Introduction
Lung cancer is the one of the leading causes of
cancer mortality in the world, and non-small-cell lung cancer
(NSCLC) accounts for nearly 85% of all lung cancers (1). Gefitinib, a typical reversible EGF
receptor tyrosine kinase inhibitor (EGFR-TKI), has a marked
therapeutic effect in NSCLC patients with EGFR mutations, and has
been used widely as the first-choice treatment for NSCLC (2,3).
However, despite the excellent initial clinical responses, almost
all responding patients eventually develop different degrees of
resistance within one year (2–4).
Hence, reversal of the resistance to EGFR-TKIs is an urgent
requirement for lung cancer therapy.
In recent years, certain Chinese herbal medicines
have exhibited notable effects on drug resistance (5). Previous studies have found that
curcumin (6), scorpion venom
(7) and bufalin (8) could delay the emergence of EGFR-TKI
resistance or inhibit drug resistance via different signaling
pathways. Clinical trials have begun to utilize Chinese herbal
medicines combined with EGFR-TKIs to treat lung cancer.
Among the Chinese herbal medicines, triptolide (TP),
a purified diterpenoid from Tripterygium wilfordii Hook.f.
(TWHF), exhibits promising potential in reversing drug resistance
(9). Previous studies confirmed
that TP has many biological properties, including immunosuppressive
and anti-inflammatory effects (10). An increasing number of preclinical
studies have demonstrated that TP has strong antitumor activities.
As an adjuvant therapeutic agent, TP has been revealed to enhance
the effect of some anticancer agents at low doses, such as
hydroxycamptothecin (11) and
fluorouracil (12), and increase
the sensitivity of drug resistant cells to chemotherapeutics
(9,13,14),
rendering the combination superior to mono-therapy. However, the
molecular mechanisms by which TP induces inhibition of drug
resistance and sensitization are unclear. Previously, we used
high-sensitivity isobaric tags for a relative and absolute
quantitation technique and observed that TP treatment caused
abnormal expression of proteins involved in a variety of biological
processes. In particular, the increase in E-cadherin was
particularly pronounced (15).
E-cadherin is a core protein of epithelial-mesenchymal transition
(EMT) and is involved in cancer invasion and metastasis (16). E-cadherin is closely related to
molecular treatment targeting EGFR resistance and sensitivity.
Increased expression of E-cadherin enhanced the sensitivity of
tumor cells to the EGFR inhibitor gefitinib, while knockdown of
E-cadherin in parental cells induced gefitinib resistance and
stemness (17–19). Thus, it was speculated that
E-cadherin may participate in the development of sensitivity or
resistance to EGFR-TKIs, and play a role in the complex
intercellular regulation.
In the present study, it was revealed that TP
combined with gefitinib had a synergistic inhibitory effect on the
proliferation, migration, and invasion of A549 cells, which are
resistant to gefitinib. The effect of TP against gefitinib
resistance was attributed to its ability to reverse EMT by
upregulating E-cadherin levels and inhibiting cell proliferation.
In addition, this combinational therapy reduced the tumor volume
more effectively than gefitinib or TP alone in a xenograft mouse
model, and this synergistic interaction was associated with the
ability of TP to reverse EMT. Thus, evidence is provided that the
combination of TP and gefitinib could overcome TKI resistance in
patients with NSCLC with EGFR mutations and could lead to the
development of new combinatorial therapies for lung cancer.
Materials and methods
Chemicals
TP was purchased from Sigma-Aldrich; Merck KGaA. The
molecular formula of TP is
C20H24O6, it has a molecular
weight of 360.4 Da, and a purity≥98%. Gefitinib was also purchased
from Sigma-Aldrich; Merck KGaA. The molecular formula of gefitinib
is C22H24ClFN4O3, it
has a molecular weight of 446.90 Da and purity≥98%. Both TP and
gefitinib were stored in dimethyl sulfoxide (DMSO) at 100 µg/ml at
−80°C and diluted to the indicated concentrations using serum-free
culture medium.
Cell line and culture
Human lung cancer A549 (American Type Culture
Collection; ATCC® CCL185™) cells were purchased from
Meixuan Biological Science Co., Ltd. (identification number
MXC026). The cells were maintained in 90% Dulbecco's modified
Eagle's medium (DMEM, Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), L-glutamine (2 mM), 1% penicillin-streptomycin
(100 U/ml penicillin and 100 µg/ml streptomycin), and HEPES (25 mM)
according to the supplier's instruction manual. Cells were
incubated in a humid incubator containing 5% CO2 at
37°C.
The gefitinib-resistant human lung adenocarcinoma
cell line (A549/G) was established by increasing the gefitinib
concentration. Briefly, A549 cells with 80% fusion were first
treated with 5 µg/ml gefitinib for 24 h. The surviving cells were
cultured for further cycles with increased doses of gefitinib (10
µg/ml and 15 µg/ml), until the A549 cells grew steadily in the
medium with 15 µg/ml gefitinib. The resistance index of A549/G was
detected using a Cell Counting Kit-8 assay (Beyotime Institute of
Biotechnology), and calculated according to the equation of the
half maximal inhibitory concentration: IC50
(A549/G)/IC50 (A549), which provided a resistance index
of 5.48.
Another gefitinib-resistant human lung
adenocarcinoma cell line (A549/siE-cad) was established using
lentivirus-mediated small hairpin RNA (shRNA) to attenuate
E-cadherin (CDH1) expression in the A549 cell line. The shRNA
targeting CDH1 was cloned into the pLV-shRNA-EGFP (2A) Puro
vector. The shRNA sequence for CDH1 was as follows:
GGATCC-GCCCACAGATCCATTTCTTGCTCGAGCAAGAAATGGATCTGTGGGTTTTT-GAATTC.
The generated vectors were confirmed by DNA sequencing.
Transfection
The 293T cells were cultured in 10-cm dishes at a
density of 4.0×106. Then, 500 µl of opti-MEM containing
5 µg of lentiviral interference vector targeting E-cadherin, 3.75
µg of pH1 vector, and 1.25 µg of pH2 vector was added into 500 µl
of opti-MEM including 20 µl of Lipofectamine 2000, the mixture was
incubated at room temperature for 20 min and added dropwise to the
293T cells. The medium was replaced with serum-free DMEM at 5 h
post-transfection. The lentiviral particles were harvested by
ultracentrifugation at 82,700 × g for 48 h after transfection.
Cells were infected with the harvested lentiviruses (siE-cad) at a
multiplicity of infection (MOI) of 20. The optimal infection
condition was confirmed by observing the cells expressing the green
fluorescent protein at 72 h after infection. Puromycin (2 µg/ml)
was used to screen for stably infected cells. The resistance index
of cells with stably interfered CDH1 (A549/siE-cad) was
detected by Cell Counting Kit-8, and calculated, according to the
equation: IC50 (A549/ siE-cad)/IC50 (A549),
providing a resistance index of 2.95.
Mouse model
Male BALB/c mice (4 weeks old) were obtained from
Shanghai SLAC Laboratory Animal Co., Ltd. [SCXK (HU) 2017-0005].
The body weight ranged from 18 to 22 g. All mice were fed with
standard mouse food and tap water and maintained under conditions
of 25°C with a 12-hour light/dark cycle. The A549/siE-cad cells
(2×106) were injected subcutaneously into the underarms
of the right forelimbs of 6-week-old BCLB/c mice. After 3 weeks,
all mice were randomly divided into four groups of five. These mice
were administered with drugs or saline (mock) via abdominal
administration every day for 4 weeks as follows: The control group
was administered with NaCl [0.01 ml/g body weight (BW)], the
TP-treated group was administered with TP (0.5 mg/kg BW), the
gefitinib-treated group (G) with gefitinib (50 mg/kg BW), the TP+G
group with TP (0.5 mg/kg BW) and gefitinib (50 mg/kg BW). The body
weight and tumor size were recorded every two days; the tumor
volume was calculated as: A × b × b/2 (mm3) (a indicated
the long diameter, and b the short diameter). Subsequently
euthanasia was performed in accordance with the Guidelines for
Euthanasia of Rodents Using Carbon Dioxide issued by the National
Institutes of Health. At the end of 4 weeks, all animals were
euthanized by carbon dioxide in their home cage placed in special
transparent chambers. The chambers were connected with compressed
carbon dioxide in a gas cylinder and flow controller. The gas
replacement rate was 28% of container volume/min. Death was further
verified by cardiac arrest and cadaveric rigidity. Their tumors
were collected, paraffin-embedded, and then serially sectioned for
subsequent hematoxylin staining and immunohistochemistry (IHC).
Cell Counting Kit-8 assays
(CCK-8)
The viability of cells treated with different drugs
was detected using CCK-8 assay (Beyotime Institute of
Biotechnology). The A549 cells were exposed to various
concentrations of TP (0,1,2,4,8,16 and 32 ng/ml) and gefitinib (0,
0.627, 1.25, 2.5, 5, 10 and 20 µg/ml), alone or in combination, for
24, 36 and 48 h, respectively, as indicated. Cell viability was
evaluated according to manufacturer's instructions. The combination
index (CI), resistance index (RI), and IC50 values were
calculated based on the data from the cell viability assays. The
optimal combined concentration of TP and gefitinib was determined
by CI<1, which indicated that the effect of two drugs was
synergistic. The RI equation was as follows: IC50
(A549/G)/IC50 (A549), or IC50
(A549/siE-cad)/IC50 (A549).
Cell migration and invasion
Cell migration and invasion were determined using a
Transwell system (product no. 3422; Corning, Inc.) with 8.0-µm
diameter pores. For the invasion assays, 45 µl diluted Matrigel was
pre-coated on the Transwell membranes. A total of 5×104
cells were seeded into the upper chamber, and 20% FBS-containing
medium was seeded into the lower chamber to induce cell migration
and invasion. After 12 or 48 h of incubation (12 h for the
migration assays and 48 h for the invasion assays), the cells on
the upper surface of the filter were removed, and then the residual
cells in the Transwell chambers were fixed with 4% paraformaldehyde
at 4°C for 10 min, stained with crystal violet (0.1%, w/v) at room
temperature for 5 min, and photographed under a microscope (Olympus
Corp.). Five fields were selected at random, and the number of
cells was counted in each field. Experiments were performed three
times independently.
Western blot analysis
The prepared cells were harvested to extract
proteins by enhanced RIPA lysis buffer (P0013B; Beyotime Institute
of Biotechnology), and its components included 50 mM Tris (pH 7.4),
150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS,
sodium orthovanadate, sodium fluoride, EDTA, leupeptin and protease
inhibitors. The concentration was determined using a BCA protein
assay kit (735094; Roche Diagnostics). The proteins (20 µg) were
separated using 8% SDS-PAGE and then transferred to a
nitrocellulose membrane (Bio-Rad Laboratories, Inc.). The membrane
was then incubated with the target primary antibodies E-cadherin
(dilution 1:1,000; product code Ab4077; Abcam) MMP9 (dilution
1:1,000; product code Ab38898; Abcam), GAPDH (dilution 1:5,000;
product code Ab8245; Abcam), caspase-3 (dilution 1:1,000; product
no. 9662; Cell Signaling Technology), vimentin (dilution 1:1,000;
product no. 3932; Cell Signaling Technology), Snail (dilution
1:1,000; product no. 3879; Cell Signaling Technology) and cleaved
caspase-3 (dilution 1:1,000; product no. 9654; Cell Signaling
Technology) at 4°C overnight. After washing three times with
Tris-buffered saline Tween-20 buffer, the membrane was incubated
with secondary antibodies: Anti-rabbit IgG HRP-linked (dilution
1:2,000; product no. 7074; Cell Signaling Technology, Inc.; for
Snail, vimentin, caspase-3 and cleaved caspase-3); goat anti-rabbit
IgG H&L (HRP) (dilution 1:6,000; product code Ab205718; Abcam;
for MMP-9 and E-cadherin); goat anti-mouse IgG H&L (HRP)
(dilution 1:6,000; product code Ab205719; Abcam; for GAPDH) at room
temperature for 2 h. Immune complexes were visualized using an
enhanced chemiluminescence detection system (EMD Millipore).
Hematoxylin-eosin (H&E)
staining
The collected tumor slides were dewaxed and
rehydrated. H&E staining was performed and then the samples
were observed under a light microscope to assess their
morphology.
Terminal deoxynulceotidyl transferase
nick-end-labeling (TUNEL)
A TUNEL kit was used (product no. 11684817910; Roche
Diagnostics) as described by the manufacturer. The collected tumor
slides were dewaxed and rehydrated. Endogenous peroxidase activity
was blocked using 3% hydrogen peroxide for 5 min. The samples were
then washed with phosphate-buffered saline (PBS) at room
temperature and incubated in the TUNEL Reaction Mixture at 37°C.
Thereafter, incubation with converter-POD solution was carried out
at 37°C. Next, the slides were incubated with diaminobenzydine
(DAB) and stained with hematoxylin. Samples were dehydrated using
graded ethanol, vitrified with dimethylbenzene and deposited in
neutral resins. Finally, the samples were observed under a light
microscope.
Immunohistochemistry
The collected tumor slides were first dewaxed and
then rehydrated. The slides were incubated in citrate buffer (0.01
mol/l, pH 6.0) for antigen retrieval. Endogenous peroxidase
activity was inhibited by using 0.3% hydrogen peroxide for 15 min.
Primary antibodies against E-cadherin (product code ab40772;
Abcam), matrix metalloproteinase-9 (MMP9) (product code ab38898;
Abcam), and caspase-3 (product number 9664; Cell Signaling
Technology, Inc.) were incubated with the slides at 4°C overnight
in a humidity chamber. After five washes with PBS, the slides were
incubated with secondary antibodies: Goat anti-rabbit IgG H&L,
HRP-linked antibody (dilution 1:5,000; product code Ab205718;
Abcam; for E-cadherin and MMP9); Signal Stain Bosst IHC detection
reagent HRP-linked antibody rabbit (dilution 1:2,000; product no.
8114; for caspase-3) at room temperature for 30 min. The tumor
samples on the slides were stained with hematoxylin, dehydrated by
graded ethanol, vitrified by dimethylbenzene, and deposited in
neutral resins. Finally, the samples were observed and quantified
under a light microscope.
Statistical analysis
The results are expressed as the means ± SD. One or
two-way ANOVA followed by Tukey's, Dunnett's and Sidak's multiple
comparisons test in GraphPad Prism 6 (GraphPad Software) were used
to analyze the statistical significance between two groups. A
P-value<0.05 was considered to indicate a statistically
significant difference.
Results
Combined treatment with TP and G
induces enhanced inhibition of A549 cell viability
First, the effect of TP or G alone was assessed on
the A549 cells. As revealed in Fig.
1A, both TP and G induced a marked dose-dependent inhibition of
A549 cell viability, and the combined treatment with TP and G
induced a significantly increased inhibition of A549 cell viability
compared to TP or G alone. The inhibitory effect of TP and G alone
gradually increased with prolonged exposure time (Fig. 1B and C). The 50% growth inhibition
(IC50) at 48 h for TP and G was 10.985 ng/ml and 5.801
µg/ml respectively. Next, the combined effect of TP and G were
evaluated, and the data revealed similar dose- and time-dependent
inhibitory effects on the A549 cells, with an IC50 for
TP 4.228 ng/ml, and an IC50 for G of 2.644 µg/ml at 48 h
(Fig. 1D). These results indicated
that TP had a similar inhibitory effect to G on the proliferation
of A549 cells, and the combined treatment comprising TP and G more
effectively inhibited A549 cell growth than TP or G alone.
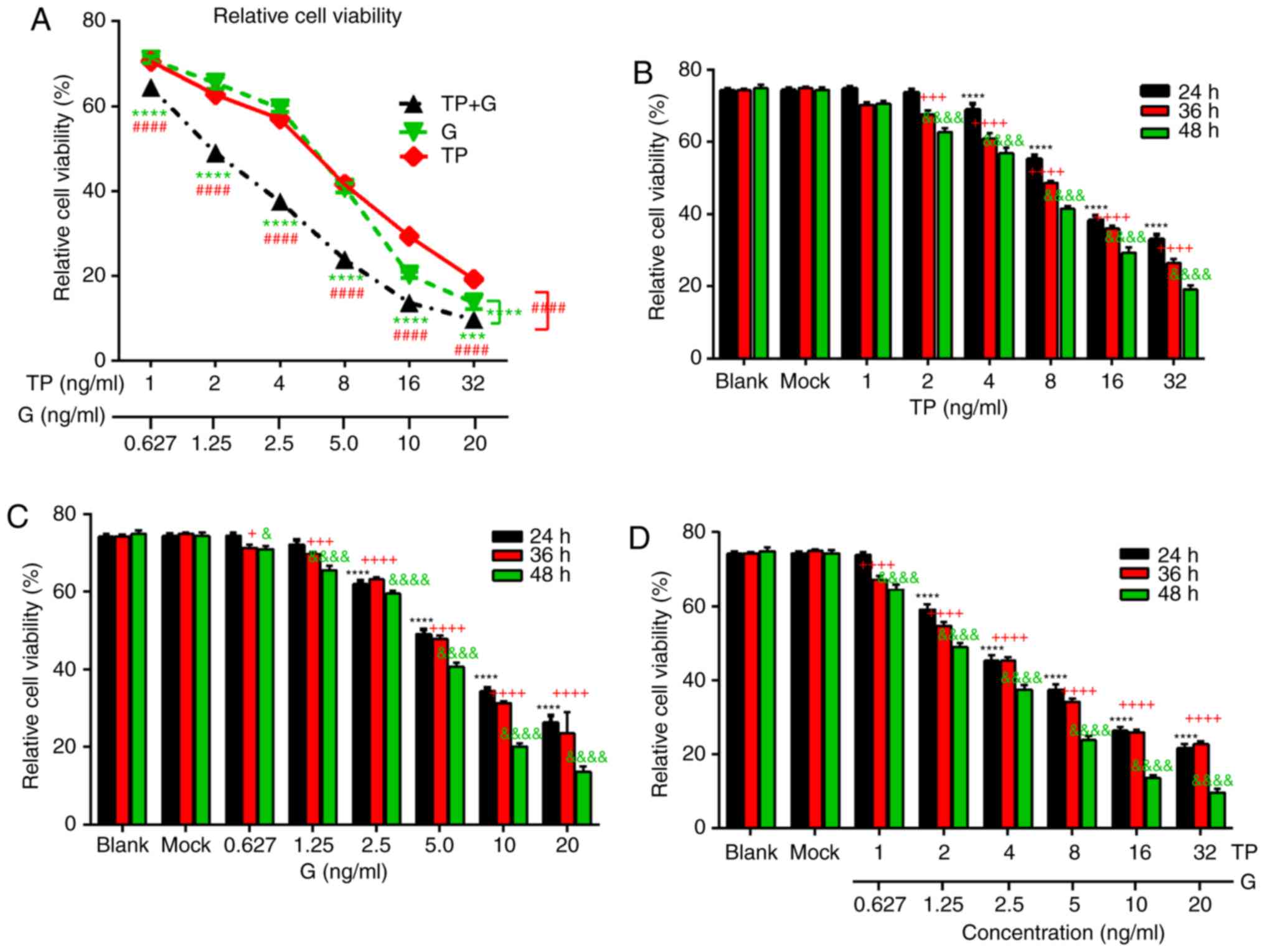 | Figure 1.Effect of TP and G, alone or in
combination, on A549 cell viability. (A) A549 cells were treated
with the indicated concentrations of TP (1, 2, 4, 8, 16, and 32
ng/ml) and G (0.627, 1.25, 2.5, 5, 10, and 20 µg/ml) alone or in
combination for 24 h, Green ‘*’ indicates statistical differences
between ‘TP+G’ and ‘G’, and the red ‘#’ represents statistical
differences between ‘TP+G’ and TP. (B) The individual effects of TP
on A549 cell growth at 24, 36, and 48 h. A549 cells were treated
with graded concentrations of TP (1, 2, 4, 8, 16, and 32 ng/ml,
respectively). Statistical differences between mock and treated
groups at 24, 36 and 48 h were respectively indicated with black
‘*’, red ‘+’ and green ‘&’. (C) The individual effects of G on
A549 cell growth at 24, 36, and 48 h. A549 cells were treated with
graded concentrations of G (0.627, 1.25, 2.5, 5, 10, and 20 µg/ml,
respectively). Statistical differences between mock and treated
groups at 24, 36 and 48 h were respectively indicated with black
‘*’, red ‘+’ and green ‘&’. (D) The combined effects of TP and
G on A549 cell growth at 24, 36, and 48 h. A549 cells were treated
with the combined concentrations of TP (1, 2, 4, 8, 16, and 32
ng/ml, respectively) and G (0.627, 1.25, 2.5, 5, 10, and 20 µg/ml,
respectively). Statistical differences between mock and treated
groups at 24, 36 and 48 h were respectively indicated with black
‘*’, red ‘+’ and green ‘&’. Two-way ANOVA followed by Tukey's
and Dunnett's multiple comparisons test were used to analyze
differences between groups. TP, triptolide; G, gefitinib. |
Then, the combination index (CI) was calculated to
estimate the synergistic effect of TP and G on A549 cells,
according to the method reported by Chou and Talalay (20), in which the synergism is defined by
CI<1. The CI values were 1.01 and 1.06 when A549 cells were
treated with TP and G for 24 and 36 h, respectively, while it was
0.84 when the A549 cells were exposed for 48 h (Table I). This demonstrated an evident
synergistic effect of TP and G on A549 cells. Based on this
evidence, the effect of TP and G on cell behavior was next
assessed. During the subsequent experiments, the combination of TP
(2 ng/ml) and G (1.25 µg/ml) were used to treat the A549 cells,
which exhibited the synergistic effect of inhibiting the growth of
A549 lung adenocarcinoma cells.
 | Table I.CI and IC50 analysis for
TP and G combination treatment in A549 cells. |
Table I.
CI and IC50 analysis for
TP and G combination treatment in A549 cells.
|
|
IC50 |
|
|---|
|
|
|
|
|---|
|
|
|
| TP+G |
|
|---|
|
|
|
|
|
|
|---|
| Exposure time
(h) | TP (ng/ml) | G (µg/ml) | TP (ng/ml) | G (µg/ml) | CI |
|---|
| 24 | 21.35 | 10.10 | 9.27 | 5.80 | 1.01 |
| 36 | 16.05 |
8.87 | 7.99 | 4.99 | 1.06 |
| 48 | 10.99 |
5.80 | 4.23 | 2.64 | 0.84 |
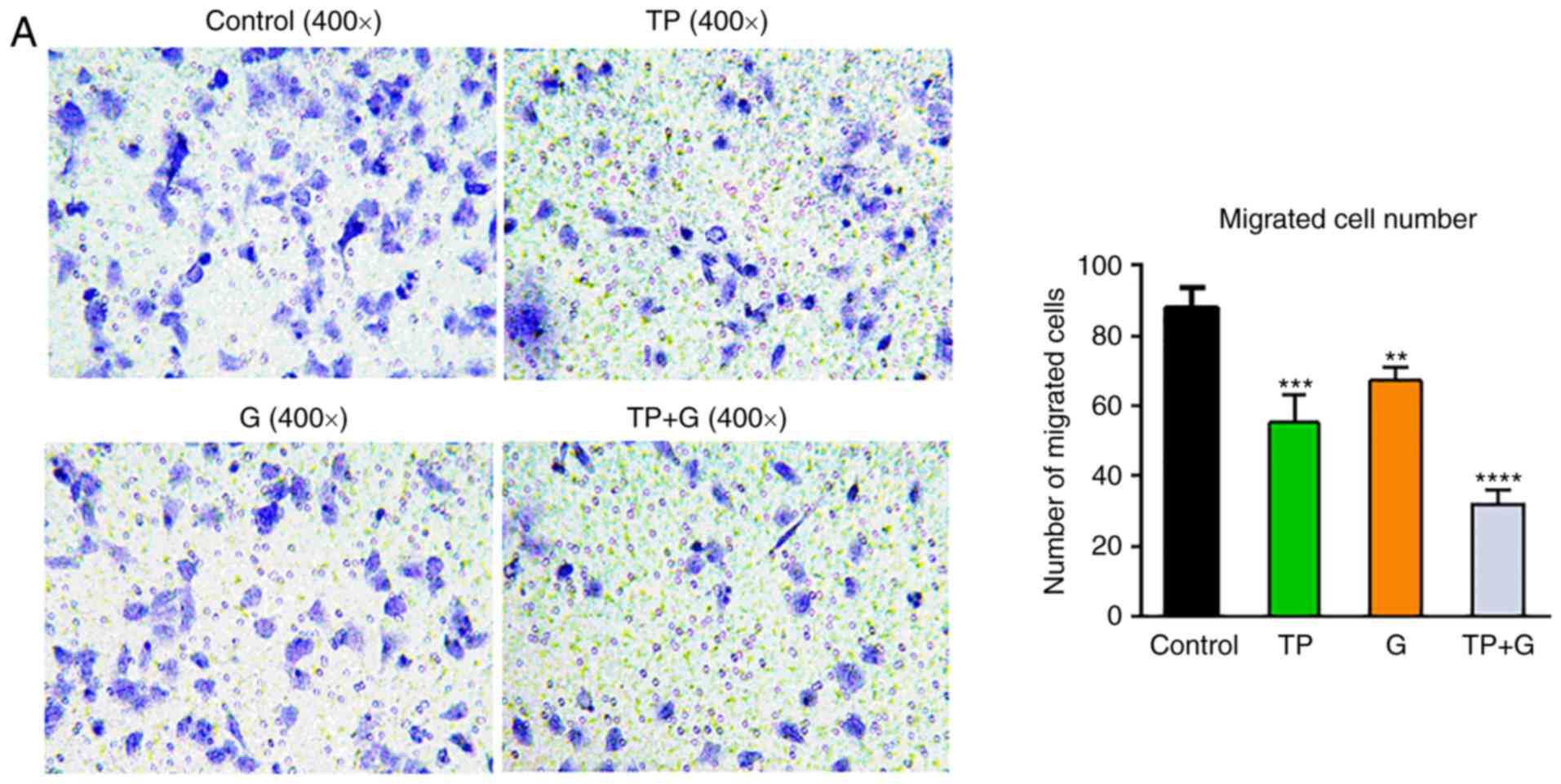
TP and G synergistically suppress cell
migration of A549 cells
Resistance to gefitinib results in cell metastasis;
therefore, the individual or synergistic effect of TP and G on the
migration of A549 cells was investigated using Transwell invasion
assays. The results in Fig. 2
revealed that treatment with TP and G alone, or in combination,
significantly decreased the number of migrated cells (P<0.01).
The number of migrated cells in the TP+G group decreased by nearly
60% (P<0.01), which was significantly lower than that in the TP-
or G-treated cells alone. These data indicated that TP played a
suppressive role with G on the migration of A549 cells, and that TP
combined with G induced a robust decrease in cell migration.
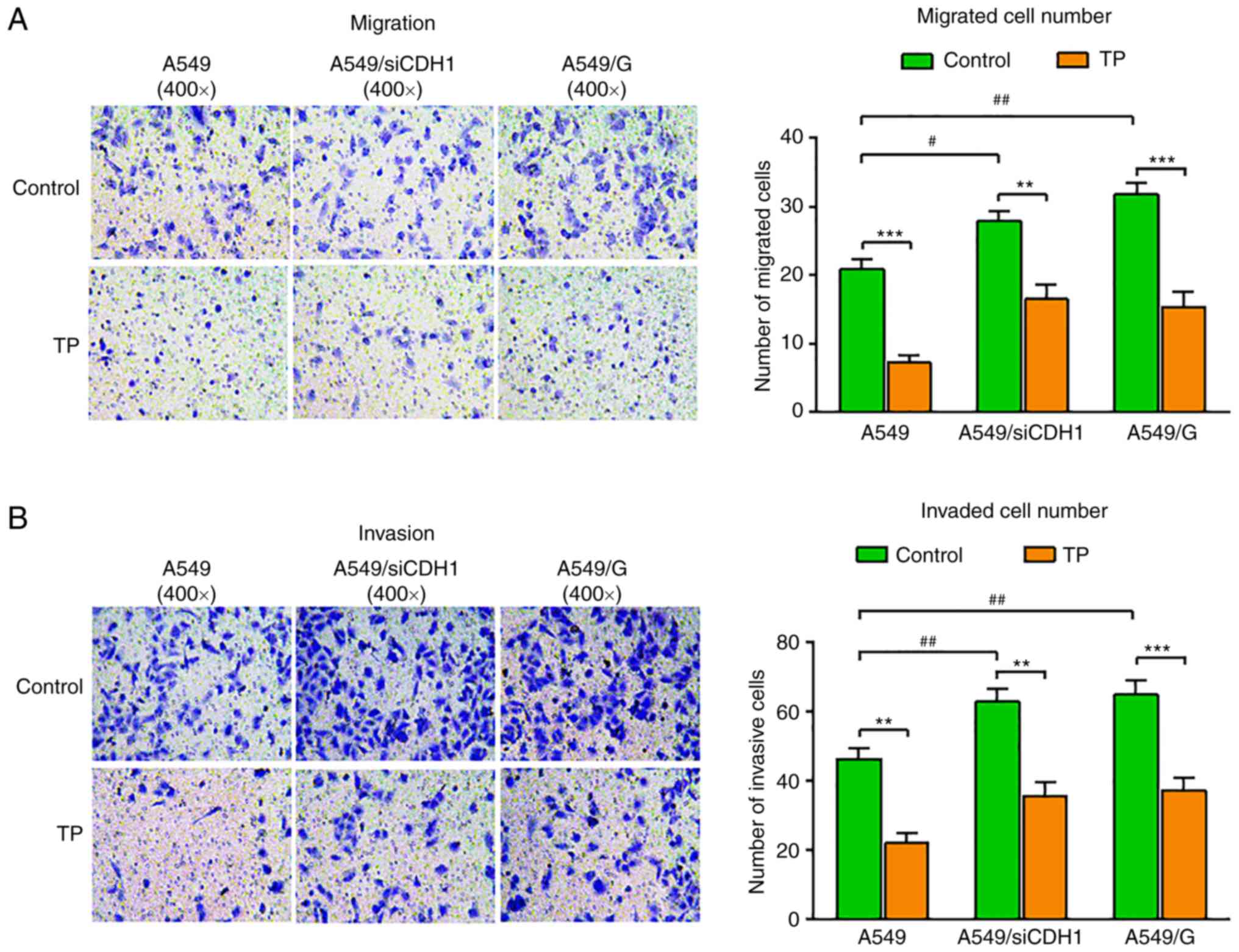
TP suppresses the migration and
invasion of gefitinib-resistant A549 cells
To demonstrate the role of TP in gefitinib
resistance, a gefitinib-resistant A549 cell line (A549/G) was first
established. Deficiency of E-cadherin is implicated in EMT,
invasion, and metastasis; therefore, an A549 cell line exhibiting
stable interference of E-cadherin expression was also established
(A549/siE-cad). Next, the A549/G, A549/siE-cad and control cells
were plated in Transwell devices and treated with 2 ng/ml of TP.
The results revealed that E-cadherin interference and gefitinib
resistance both led to increased cell migration across the
Transwell filters; however, TP markedly suppressed the migration of
A549, A549/siE-cad, and A549/G cells (Fig. 3A). The cell invasion assay results
revealed that the number of invading A549/siE-cad and A549/G cells
was markedly higher than that of the control cells. TP treatment
significantly impaired cell invasion of the control, A549/siE-cad,
and A549/G cells (Fig. 3B). These
results indicated that TP could inhibit cell migration and
invasion, and reverse the gefitinib resistance phenotype of
A549/siE-cad cells and A549/G cells.
TP reverses the gefitinib resistance
of A549 cells by regulating E-cadherin and MMP9
To determine the underlying molecular mechanism,
proteins implicated in EMT were detected using western blotting.
The levels of E-cadherin were significantly increased in the
drug-treated cells. The E-cadherin expression in the TP+G-treated
cells was nearly twice that in the control cells (Fig. 4A). In addition, the matrix
metalloproteinase-9 (MMP9), Snail family transcriptional repressor
1 (SNAIL) and vimentin protein levels significantly decreased in
the TP- and TP+G-treated cells compared with those in the control,
and the G treatment induced a modest reduction of MMP9, SNAIL and
vimentin. These results indicated that TP significantly upregulated
E-cadherin and downregulated MMP9, SNAIL and vimentin protein
expression, and treatment with TP+G had a synergistic effect on the
levels of E-cadherin.
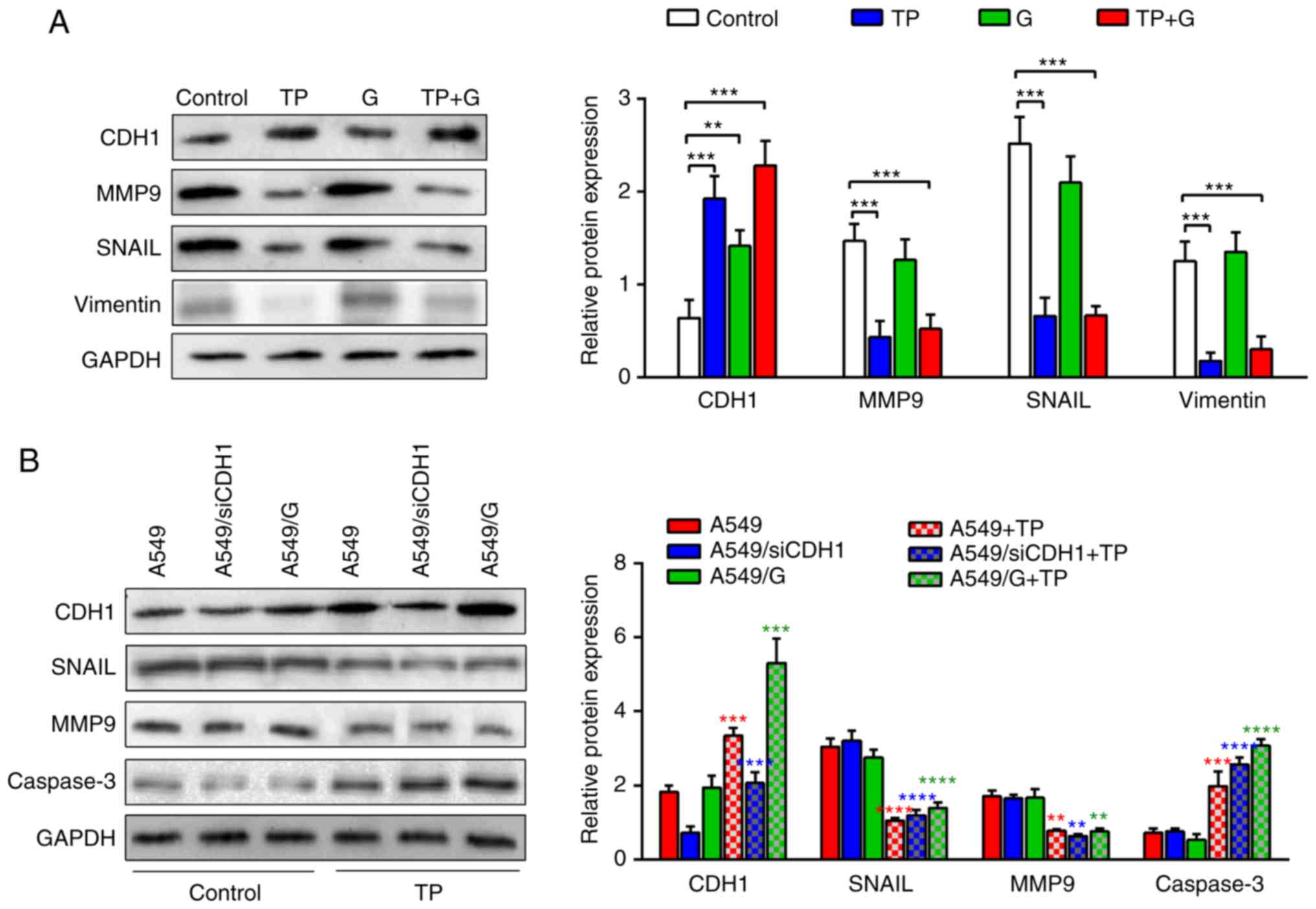 | Figure 4.TP regulates CDH1, MMP9, and
caspase-3 protein expression. (A) CDH1, MMP9, SNAIL and vimentin
proteins in A549 cells treated with TP (2 ng/ml) and G (1.25 µg/ml)
and their combination were analyzed using western blot analysis.
The data for CDH1, MMP9, SNAIL and vimentin protein levels are
expressed as the mean ± SD. Two-way ANOVA and Dunnett's test were
used to analyze the statistical differences between the control and
drug-treated cells. Statistical differences between the control and
treated groups were represented by black ‘*’. (B) CDH1, MMP9, SNAIL
and caspase-3 protein levels in A549, A549/siE-cad and A549/G cells
treated with TP (2 ng/ml) were detected by western blotting
analysis. The data of protein levels are expressed as the mean ±
SD. GAPDH was used as a loading control. The statistical
differences between groups were analyzed by two-way ANOVA combined
with Tukey's multiple comparisons test. Statistical differences
between control A549 and TP-treated A549 were represented by red
‘*’, statistical differences between A549/siE-cad and TP-treated
A549/siE-cad were represented blue ‘*’, statistical differences
between A549/G and TP-treated A549/G were represented green ‘*’.
TP, triptolide; CDH1, E-cadherin; MMP9, matrix metalloproteinase-9;
G, gefitinib. |
To determine the role of E-cadherin in TP-treated
cells, the E-cadherin levels in A549, A549/siE-cad, and A549/G
cells were detected. The results revealed that the E-cadherin level
decreased significantly in the A549/siE-cad cells compared with
that in the A549 cells and A549/G cells. TP treatment increased the
E-cadherin in all three groups, especially in A549 cells (Fig. 4B). The protein expression of MMP9
and SNAIL in A549, A549/siE-cad and A549/G cells significantly
decreased after TP treatment compared with those in their
respective controls. Concurrent, TP treatment significantly
increased caspase-3 in A549, A549/siE-cad and A549/G cells. These
results indicated that TP suppressed cell migration and invasion of
A549, A549/siE-cad, and A549/G cells by regulating the E-cadherin
and MMP9 signaling pathway, and induced apoptosis of these cells by
increasing the levels of caspase-3.
Combination of TP and G
synergistically inhibits A549/siE-cad cell xenografts by inducing
apoptosis
To further verify the effect of TP on A549 cell
tumor induction, an A549/siE-cad tumor-bearing mouse model was
produced, and TP, G, and TP/G were administrated to these mice. The
results revealed that there were no significant differences in body
weight (BW) among the TP, G and TP+G-treated groups. The BW of the
three treated groups was slightly heavier than that of the control
group, while there was no statistical difference between the
treated and control groups (Fig.
5A). With regards to the tumor volume, TP, G and TP/G treatment
significantly reduced the tumor volume in a time-dependent manner
(Fig. 5B). The tumor volume of the
TP+G group was significantly smaller than that of TP, G and control
groups. These results indicated that both TP and G have inhibitory
effects on tumorigenesis of A549/siE-cad tumor-bearing mice in
vivo, and that the inhibitory effect of TP+G treatment was
greater than that of either T or G alone.
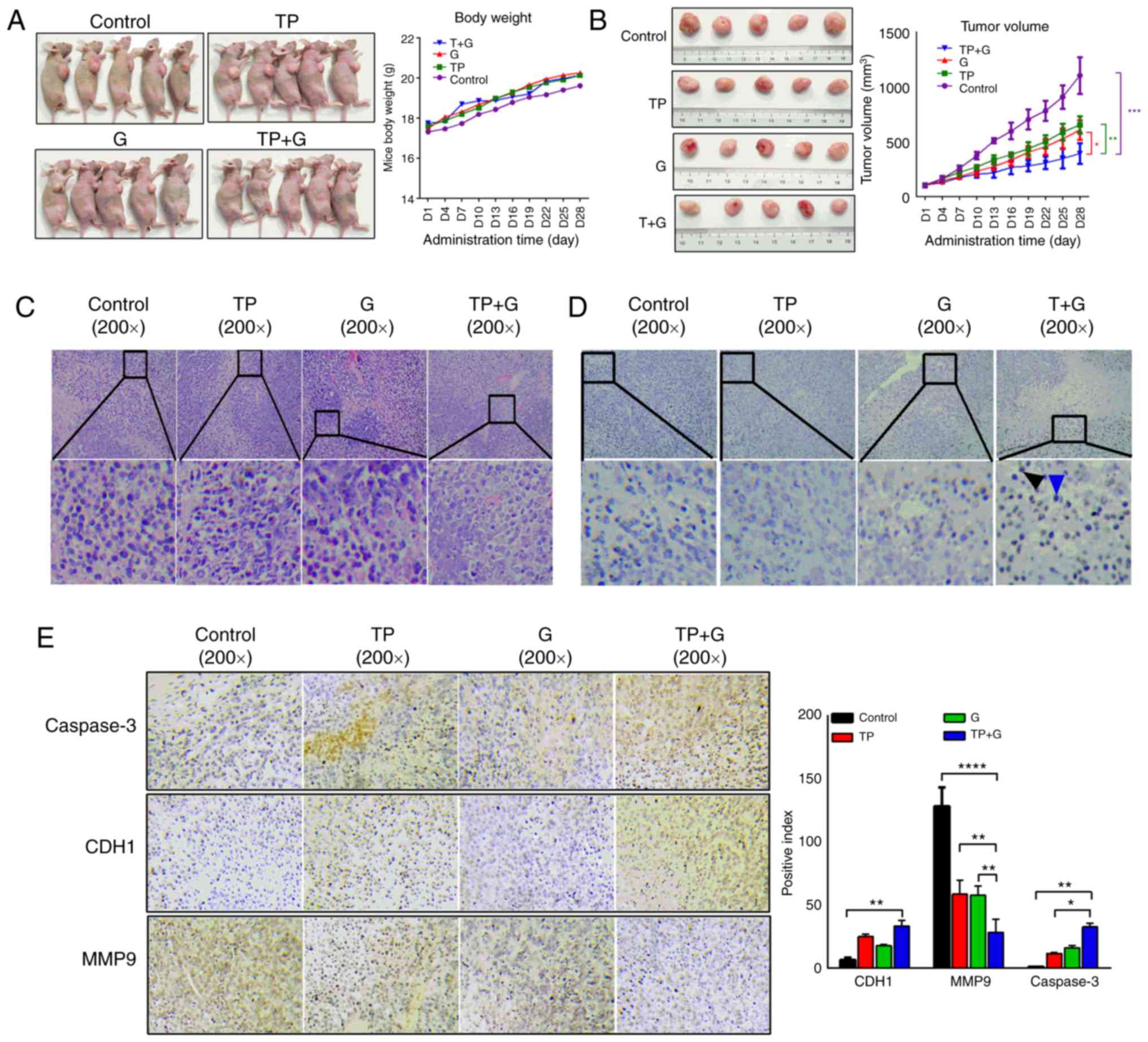 | Figure 5.TP and G combination reveals an
enhanced inhibition of A549/siE-cad-induced tumors. (A) Mice
bearing A549/siE-cad tumors were divided into four groups of five
mice each: The control group [NaCl, 0.1 ml/10 g body weight (BW)],
the TP-treated group (TP, 0.5 mg/kg BW), the G-treated group (G, 50
mg/kg BW) and the TP+G-treated group (TP, 0.5 mg/kg BW; G, 50 mg/kg
BW). Each group was treated with NaCl, TP, G, and TP+G,
respectively once a day for 4 weeks. The body weight was recorded
every three days. (B) The tumor volume was recorded every two days,
TP and G, alone or in combination, inhibited the tumor volume at
the end of the experiment. Statistical difference between TP+G and
control was represented purple “*”, statistical difference between
TP+G and TP was represented green “*”, statistical difference
between TP+G and G was represented red “*”. (C) Representative
images of H&E staining were obtained for the control, TP, G and
TP+G groups. (D) TUNEL assays were performed to detect the
apoptosis of tumors in the control, TP, G and TP+G groups. The
number of apoptotic cells from the control, TP, G and TP+G groups
is expressed as the mean ± SD (n=3). (E) The representative images
of IHC staining for CDH1, MMP9 and caspase-3 from the control, TP,
G and TP+G groups. The positive index of CDH1, MMP9, and caspase3
are showed as the mean ± SD (n=3). The statistical differences
between groups were analyzed by two-way ANOVA combined with Tukey's
multiple comparisons test. TP, triptolide; G, gefitinib; CDH1,
E-cadherin; H&E, hematoxylin and eosin; TUNEL, terminal
deoxynulceotidyl transferase nick-end-labeling; IHC,
immunohistochemistry. |
Next, H&E analysis was performed to investigate
the pathological changes in the tumors of each group. Treatment
with T or G alone induced nuclear aberration, cell degradation, and
necrosis, and treatment with T+G enhanced these effects (Fig. 5C). TUNEL staining revealed that
treatment with T or G alone induced significant tumor cell
apoptosis, and this apoptosis was enhanced by treatment with the
TP+G combination (Fig. 5D).
Combination of TP and G induces an
apoptotic pathway in gefitinib-resistant A549 cells that induces
tumors by regulating MMP9 and caspase-3
IHC assays were conducted to investigate the
signaling pathways involved in TP-induced inhibition of gefitinib
resistance in A549 cells during tumor induction. As revealed in
Fig. 5E, the protein level of
E-cadherin was significantly increased after treatment with TP+G,
it was also increased in the TP- and G-treated tumor groups, while
the statistical difference between the control and monotherapy, or
monotherapy and combination therapy was not significant. The
protein level of MMP9 was significantly decreased after TP, G and
TP+G treatment, and the inhibition in TP+G was greater than that in
TP or G alone. In addition, caspase-3 protein expression increased
in the TP-, G- and TP+G-treated tumors, and the increase in the
TP+G group was higher than that in the TP or G treatment groups.
These results indicated that both TP and G could upregulate
E-cadherin and caspase-3 expression levels, and downregulate MMP9
protein levels in A549/siE-cad tumor-bearing mice in vivo,
however, treatment with TP+G exhibited a more potent effect.
Discussion
Currently, molecular targeted therapy is the
preferred treatment for NSCLC. Gefitinib is a typical
representative of molecular targeted drugs for lung cancer and
belongs to the EGFR-TKI class of drugs (21). However, nearly all patients treated
with gefitinib deteriorate due to the emergence of EGFR-TKI
acquired resistance, for which no effective therapy is currently
available (22,23). Emerging studies have reported that
certain Chinese medicines could reduce gefitinib-induced drug
resistance, including TP (24,25).
However, the role and mechanism of TP in gefitinib-induced drug
resistance in NSCLC is unclear. In the present study, it was
confirmed that TP inhibited the migration and invasion of A549
cells to a greater extent than G, and this effect was further
enhanced using the combination of G and TP, indicating that TP and
gefitinib have a synergistic effect on A549 cells. Subsequently, a
gefitinib-resistant A549 cell line (A549/G) was established to
investigate whether TP regulated drug resistance. The migration and
invasion abilities of gefitinib-resistant cells were enhanced
compared with their parental gefitinib-sensitive A549 cells. TP
treatment effectively inhibited the migration and invasion of the
gefitinib-resistant cells, indicating that TP suppressed gefitinib
resistance. These results verified that TP acts as an adjuvant
therapeutic agent at low doses to enhance anticancer effectiveness
(9,26,27).
Increasing numbers of in vitro and in
vivo studies have reported various possible molecular
mechanisms of drug resistance to gefitinib, among which EGFR
gene amplification and EMT are the most studied (28–30).
Rho et al reported that EMT resulting from repeated exposure
to gefitinib blunted the sensitivity of A549 cells to EGFR
inhibitors (31). Induction of EMT
contributed to the decreased efficacy of therapy in primary and
acquired resistance to gefitinib (32). E-cadherin is involved in the
formation of cell-to-cell adherens junctions that assemble adjacent
epithelial cells and maintains their quiescence. Downregulation of
E-cadherin is considered the core element of EMT (33,34).
Herein, it was revealed that TP significantly increased the levels
of E-cadherin both in gefitinib-sensitive and gefitinib-resistant
A549 cells, indicating that TP inhibited EMT and increased
gefitinib sensitivity by increasing E-cadherin levels. SNAIL, as
the core transcription factor regulating EMT, inhibits the
expression of CDH1 by competitive binding to the E-box
sequence in the CDH1 promoter region, and induces
mesenchymal proteins such as vimentin and MMP-9, which further
promote EMT (35,36). Vimentin was originally identified as
a specific marker of mesenchymal tumors; however, it was later
revealed to be associated with cancer cell expression and prognosis
in patients, and its expression is widely considered as a necessary
condition to enhance cancer invasion and metastasis (37). MMP9 is a matrix metalloproteinase
secreted by tumor cells that weakens the natural barrier and
promotes tumor cell metastasis by degrading the tumor extracellular
matrix (ECM). Therefore, a decrease of MMP9 expression plays an
important role in inhibiting EMT (38,39).
Given the role of SNAIL in the regulation of EMT markers, it was
speculated that the decrease in SNAIL levels induced by TP led to a
reduction in MMP9 and vimentin, and increased the levels of
E-cadherin. The combined effect of these changes resulted in EMT
inhibition.
Given the beneficial effects of TP in inhibiting EMT
and increasing gefitinib sensitivity, gefitinib-resistant A549
cells were established by interfering with E-cadherin expression
and continuous exposure to gefitinib to confirm the ability of TP
to increase the sensitivity of gefitinib-resistant cells. The
results revealed that the migration and invasion of
gefitinib-resistant cells was significantly enhanced.
Immunoblotting assays confirmed the loss of E-cadherin, and gain of
MMP9 and caspase-3. In addition, TP combined with gefitinib still
exhibited an inhibitory effect on the tumors derived from
A549/siE-cad cells by upregulating the levels of E-cadherin and
caspase-3, and decreasing those of MMP9. Caspase-3 is a pivotal
junction protein in apoptotic pathways, which can be activated by
other activated caspases and then induces apoptosis (37,38).
The present results were consistent with previous studies which
revealed that E-cadherin expression potentiates sensitivity to the
apoptotic effects of gefitinib (31,32).
The increase of E-cadherin is accompanied by the occurrence of
apoptosis, which synergistically inhibits tumor growth (40). In a previous study MMP9 inhibition
by tetramethylpyrazine was related to vascular endothelial cell
apoptosis (41). As for vimentin,
it was involved in the emergence of apoptosis induced by
peptidylarginine deiminase 2 and TNF-α (42,43).
Furthermore, the interference with SNAIL signaling by TPD52L2
induced apoptosis of glioblastoma cells (44). In fact, the inhibition of MMP9,
SNAIL, and vimentin proteins always coexists with apoptosis
(45). In conclusion, these results
indicated that TP could reverse gefitinib resistance of A549 cells
by suppressing the EMT signaling pathway and inducing
apoptosis.
Acquired resistance to EGFR-TKIs is almost
inevitable in patients with NSCLC with EGFR mutations.
Management of TKI resistance has become the focus of research to
increase the overall survival of these patients. In the present
study, it was demonstrated, for the first time to the best of our
knowledge, that TP treatment overcomes TKI resistance in
vitro and in vivo by reverting EMT. Further studies are
required to develop the synergistic anticancer action and drug
resistance reversal effect of TP combined with gefitinib for
clinical use.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Natural Science
Foundation of Zhejiang Province (grant. no. LQ17H290001) and the
Natural Science Foundation of China (grant. no. 81774026).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
FL, JW and WW conceived and designed the
experiments. FL, JW and XG performed the experiments. HC and XJ
collected, analyzed and interpret the data. FL drafted the
manuscript. JW and FL revised the manuscript, WW and XG provided
important intellectual content. All authors read and approved the
final manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animal treatment protocols were approved by the
Institutional Animal Care and Use Committee of The Tongde Hospital
of Zhejiang Province.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing financial
interests.
References
|
1
|
Malhotra J, Jabbour SK and Aisner J:
Current state of immunotherapy for non-small cell lung cancer.
Transl Lung Cancer Res. 6:196–211. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liao BC, Lin CC, Lee JH and Yang JC:
Optimal management of EGFR-mutant non-small cell lung cancer with
disease progression on first-line tyrosine kinase inhibitor
therapy. Lung Cancer. 110:7–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu X, Lu X, Zhen F, Jin S, Yu T, Zhu Q,
Wang W, Xu K, Yao J and Guo R: LINC00665 induces acquired
resistance to gefitinib through recruiting EZH2 and activating
PI3K/AKT pathway in NSCLC. Mol Ther Nucleic Acids. 16:155–161.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y, Wu Z, Yu H, Wang H, Liu G, Wang S
and Ji X: Chinese herbal medicine wenxia changfu formula reverses
cell adhesion--mediated drug resistance via the integrin
β1-PI3K-AKT pathway in lung cancer. J Cancer. 10:293–304. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen P, Huang HP, Wang Y, Jin J, Long WG,
Chen K, Zhao XH, Chen CG and Li J: Curcumin overcome primary
gefitinib resistance in non-small-cell lung cancer cells through
inducing autophagy-related cell death. J Exp Clin Cancer Res.
38:2542019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Almaaytah A, Farajallah A, Abualhaijaa A
and Al-Balas Q: A3, a Scorpion venom derived peptide analogue with
potent antimicrobial and potential antibiofilm activity against
clinical isolates of multi-drug resistant gram positive bacteria.
Molecules. 23:E16032018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao F, Gong YB, Kang XH, Lu ZH, Wang Y,
Zhao KL, Miao ZH, Liao MJ and Xu ZY: Degradation of MCL-1 by
bufalin reverses acquired resistance to osimertinib in EGFR-mutant
lung cancer. Toxicol Appl Pharmacol. 379:1146622019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou ZY, Tong XP, Peng YB, Zhang BK and Yan
M: Broad targeting of triptolide to resistance and sensitization
for cancer therapy. Biomed Pharmacother. 104:771–780. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu D and Kao PN: Immunosuppressive and
anti-inflammatory mechanisms of triptolide, the principal active
diterpenoid from the Chinese medicinal herb Tripterygium
wilfordii Hook. f. Drugs R D. 4:1–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meng G, Wang W, Chai K, Yang S, Li F and
Jiang K: Combination treatment with triptolide and
hydroxycamptothecin synergistically enhances apoptosis in A549 lung
adenocarcinoma cells through PP2A-regulated ERK, p38 MAPKs and Akt
signaling pathways. Int J Oncol. 46:1007–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu B, Guo X, Mathew S, Armesilla AL,
Cassidy J, Darling JL and Wang W: Triptolide simultaneously induces
reactive oxygen species, inhibits NF-kappaB activity and sensitizes
5-fluorouracil in colorectal cancer cell lines. Cancer Lett.
291:200–208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie CQ, Zhou P, Zuo J, Li X, Chen Y and
Chen JW: Triptolide exerts pro-apoptotic and cell cycle arrest
activity on drug-resistant human lung cancer A549/Taxol cells via
modulation of MAPK and PI3K/Akt signaling pathways. Oncol Lett.
12:3586–3590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ho JN, Byun SS, Lee S, Oh JJ, Hong SK, Lee
SE and Yeon JS: Synergistic antitumor effect of triptolide and
cisplatin in cisplatin resistant human bladder cancer cells. J
Urol. 193:1016–1022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li F, Zhao D, Yang S, Wang J, Liu Q, Jin X
and Wang W: ITRAQ-Based proteomics analysis of triptolide on human
A549 lung adenocarcinoma cells. Cell Physiol Biochem. 45:917–934.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kourtidis A, Lu R, Pence LJ and
Anastasiadis PZ: A central role for cadherin signaling in cancer.
Exp Cell Res. 358:78–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC,
Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, et al:
Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per
se is a common mechanism for acquired resistance to EGFR TKI.
Oncogene. 38:455–468. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rastogi I, Rajanna S, Webb A, Chhabra G,
Foster B, Webb B and Puri N: Mechanism of c-Met and EGFR tyrosine
kinase inhibitor resistance through epithelial mesenchymal
transition in non-small cell lung cancer. Biochem Biophys Res
Commun. 477:937–944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iderzorig T, Kellen J, Osude C, Singh S,
Woodman JA, Garcia C and Puri N: Comparison of EMT mediated
tyrosine kinase inhibitor resistance in NSCLC. Biochem Biophys Res
Commun. 496:770–777. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yin J, Hu W, Pan L, Fu W, Dai L, Jiang Z,
Zhang F and Zhao J: Let7 and miR17 promote self-renewal and drive
gefitinib resistance in non-small cell lung cancer. Oncol Rep.
42:495–508. 2019.PubMed/NCBI
|
|
22
|
Fiore M, Trecca P, Perrone G, Amato M,
Righi D, Trodella L, D'Angelillo RM and Ramella S: Histologic
transformation to small-cell lung cancer following gefitinib and
radiotherapy in a patient with pulmonary adenocarcinoma. Tumori.
105:NP12–NP16. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Sun L, Cui J, Wang J, Liu X, Aung
TN, Qu Z, Chen Z, Adelson DL and Lin L: Yiqi chutan tang reduces
gefitinib-induced drug resistance in non-small-cell lung cancer by
targeting apoptosis and autophagy. Cytometry A. 97:70–77. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng J, Chang C, Chen Y, Bi F, Ji C and
Liu W: EGCG overcomes gefitinib resistance by inhibiting autophagy
and augmenting cell death through targeting ERK phosphorylation in
NSCLC. Onco Targets Ther. 12:6033–6043. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang R, Ma X, Su S and Liu Y: Triptolide
antagonized the cisplatin resistance in human ovarian cancer cell
line A2780/CP70 via hsa-mir-6751. Future Med Chem. 10:1947–1955.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han Y, Huang W, Liu J, Liu D, Cui Y, Huang
R, Yan J and Lei M: Triptolide inhibits the AR signaling pathway to
suppress the proliferation of enzalutamide resistant prostate
cancer cells. Theranostics. 7:1914–1927. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schueler J, Tschuch C, Klingner K, Bug D,
Peille AL, de Koning L, Oswald E, Klett H and Sommergruber W:
Induction of acquired resistance towards EGFR inhibitor gefitinib
in a patient-derived xenograft model of non-small cell lung cancer
and subsequent molecular characterization. Cells. 8:E7402019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suda K, Mizuuchi H, Maehara Y and
Mitsudomi T: Acquired resistance mechanisms to tyrosine kinase
inhibitors in lung cancer with activating epidermal growth factor
receptor mutation-diversity, ductility, and destiny. Cancer
Metastasis Rev. 31:807–814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jakobsen KR, Demuth C, Sorensen BS and
Nielsen AL: The role of epithelial to mesenchymal transition in
resistance to epidermal growth factor receptor tyrosine kinase
inhibitors in non-small cell lung cancer. Transl Lung Cancer Res.
5:172–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asakura T, Yamaguchi N, Ohkawa K and
Yoshida K: Proteasome inhibitor-resistant cells cause EMT-induction
via suppression of E-cadherin by miR-200 and ZEB1. Int J Oncol.
46:2251–2260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gloushankova NA, Zhitnyak IY and Rubtsova
SN: Role of epithelial-mesenchymal transition in tumor progression.
Biochemistry (Mosc). 83:1469–1476. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Papiewska-Pajak I, Kowalska MA and Boncela
J: Expression and activity of SNAIL transcription factor during
epithelial to mesenchymal transition (EMT) in cancer progression.
Postepy Hig Med Dosw (Online). 70:968–980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimokawa M, Haraguchi M, Kobayashi W,
Higashi Y, Matsushita S, Kawai K, Kanekura T and Ozawa M: The
transcription factor Snail expressed in cutaneous squamous cell
carcinoma induces epithelial-mesenchymal transition and
down-regulates COX-2. Biochem Biophys Res Commun. 430:1078–1082.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Richardson AM, Havel LS, Koyen AE, Konen
JM, Shupe J, Wiles WG IV, Martin WD, Grossniklaus HE, Sica G,
Gilbert-Ross M and Marcus AI: Vimentin is required for lung
adenocarcinoma metastasis via heterotypic tumor
cell-cancer-associated fibroblast interactions during collective
invasion. Clin Cancer Res. 24:420–432. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang M and Xin W: Matrine inhibiting
pancreatic cells epithelial-mesenchymal transition and invasion
through ROS/NF-κB/MMPs pathway. Life Sci. 192:55–61. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun Q, Yang Z, Li P, Wang X, Sun L, Wang
S, Liu M and Tang H: A novel miRNA identified in GRSF1 complex
drives the metastasis via the PIK3R3/AKT/NF-κB and TIMP3/MMP9
pathways in cervical cancer cells. Cell Death Dis. 10:6362019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han B, Zhang YY, Xu K, Bai Y, Wan LH, Miao
SK, Zhang KX, Zhang HW, Liu Y and Zhou LM: NUDCD1 promotes
metastasis through inducing EMT and inhibiting apoptosis in
colorectal cancer. Am J Cancer Res. 8:810–823. 2018.PubMed/NCBI
|
|
41
|
Hu JZ, Wang XK, Cao Y, Li DZ, Wu TD, Zhang
T, Xu DQ and Lu HB: Tetramethylpyrazine facilitates functional
recovery after spinal cord injury by inhibiting MMP2, MMP9, and
vascular endothelial cell apoptosis. Curr Neurovasc Res.
14:110–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu PC, Liao YF, Lin CL, Lin WH, Liu GY
and Hung HC: Vimentin is involved in peptidylarginine deiminase
2-induced apoptosis of activated Jurkat cells. Mol Cells.
37:426–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang L, Tang L, Dai F, Meng G, Yin R, Xu X
and Yao W: Raf-1/CK2 and RhoA/ROCK signaling promote TNF-α-mediated
endothelial apoptosis via regulating vimentin cytoskeleton.
Toxicology. 389:74–84. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qiang Z, Jun-Jie L, Hai W, Hong L, Hong L,
Bing-Xi L, Lei C, Wei X, Ya-Wei L, Huang A, Song-Tao Q and Yun-Tao
L: TPD52L2 impacts proliferation, invasiveness and apoptosis of
glioblastoma cells via modulation of wnt/β-catenin/snail signaling.
Carcinogenesis. 39:214–224. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bao X, Ren T, Huang Y, Ren C, Yang K,
Zhang H and Guo W: Bortezomib induces apoptosis and suppresses cell
growth and metastasis by inactivation of Stat3 signaling in
chondrosarcoma. Int J Oncol. 50:477–486. 2017. View Article : Google Scholar : PubMed/NCBI
|