide in combination
with enzalutamide in men with castration-resistant prostate cancer.
PLoS One. 13:e01983892018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu X, Liu F, Zeng L, He F, Zhang R, Yan S,
Zeng Z, Shu Y, Zhao C, Wu X, et al: Niclosamide exhibits potent
anticancer activity and synergizes with sorafenib in human renal
cell cancer cells. Cell Physiol Biochem. 47:957–971. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu K, Wang T, Li Y, Wang C, Wang X, Zhang
M, Xie Y, Li S, An Z and Ye T: Niclosamide induces apoptosis
through mitochondrial intrinsic pathway and inhibits migration and
invasion in human thyroid cancer in vitro. Biomed Pharmacother.
92:403–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
37
|
Furtek SL, Matheson CJ, Backos DS and
Reigan P: Evaluation of quantitative assays for the identification
of direct signal transducer and activator of transcription 3
(STAT3) inhibitors. Oncotarget. 7:77998–78008. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yen CJ, Izzo JG, Lee DF, Guha S, Wei Y, Wu
TT, Chen CT, Kuo HP, Hsu JM, Sun HL, et al: Bile acid exposure
up-regulates tuberous sclerosis complex 1/mammalian target of
rapamycin pathway in Barrett's-associated esophageal
adenocarcinoma. Cancer Res. 68:2632–2640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang PW, Hung MC, Hsieh CY, Tung EC, Wang
YH, Tsai JC and Lee JM: The effects of Photofrin-mediated
photodynamic therapy on the modulation of EGFR in esophageal
squamous cell carcinoma cells. Lasers Med Sci. 28:605–614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang C, Ma Q, Shi Y, Li X, Wang M, Wang
J, Ge J, Chen Z, Wang Z and Jiang H: A novel
5-fluorouracil-resistant human esophageal squamous cell carcinoma
cell line Eca-109/5-FU with significant drug resistance-related
characteristics. Oncol Rep. 37:2942–2954. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu J, Wang L, Du X, Sun Q, Wang Y, Li M,
Zang W, Liu K and Zhao G: α-solanine enhances the chemosensitivity
of esophageal cancer cells by inducing microRNA-138 expression.
Oncol Rep. 39:1163–1172. 2018.PubMed/NCBI
|
|
43
|
Zhang X, Wu X, Zhang F, Mo S, Lu Y, Wei W,
Chen X, Lan L, Lu B and Liu Y: Paclitaxel induces apoptosis of
esophageal squamous cell carcinoma cells by downregulating STAT3
phosphorylation at Ser727. Oncol Rep. 37:2237–2244. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Andrews P, Thyssen J and Lorke D: The
biology and toxicology of molluscicides, bayluscide. Pharmacol
Ther. 19:245–295. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wieland A, Trageser D, Gogolok S, Reinartz
R, Höfer H, Keller M, Leinhaas A, Schelle R, Normann S, Klaas L, et
al: Anticancer effects of niclosamide in human glioblastoma. Clin
Cancer Res. 19:4124–4136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang C, Wang L, Yang X, Lai L, Chen D and
Duan C: Expression of activated signal transducer and activator of
transcription-3 as a predictive and prognostic marker in advanced
esophageal squamous cell carcinoma. World J Surg Oncol. 13:3142015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Z, Zhu S, Shen M, Liu J, Wang M, Li
C, Wang Y, Deng A and Mei Q: STAT3 is involved in esophageal
carcinogenesis through regulation of Oct-1. Carcinogenesis.
34:678–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Timme S, Ihde S, Fichter CD, Waehle V,
Bogatyreva L, Atanasov K, Kohler I, Schöpflin A, Geddert H, Faller
G, et al: STAT3 expression, activity and functional consequences of
STAT3 inhibition in esophageal squamous cell carcinomas and
Barrett's adenocarcinomas. Oncogene. 33:3256–3266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yoshikawa T, Miyamoto M, Aoyama T, Soyama
H, Goto T, Hirata J, Suzuki A, Nagaoka I, Tsuda H, Furuya K and
Takano M: JAK2/STAT3 pathway as a therapeutic target in ovarian
cancers. Oncol Lett. 15:5772–5780. 2018.PubMed/NCBI
|
|
50
|
Yoon MK, Mitrea DM, Ou L and Kriwacki RW:
Cell cycle regulation by the intrinsically disordered proteins p21
and p27. Biochem Soc Trans. 40:981–988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xiang M, Chen Z, Yang D, Li H, Zuo Y, Li
J, Zhang W, Zhou H, Jiang D, Xu Z and Yu Z: Niclosamide enhances
the antitumor effects of radiation by inhibiting the
hypoxia-inducible factor-1α/vascular endothelial growth factor
signaling pathway in human lung cancer cells. Oncol Lett.
14:1933–1938. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zuo Y, Yang D, Yu Y, Xiang M, Li H, Yang
J, Li J, Jiang D, Zhou H, Xu Z and Yu Z: Niclosamide enhances the
cytotoxic effect of cisplatin in cisplatin-resistant human lung
cancer cells via suppression of lung resistance-related protein and
c-myc. Mol Med Rep. 17:3497–3502. 2018.PubMed/NCBI
|
|
53
|
Shi L, Zheng H, Hu W, Zhou B, Dai X, Zhang
Y, Liu Z, Wu X, Zhao C and Liang G: Niclosamide inhibition of STAT3
synergizes with erlotinib in human colon cancer. Onco Targets Ther.
10:1767–1776. 2017. View Article : Google Scholar : PubMed/NCBI
|