Introduction
Pancreatic cancer remains the fourth most common
cause of cancer-associated deaths of the digestive system and the
fourth leading cause of cancer-related deaths in America (1). It was reported that 18.1 million new
cancer cases and 9.6 million cancer-related deaths occurred in
2018. PDAC has been revealed to account for approximately 2.5% of
new cancer cases and 4.5% of cancer-related deaths in 185 countries
(2). Moreover, the mortality of
pancreatic cancer was revealed to be nearly 94.1%, and the 5-year
relative survival rate less than 10%, which is the lowest rate
among digestive system malignancies (2–4).
Pancreatic cancer was reported as the seventh leading cause of
cancer-related deaths in patients regardless of sex worldwide in
2018 and may become the second leading cause of cancer-related
deaths in America by 2030 (2,5). PDAC
has a poor prognosis owing to several factors, including
inefficient diagnosis due to the absence of evident early
pathognomonic symptoms and propensity to develop drug resistance
(6–7). In the advanced stages, local
infiltration or distant metastasis are observed among most PDAC
patients. Tumor metastasis renders treatment strategies such as
surgery and chemotherapy rarely effective (8–10).
However, the underlying mechanisms governing PDAC carcinogenesis
and metastasis remain largely unclear. Therefore, there is an
urgent need to identify both novel early biomarkers for diagnosing
and sufficiently effective therapeutic targets to improve the
prognosis of pancreatic cancer.
Epithelial-mesenchymal transition (EMT) is a gradual
developmental process in which epithelial cells undergo various
biochemical changes; during this process, epithelial cells
gradually lose the epithelial phenotype and acquire mesenchymal
features (11,12). Evidence suggests that the invasion
and dissemination of cancer cells are due to the transformation of
epithelial cells (13).
Transforming growth factor-β (TGF-β) is a crucial driver of EMT and
usually promotes the process of EMT (14). TGF-β has also been demonstrated to
have an essential role in the regulation of cell invasion and EMT
progression in pancreatic cancers (15–16).
The poor prognosis of pancreatic cancer is closely related to its
early metastasis, and EMT is a critical event in the process of
cancer metastasis. Thus, it is important to understand the
potential role of TGF-β and Girdin in the process of EMT in
PDAC.
Girdin, also known as Akt phosphorylation enhancer,
Hook-related protein 1 and G-interacting vesicle-associated
protein, is a novel actin protein containing 1870 amino acids in
humans with a molecular mass of 220–250 kDa; it was independently
reported by four groups in 2005 (17–20).
Considering its role in regulating intracellular microfilaments and
actin organization and cell motility, it is referred to as GIRDers
of actIN filament (21). Recently,
Girdin was revealed to be aberrantly expressed in a variety of
malignancies, such as glioma, lung cancer, breast cancer,
esophageal squamous cell carcinoma and colon cancer (22–26).
There is evidence suggesting that Girdin plays a key role in
promoting carcinogenesis and development in lung tumors, regulating
invasion and metastasis in breast cancer and glioma cancer and
leading to poor prognosis of cancer patients (23,24,27).
Moreover, Girdin was also revealed to be involved in angiogenesis
and autophagy via its regulation of signaling pathways (28–29).
Previous research by our group revealed that Girdin was highly
expressed in PDAC and was closely related to the pathological grade
of PDAC patients (30). These
studies indicated that Girdin may be a promising biomarker for some
cancer patients and an important participant in the carcinogenesis
and progression of cancer. However, the underlying mechanisms of
Girdin in the metastasis and TGF-β-induced EMT of PDAC have not
been well demonstrated.
In the present study, it was revealed that Girdin
was highly expressed in PDAC cells and that high Girdin expression
was related to poor prognosis in PDAC patient data from GEPIA.
Inhibiting the expression of Girdin inhibited the proliferation,
migration and invasion of pancreatic cancer cells in vitro.
In addition, knockdown of Girdin suppressed PDAC growth and
metastasis in vivo. The molecular mechanism assays indicated
that Girdin may promote the invasion and migration of pancreatic
cancer cells via the PI3K/AKT signaling pathway. Mechanistic
investigations also elucidated that Girdin may interact with
vimentin to suppress TGF-β-induced EMT in pancreatic cancer.
Materials and methods
Cell culture
Human pancreatic nestin-expressing (hTERT-HPNE)
cells were separated from human pancreatic cells and used as normal
human pancreatic ductal cells. The three human PDAC cell lines
(AsPC-1, PANC-1 and BxPC-3) and the normal human pancreatic ductal
cell line (HPNE) were purchased from the American Type Culture
Collection (ATCC). The human PDAC cell lines PANC-1 and BxPC-3 and
the control HPNE cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) (Gibco; Thermo Fisher Scientific, Inc.). The medium
contained 4.5 g/l D-glucose, and 10% fetal bovine serum (FBS)
(Wisent, Inc.) was added. AsPC-1 cells were maintained in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc.) with 10% FBS. The
cells aforementioned were all cultured in a sterile cell culture
incubator, which contained 5% CO2 and maintained in a
humidified environment at 37°C.
Cell infection and TGF-β
treatment
Recombinant adenoviruses were constructed containing
a specific short hairpin RNA of Girdin (sh-Girdin) or a
nontargeting short hairpin RNA designed as the control (sh-NC). The
vector containing pAd-U6-CMV-GFP was used for synthesizing
recombinant adenoviruses. After the recombinant adenovirus was
amplified and purified from 293A cells, it was used to transfect
the experimental cells. Untreated cells were also used as the
normal group (N) and compared with the experimental control. When
the cells grew to 70–80% confluence, recombinant adenovirus was
added to the six-well plate with serum-free medium for 4–6 h. After
the cells were washed twice with PBS, the cells were cultured for
24 or 48 h in medium with or without serum for the experiments.
Western blot analysis and q-PCR analysis were used to confirm the
infection efficiency. The TGF-β treatment time was 24 h, and the
concentration was 10 µM; the cells were co-incubated with TGF-β and
sh-NC or sh-Girdin.
Cell proliferation assay
The cells of various treatment groups (N, sh-NC and
sh-Girdin) were adjusted to a density of 5×104 cells/l,
and then a 100-µl volume of the suspension (5×103 cells)
was added to a 96-well plate. After the virus effectively
transfected the cells, the cells were replated, and the cell
viability was detected by sulforhodamine B (SRB) (Shanghai Sixin
Biological Technology Co., Ltd.) analysis at four different
time-points of incubation (24, 48, 72, and 96 h).
Subsequently, a 96-well culture plate was removed at
the corresponding time and fixed with 1% trichloroacetic acid (TCA)
(Sinopharm Chemical Reagent Co., Ltd.). The plate was left in a
refrigerator at 4°C overnight or at room temperature for 3 h and
then washed 5 times with PBS. When the 96-well plate was dry, it
was stained with 100 µl SRB solution (0.4% w/v) for another 15 min
in a place protected from light at room temperature. Then, 1%
glacial acetic acid was utilized to wash the wells 5 times, and the
plate was dried overnight under a fume hood. The protein-bound SRB
stain was solubilized by adding 100 µl Tris-base (10 mM, pH=10.5)
(Sinopharm Chemical Reagent Co., Ltd.) into every well. Ultimately,
the cell proliferation rate was detected by a spectrophotometer
that measured the absorbance at 490 nm.
Colony formation assay
The sh-RNA-transfected or untreated cells were
cultured for 24 h, and 300 cells were seeded in 6-well plates.
Cells were cultured for two weeks, and when colonies were clearly
visible, 4% paraformaldehyde was used to fix the cells overnight at
4°C; the wells were washed two times with PBS and stained with 0.4%
crystal violet staining solution for 15 min at room temperature.
Colony formation was quantified by counting with the naked eye. The
number of colonies is presented in the figures.
Transwell migration and invasion
assay
PANC-1 and BxPC-3 cells in a 60×15 mm cell culture
dish were treated with or without recombinant adenovirus for 6 h
and then stimulated with or without LY294002 (10 µM/ml) (Cell
Signaling Technology, Inc.) for 24 h. Cell migration was assessed
by Transwell assay, and the various groups (N, sh-NC, sh-Girdin,
sh-NC+DMSO, sh-NC+LY294002, sh-Girdin+DMSO, sh-Girdin+LY294002) of
cells (1×104/well) were seeded into the upper chamber of
a Transwell (8 µm PET, Millicell; EMD Millipore) with 200 µl
serum-free DMEM. For the invasion assay, Matrigel (BD Biosciences)
mixed with serum-free DMEM (1:5) was added to the 24-Well Hanging
Inserts of the upper chamber, and the volume of the mixed liquid
was 60 µl. Then, the chambers were placed in the incubator for 8 h
at 37°C. A volume of 800 µl DMEM supplemented with 10% FBS (Wisent,
Inc.) was added to the lower chambers before adding cells to the
upper chamber. Thereafter, the cells added into the chambers were
placed in a sterile incubator to invade for 24 h. The
migrated/invaded cells were fixed with 800 µl 4% paraformaldehyde
in a 4°C freezer overnight and stained with 0.4% crystal violet
staining solution for 15 min at room temperature. The cells on the
top of the membranes were removed with swabs, and then the
remaining cells were washed twice with PBS. The migrated/invaded
cells were imaged and counted in five separate fields under a light
microscope (Olympus Corporation; magnification, ×10), and the
experiment was performed at least 3 times.
RNA isolation and real-time
quantitative PCR (RT-qPCR)
TRIzol® reagent (Tiangen Biotech Co.,
Ltd.) was used to extract the total RNA from the PDAC cell lines,
and according to the manufacturer's operating instructions, the
PrimeScript™ RT reagent kit (Takara Bio, Inc.) was used to
reverse-transcribe it into cDNA. The mRNA levels of Girdin were
normalized to those of β-actin and examined by the following
primers: Girdin forward, 5′-AGGAAATGGGACCAACCTTGA-3′ and reverse,
5′-GTGCATTCTAAGTGAGGCATCAT-3′; β-actin forward,
5′-ATCGTGCGTGACATTAAGGAGAAC-3′ and reverse,
5′-AGGAAGGAAGGCTGGAAGAGTG-3′. Girdin mRNA was amplified by RT-qPCR.
SYBR Green and the iQ5 Multicolor Real Time Detector System (both
from Bio-Rad Laboratories, Inc.) were used according to the
manufacturer's protocol. The thermocycling conditions were as
follows: Reverse transcription at 50°C for 2 min, denaturation at
95°C for 10 min, followed by 40 cycles of amplification (15 sec at
95°C, 1 min at 60°C and 1 min at 72°C). Relative levels of mRNA
expression were obtained with the 2−ΔΔCq method
(31).
Western blot analysis
PDAC cells were washed with PBS and then lysed with
phenylmethanesulfonyl fluoride (PMSF) and RIPA lysate buffer at a
ratio of 1:100 (Thermo Fisher Scientific, Inc.). The bicinchoninic
acid assay was applied to examine the protein concentration in the
various groups. Equal amounts of protein (30 µg) were separated by
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) at 6% or 8% concentrations. Polyvinylidene fluoride
(PVDF) (0.22-µm diameter) (EMD Millipore) membranes were used to
transfer the protein for 1.5 or 3 h at 95 volts. Then, the
membranes were blocked with a total volume of 25 ml dry skimmed
milk (5%, w/v) or bovine serum albumin (5%, w/v), which was
dissolved in Tris-buffered saline (TBS) containing 0.05% Tween-20
(TBST) for 2 h at room temperature. Before placing them on a
horizontal shaker in a four-degree refrigerator overnight, the
membranes were incubated with the following primary antibodies:
Anti-Akt (1:1,000; product code ab32505), anti-PI3k (1:1,000;
product code ab133595), anti-Girdin (1:1,000; product code
ab179481) were purchased from Abcam; anti-p-AKT (1:500; cat. no.
AF0016), anti-p-PI3K (1:500; cat. no. AF3241) were obtained from
Affinity Biosciences.; anti-MMP2 (1:1,000; cat. no. 10373-2-AP),
anti-MMP9 (1:1,000; cat. no. 10375-2-AP), anti-E-cadherin (1:1,000;
cat. no. 20874-1-AP), anti-N-cadherin (1:1,000; cat. no.
22018-1-AP), anti-vimentin (1:1,000; cat. no. 60330-1-Ig) and
anti-HRP-conjugated beta-actin mouse McAb (HRP-60008) were acquired
from ProteinTech Group, Inc. TBST buffer was used to wash the
membrane three times for 30 min, and then the membranes were
incubated with the respective homologous horseradish
peroxidase-conjugated secondary antibodies: goat anti-mouse IgG
(H+L) (1:8,000; cat. no. 33201ES60) was purchased from Shanghai
Yesheng Biotechnology Co., Ltd. and goat anti-rabbit IgG-HRP
antibodies (1:8,000; sc2004) were acquired from Santa Cruz
Biotechnology, Inc. The blots were incubated at room temperature on
a low-speed centrifuge shaker for 2 h. The blots were washed three
times with TBST for 45 min and Western Bright™ ECL (cat. no.
R-03031-D25), which was purchased from Advansta, Inc., was applied
following the manufacturer's protocol. The blots were observed and
imaged through by Chemiluminescence Gel Imager (Tanon-5200Multi;
Shanghai Tanon Technology Co., Ltd.).
Immunofluorescence
Before adding sterile confocal glass slides (BD
Biosciences) to 24-well plates, a small amount of serum-free DMEM
was added to enhance the adsorption capacity of the slides. PANC-1
or BxPC-3 cells were seeded into 24-well plates and cultured for 24
h. Then, the cells were fixed with 4% paraformaldehyde on glass
slides overnight at 4°C. After washing with PBS, the cells were
permeabilized with 0.1% Triton X-100 for 15 min and then washed
three times for 15 min. Next, 3% bovine serum albumin was used to
block the cells for 30 min at 37°C. Then, the primary antibody,
anti-Girdin (1:200; product code ab179481) or anti-vimentin (1:200;
cat. no. 60330-1-Ig), was added to the glass slides overnight;
next, the cells were washed with PBS 3 times for 15 min. The cells
were then incubated with fluorescent secondary antibodies Alexa
Fluor® 594 donkey anti-rabbit IgG (H+L) (1:1,000;
product code ab150076) and Alexa Fluor® 488 goat
anti-mouse IgG (H+L) (1:1,000; product code ab150113; both from
Abcam) for ~30 min at 37°C. Finally, DAPI (blue) was used to
counterstain cell nuclei for 1 min at room temperature, and the
cells were examined by fluorescence microscopy (Olympus
Corporation) at a magnification of ×100.
Co-immunoprecipitation
The Girdin immunocomplex was isolated by
coimmunoprecipitation (Co-IP). PANC-1 and BxPC-3 cells were lysed
in Nonidet P-40 buffer (0.5% Nonidet P-40, 0.25% deoxycholate, 50
mM Tris-HCl (pH 8.0), 5 mM EDTA and 150 mM NaCl). Total cell
lysates (1,000 ml) containing a protein weight of 1,000 µg were
incubated with 3–5 µg of specific antibodies (anti-Girdin or
anti-vimentin) overnight at 4°C. Protein G beads (EMD Millipore)
were washed three times with Nonidet P-40 buffer before incubation
with the cell lysate. One milligram of precleared lysate was
incubated with 200 µl of Protein G beads, which were washed with 1
ml of lysis buffer three times and cross-linked with the indicated
antibody. The beads were added to the lysate and incubated for ~6 h
at 4°C. Then, the beads were extensively washed with 1 ml of lysis
buffer three times and 1 ml of PBS one time. SDS-PAGE 2X loading
buffer (50 µl) was applied to obtain the proteins from the beads.
The immunoprecipitated protein complexes resolved by 6% SDS-PAGE
were analyzed by western blotting.
In vivo tumor formation and metastasis
assays
The animal experiments strictly complied with the
Guidelines for the Care and Use of Laboratory Animals of the
National Institutes of Health [Eighth Edition (2011)]. The protocol
of this experiment was approved by the Animal Ethics and Welfare
Committee (AEWC) of Nanjing Medical University (IACUC-1601161). A
total of 20 six-week-old BALB/c male mice were purchased from the
Animal Center of Nanjing University (Nanjing, China). The mice were
reared under certain pathogen-free conditions of laboratory
animals, and housed (temperature: 25°C and humidity: 55%) in
microisolator individually ventilated cages with ad libitum
water and food. One cage for every five mice. Every three days, the
health of the mice was observed, and water, food, and litter were
changed. BxPC-3 cells were stably transfected with recombinant
adenovirus that contained sh-Girdin or empty vector. Then, the
cells were washed and harvested with PBS. Cells were resuspended in
PBS at 5×107 cells/ml. After the mice were anesthetized
by chloral hydrate (400 mg/kg of body weight) through
intraperitoneal injection, then the cells (1×107 cells,
0.2 ml PBS) were injected into the right side of the ventral
anterior of 10 athymic male nude mice. After six days, the tumor
size of mice was calculated and then calculated again every 3 days;
tumor volumes were calculated with the formula:
V=0.5×Dxd2 (V, volume; D, longitudinal diameter; and d,
latitudinal diameter). The mice were euthanized by injecting a
concentration of 100% carbon dioxide into a sealed box at a flow
rate of 20% per min for 5 min. The death of mice was confirmed by
observing the absence of breathing and a heartbeat, or dilated
pupils. The size and weight of tumors in all mice were also
determined and analyzed.
Exploring the role of Girdin in tumor
metastasis in vivo
Transfected PDAC cells (5×106 cells, 0.1
ml PBS) were injected into nude mice through the tail vein when the
10 mice were anesthetized. After the tail vein injection, the
health status and diet of the nude mice were regularly observed. At
six weeks, one of the mice in the sh-Girdin group exhibited weight
loss and diet reduction. The experiment was terminated and the 10
nude mice were euthanized with carbon dioxide as aforementioned.
Subsequently, the lungs of nude mice in sh-NC group and sh-Girdin
group were surgically dissected. Half of the tumor samples and the
lung metastasis nodules were fixed for immunohistochemical
staining, and the other half was stored at −80°C for protein
detection.
Immunohistochemical staining
The tumor samples and the lung metastasis nodules
were fixed in 4% paraformaldehyde for ~2 days at room temperature
and embedded in paraffin. The thickness of the sections was ~5 µm.
Tissue sections were stained with hematoxylin staining solution for
5 min and stained with 1% eosin for 2 min at room temperature for
hematoxylin and eosin (H&E) staining. The sections of tumor
samples were incubated with the anti-Ki67 antibody (1:200; product
code ab92742) or anti-Girdin (1:200; product code ab179481; both
from Abcam) at 4°C overnight and a goat anti-rabbit IgG-HRP
antibodies (1:1,000; sc2004) at room temperature for 30 min. Then,
a DAB Substrate Kit (Maxin) was used for the following
immunohistochemical staining according to the manufacturer's
instructions. The staining results were observed and imaged through
light microscopy (Olympus Corporation) at a magnification of ×4 and
×40.
Statistical analysis
GraphPad Prism 8.0 software (GraphPad Software,
Inc.) was used for statistical analyses Data were statistically
analyzed using Student's t-test for comparisons of two groups and
ANOVA followed by Dunnett's post hoc test for comparisons of more
than two groups. Kaplan-Meier plotter was used to analyze the
prognostic role of Girdin in pancreatic cancer in GEPIA data
(http://gepia.cancer-pku.cn/). Log-rank
tests were performed for the survival time probability of PDAC
patients with high and low expression of Girdin expression. The
experiment results were independently reproduced three times, and
the experimental data are presented as the means ± SD. A two-sided
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
Expression levels of Girdin in PDAC
cell lines and Girdin protein are associated with poor
survival
To explore the expression level of Girdin in PDAC,
the mRNA level of Girdin was analyzed by RT-qPCR. Western blotting
was used to investigate the protein expression of Girdin in three
PDAC cell lines (PANC-1, AsPC-1 and BxPC-3) and a normal human
pancreatic ductal cell line (hTERT-HPNE). The results demonstrated
that the level of Girdin was significantly enhanced in PDAC cell
lines compared to hTERT-HPNE (Fig.
1A-C). PANC-1 and AsPC-1 are highly invasive and poorly
differentiated cell lines; however, BxPC-3 has low invasion
abilities and is highly differentiated (32). The results revealed that the
expression of Girdin in PANC-1 cells was higher than that in any
other cell line, and BxPC-3 was poorly expressed among PDAC cell
lines (Fig. 1B). A histogram of
Girdin protein expression in PDAC cell lines is presented in
Fig. 1C. Kaplan-Meier plotter
analysis of the GEPIA data indicated that a high level of Girdin
was associated with poor survival (Fig.
1D). Collectively, these results indicated that the high
expression of Girdin in PDAC patients may be a candidate biomarker
for improving treatment.
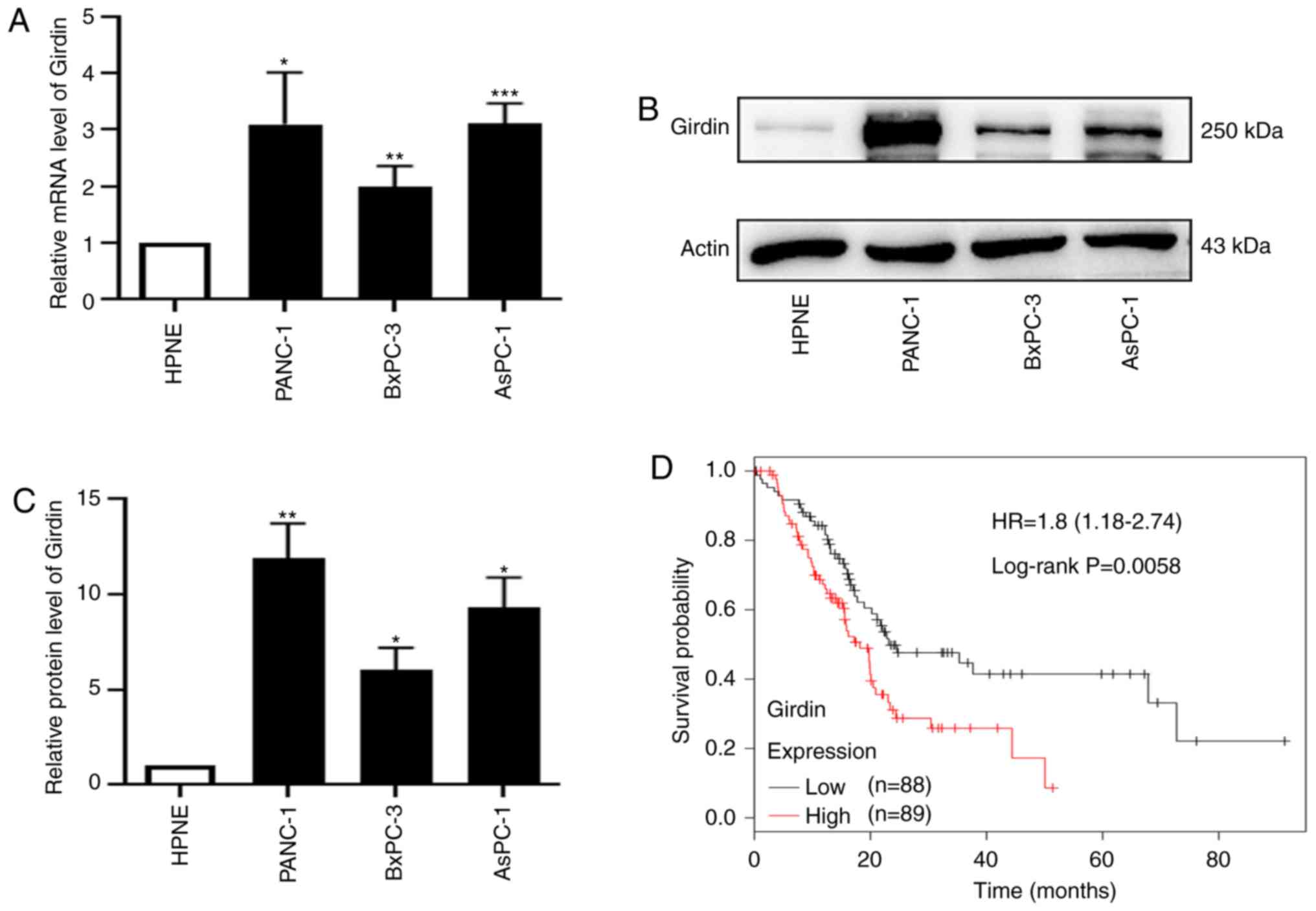 | Figure 1.Girdin is upregulated in PDAC, and
high expression of Girdin is associated with poor survival. (A)
Relative mRNA expression of Girdin in three PDAC cell lines,
PANC-1, BxPC-3 and AsPC-1, compared with the normal human
pancreatic ductal cell line hTERT-HPNE was assessed by RT-qPCR. (B)
Protein levels of Girdin in the PDAC cell lines PANC-1, BxPC-3 and
AsPC-1 and the normal human pancreatic ductal cell line hTERT-HPNE
were confirmed by western blotting. (C) Histogram of the protein
expression of Girdin in the PDAC cell lines PANC-1, BxPC-3 and
AsPC-1 and the normal human pancreatic ductal cell line hTERT-HPNE.
(D) Kaplan-Meier plotter analysis of GEPIA(http://gepia.cancer -pku.cn/) data indicated that a
high level of Girdin was associated with poor survival; high (n=88)
and low (n=89). Error bars were expressed as the mean ± SD, and the
experiment was performed at least in triplicate. *P<0.05,
**P<0.01, and ***P<0.001 compared to HPNE. PDAC, pancreatic
ductal adenocarcinoma; RT-qPCR, real-time quantitative polymerase
chain reaction; Actin, internal reference; GEPIA, Gene Expression
Profiling Interactive Analysis. |
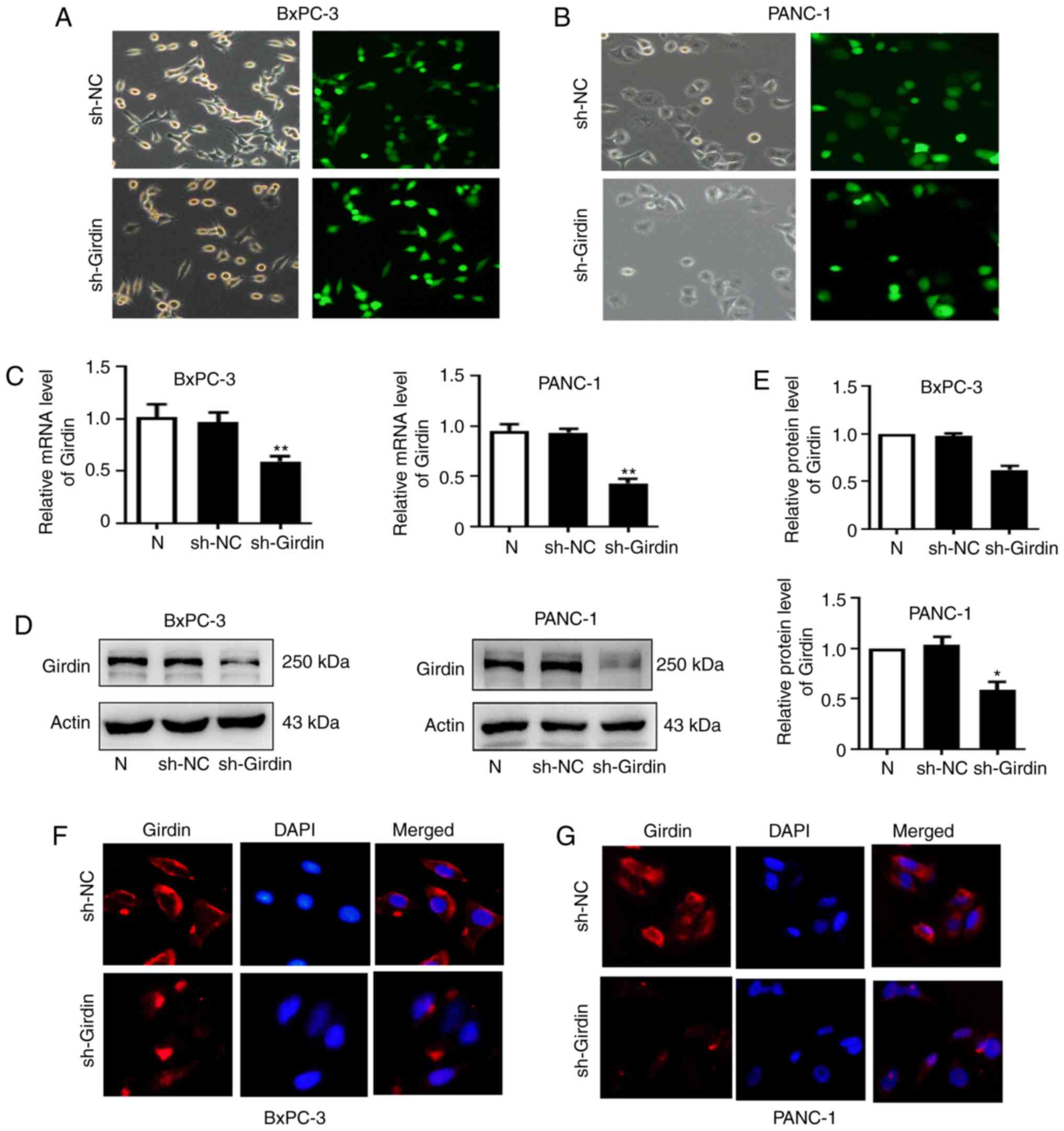
Knockdown of Girdin expression in
PANC-1 and BxPC-3 cells by shRNA
The negative control (sh-NC) and Girdin-shRNA
(sh-Girdin) recombinant adenoviruses were transfected into PDAC
cells (PANC-1 and BxPC-3). The infection efficiency was first
verified by GFP expression with fluorescence microscopy after
infection with adenoviruses for 48 h (Fig. 2A and B). RT-qPCR and western
blotting were performed to evaluate the expression level of Girdin
in cells infected with Girdin-shRNA for 24 and 48 h. The mRNA
expression levels of Girdin were significantly decreased in the
sh-Girdin group compared with the untreated (N) or the negative
control (Fig. 2C) groups.
Similarly, western blot analysis was also used and a significant
decrease in Girdin protein levels after transfection with sh-Girdin
(Fig. 2D and E) was detected.
Furthermore, the expression of Girdin was detected by
immunofluorescence staining and the results obtained were similar
to those aforementioned (Fig. 2F).
These results indicated that following sh-Girdin infection, the
expression of Girdin in PDAC cells was successfully suppressed.
Girdin-shRNA treatment inhibits the
proliferation, invasion, and migration of PDAC cells
To identify the potential role of Girdin in PDAC
progression, Girdin-knockdown PANC-1 and BxPC-3 cell lines were
established by adenovirus transfection. Then, SRB experiments and
colony formation assays were performed to evaluate the effect of
Girdin on the growth of pancreatic cancer in vitro. Girdin
knockdown significantly decreased PANC-1 and BxPC-3 cell viability
compared with that in the sh-NC group (Fig. 3A and B). The colony-forming ability
of pancreatic cancer cells was also significantly reduced by
sh-Girdin (Fig. 3C). There is
evidence that the expression of Girdin in breast cancer tissues is
high, and a high level of Girdin is associated with a high risk of
developing metastasis (12).
However, whether Girdin has a biological function in the migration
and invasion of PDAC has not been reported previously. To further
determine whether Girdin is related to the metastasis of PDAC,
Transwell experiments were performed to demonstrate that Girdin may
regulate the migration and invasion of BxPC-3 and PANC-1 cells. The
results revealed that knockdown of Girdin suppressed cell migration
and invasion (Fig. 3D and E).
Collectively, these findings indicated that Girdin could promote
the growth and metastasis of PDAC cells in vitro.
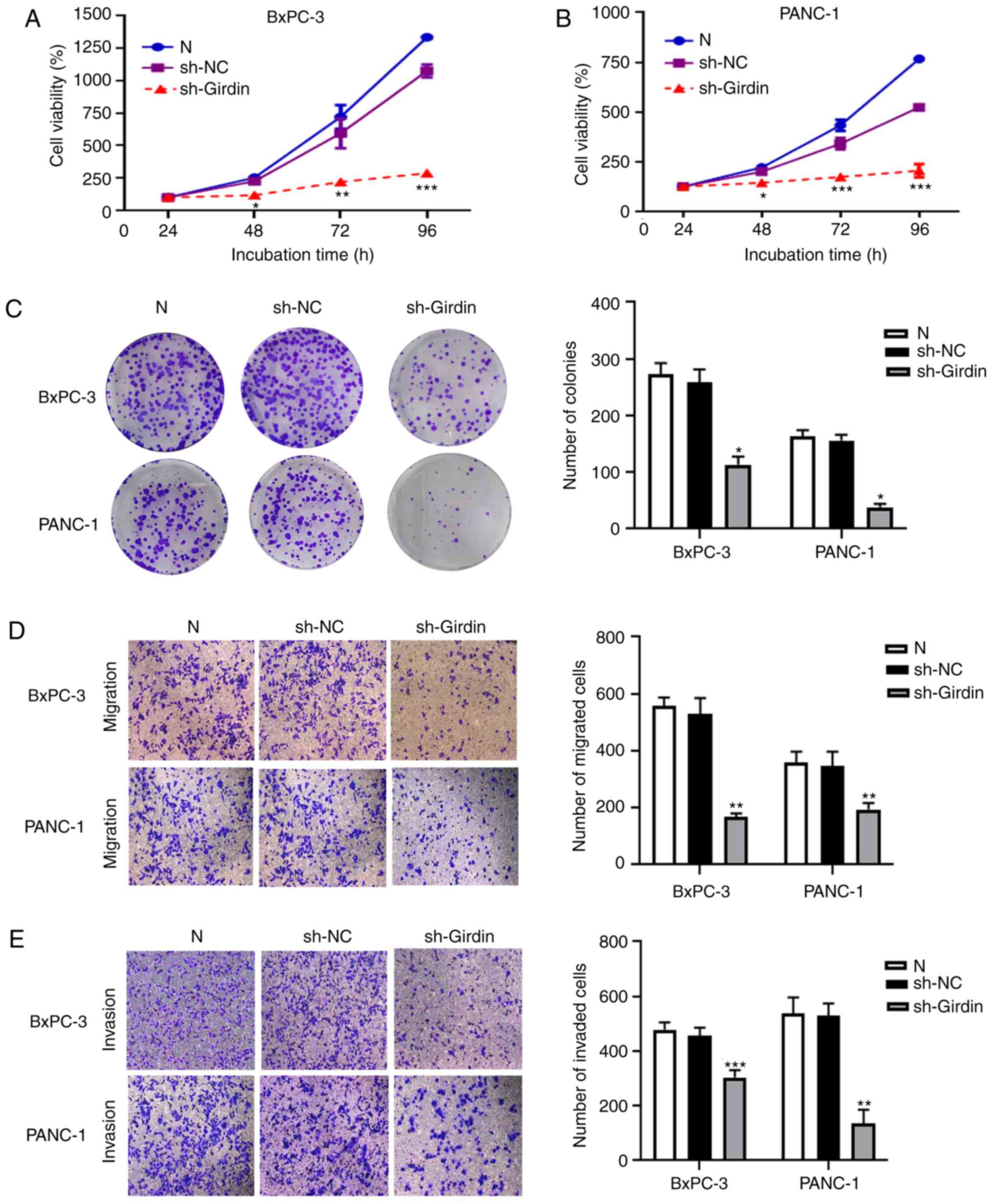 | Figure 3.Effects of sh-Girdin on the
proliferation, migration and invasion of pancreatic cancer cells.
(A) The absorbance value of control cells and BxPC-3 cells
transfected with sh-Girdin incubated for various time periods (24,
48,72 and 96 h) was measured by SRB assay. Knockdown of Girdin
significantly reduced proliferation after incubation for 48 h. (B)
The cell viability of BxPC-3 cells was measured by SRB assay at an
absorbance of 490 nm. The proliferation of PANC-1 cells was
inhibited by sh-Girdin. (C) The number of colonies was used to
investigate proliferation after the cells were treated for 14 days,
and knockdown of Girdin suppressed the formation of colonies in
PDAC. (D and E) Transwell assays were used to confirm the
metastasis of PDAC cells in vitro. Treatment with sh-Girdin
reduced the number of migrating and invading cells in the lower
chambers, scale bar, 100 µm. Three experiments were independently
performed. The data are presented as the mean ± SD. *P<0.05,
**P<0.01, and ***P<0.001 compared with sh-NC. PDAC,
pancreatic ductal adenocarcinoma; SRB, sulforhodamine B; sh-NC,
non-targeting control short hairpin RNA; sh-Girdin, a specific
short hairpin RNA of Girdin. |
Girdin may interact with vimentin to
affect TGF-β-induced EMT
EMT has been proposed to have various tumor
functions, such as tumor initiation, malignant progression, tumor
migration, metastasis and resistance to therapy (18). To further explore the underlying
role of Girdin in the migration of PDAC cells, 48 h after
transfection with sh-Girdin in PANC-1 and BxPC-3 cell lines, the
metastasis-associated proteins MMP2/9 were detected by western
blotting. As revealed from the results, the protein levels of MMP2
or MMP9 were decreased in PDAC cells transfected with sh-Girdin
(Fig. 4A and B). These results were
in accordance with the function of Girdin in cell migration. There
is evidence that EMT is involved in PDAC progression (19–21).
The Girdin/β-catenin interaction may be a potential mechanism by
which Girdin is involved in cell migration and invasion, and the
Girdin/E-cadherin interaction may explain its involvement in
cell-cell adhesion in skin cancer cells (11). Therefore, it was speculated that
Girdin may be involved in the EMT pathway to affect the progression
of pancreatic cancer. To confirm the mechanism by which Girdin
mediates PDAC cell motility, a mass spectrometric shotgun was used
to distinguish Girdin immunocomplexes isolated from the lysate of
PDAC cancer cells by tandem affinity purification. The EMT
biomarker vimentin, which has a critical role in tumor progression,
was identified in our experiment. Immunoprecipitation (IP) results
demonstrated that vimentin was co-immunoprecipitated with the
Girdin antibody; in addition, Girdin was co-immunoprecipitated with
the vimentin antibody in PDAC cancer cells (Fig. 4C). Immunofluorescence staining of
PANC-1 cells clearly revealed the co-localization of vimentin and
Girdin in cells. In addition, similar phenomena were also observed
in BxPC-3 cells (Fig. 4D). These
results indicated that Girdin may interact with vimentin to affect
the EMT of PDAC. The involvement of Girdin in regulating EMT
biomarkers was investigated by examining the protein expression
levels of E-cadherin, N-cadherin and vimentin. TGF-β was utilized
to induce EMT in PDAC cells. The TGF-β treatment time was 24 h, and
the concentration was 10 µM. Compared with the untreated group, the
expression level of E-cadherin, an epithelial phenotype biomarker,
was effectively increased, and the expression levels of N-cadherin
and vimentin, mesenchymal phenotype biomarkers, were significantly
reduced in the sh-Girdin group (Fig. 4E
and F). Although the EMT biomarker E-cadherin was not
significantly reduced in PANC-1 cells, N-cadherin and vimentin
expression was significantly increased by TGF-β in the PANC-1 and
BxPC-3 cells treated by sh-NC with TGF-β compared to the sh-NC
group. It was also revealed that TGF-β-induced EMT was reversed by
sh-Girdin in the PANC-1 and BxPC-3 cells in the sh-NC with TGF-β
group compared to the sh-Girdin with TGF-β (Fig. 4E and F). Collectively, the potential
mechanism by which Girdin affects cell migration may occur through
its interaction with vimentin.
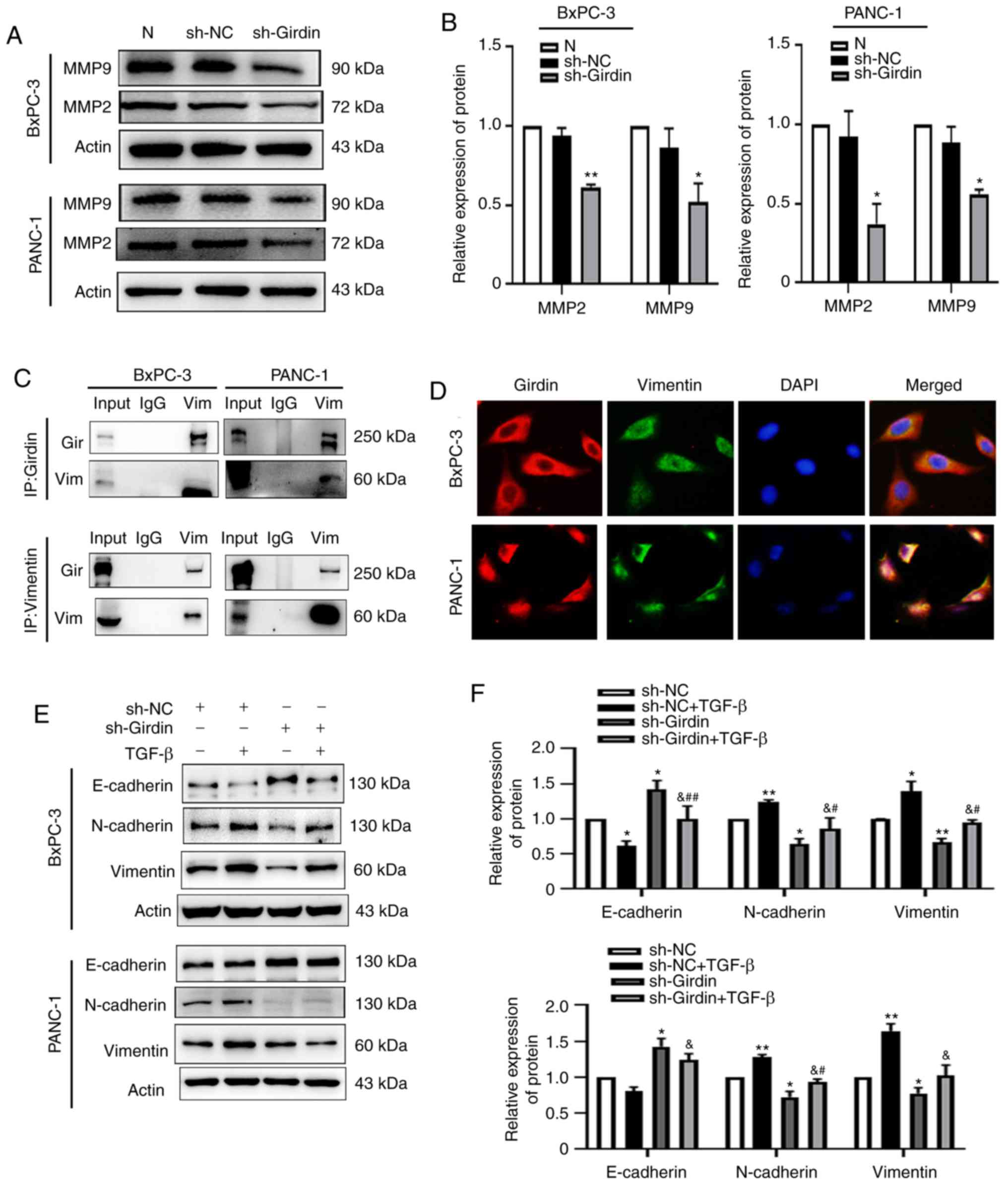 | Figure 4.Girdin interacts with vimentin to
reverse TGF-β-induced EMT. (A and B) Girdin was knocked down by
sh-Girdin, and the cells were treated for 48 h; western blotting
was used to analyze the protein levels of MMP2 and MMP9 in the PDAC
cell lines. (C) Girdin-interacting proteins were assessed in PANC-1
and BxPC-3 cells by co-IP, which revealed the endogenous
Girdin-vimentin interaction. The lysates were analyzed by western
blotting. (D) Endogenous Girdin and vimentin in PANC-1 and BxPC-3
cells was detected by immunofluorescence staining. The cells were
not treated before fixation with 4% paraformaldehyde, and the
cellular location of Girdin (red) and vimentin (green) was detected
by immunofluorescence staining using anti-Girdin and anti-vimentin
antibodies. The nuclei were stained with DAPI (blue) and visualized
by fluorescence microscopy at a scale bar of 100 µm. (E and F) The
EMT markers E-cadherin, N-cadherin and vimentin were evaluated by
western blotting. As revealed, sh-Girdin reduced the protein levels
of N-cadherin and vimentin compared with those in the sh-NC group,
and the TGF-β-induced change in the expression of these markers was
reversed by sh-Girdin. Three experiments were independently
performed. The data are presented as the mean ± SD. *P<0.05, and
**P<0.01 compared with sh-NC; &P<0.05,
compared with sh-NC co-incubated with TGF-β; #P<0.05,
and ##P<0.01 compared with sh-Girdin. PDAC,
pancreatic ductal adenocarcinoma; Gir, Girdin; Vim, Vimentin;
TGF-β, transforming growth factor-β; EMT, epithelial-mesenchymal
transition; sh-NC, non-targeting control short hairpin RNA;
sh-Girdin, a specific short hairpin RNA of Girdin. |
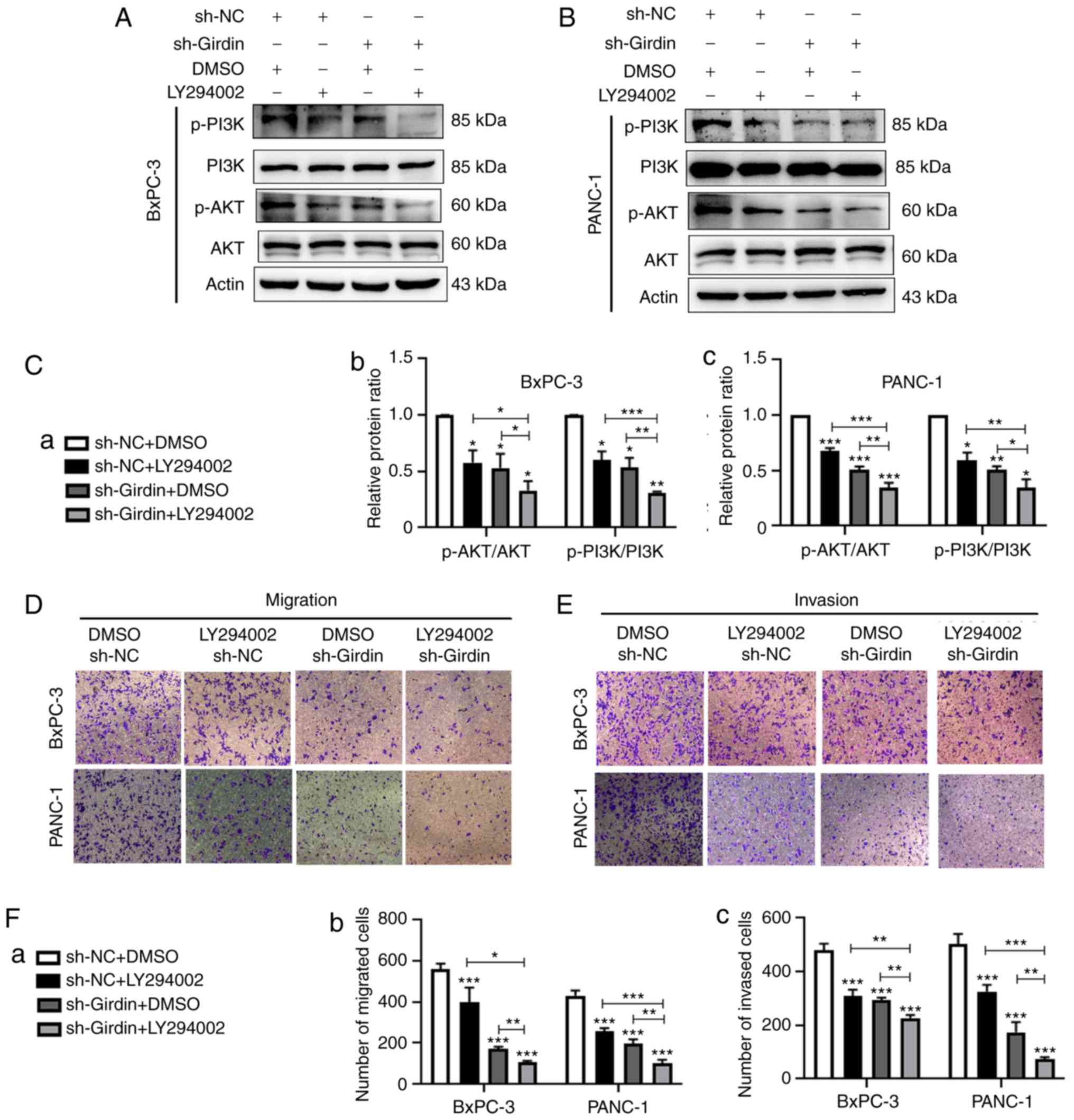
Girdin inhibition suppresses cell
migration and invasion through the PI3K/AKT signaling pathway
There is evidence suggesting that Girdin is a
substrate of AKT and has an important role in regulating the
migration of cancer cells, such as colon cancer cells (18,22,25).
To the best of our knowledge, we were the first to further explore
the potential mechanism by which sh-Girdin inhibits pancreatic
cancer cell migration and invasion. Thus, the effect of Girdin on
PI3K/Akt pathway activation in PDAC cells was examined. Western
blotting was performed to analyze the level of phosphorylation of
PI3K and AKT, and the total protein level of PI3K and AKT.
Consistent with previous results, knockdown of Girdin significantly
inhibited the phosphorylation of PI3K and AKT in PANC-1 and BxPC-3
cells compared with that in the negative control cells (Fig. 5A-C). These results revealed that
Girdin knockdown inactivated the PI3K/Akt signaling pathway.
Furthermore, it was demonstrated that treatment with the PI3K
inhibitor LY294002 significantly enhanced the inhibitory effects of
sh-Girdin on PDAC cells (Fig.
5A-C). In addition, a Transwell assay was used to confirm that
Girdin regulates PDAC cell migration and invasion via the PI3K/AKT
pathway. As the results revealed, the PI3K inhibitor LY294002
further increased the inhibitory effect of sh-Girdin on PANC-1 cell
migration and invasion in the sh-Girdin with LY294002 group
compared to the sh-Girdin with DMSO group. Similar results were
observed in BxPC-3 cells (Fig.
5D-F). These results indicated that Girdin knockdown may
suppress pancreatic cell migration and invasion by inactivating the
PI3K/Akt signaling pathway.
Knockdown of Girdin inhibits the
growth and metastasis of PDAC cells in vivo
A xenograft mouse model was further applied to
confirm the aforementioned in vitro findings. BxPC-3 cells
were stably transfected with sh-Girdin or negative control sh-NC.
Then, control cells and BxPC-3 cells with stable knockdown of
Girdin were inoculated into male nude mice to determine whether
Girdin affects PDAC cell tumorigenesis in vivo. In this
study, On the 18th day after injection, compared with
the negative control containing the sh-RNA vector (sh-NC), the
tumors formed by BxPC-3 cells with Girdin-shRNA (sh-Girdin) clearly
grew significantly slower in male nude mice (Fig. 6A and B). Moreover, the weights of
tumors from the sh-Girdin group were significantly lighter than
those from the sh-NC group (Fig.
6C). Images from these experiments revealed H&E and Ki-67
staining along with Girdin staining of xenograft tumors from the
sh-NC and sh-Girdin groups. Tumors formed from BxPC-3 cells
transfected with sh-Girdin exhibited decreased expression of Ki-67
compared with that in tumors formed from sh-NC cells. Similarly, a
positive decrease in Girdin was confirmed in the tumors formed from
the sh-Girdin group by immunohistochemistry (Fig. 6D). Furthermore, the protein level of
Girdin was also inhibited by sh-Girdin compared with the sh-NC
group in vivo (Fig. 6E). To
further investigate whether sh-Girdin affected tumor cell migration
in nude mice, western blotting was also used to detect EMT
biomarkers. Consistent with the function in vitro, the
protein expression of N-cadherin and vimentin was suppressed, and
the expression of E-cadherin was increased in vivo (Fig. 6E). Moreover, the lung metastasis of
nude mice was assessed to demonstrate that Girdin is involved in
malignant tumor metastasis. In the present study, stable
Girdin-knockdown BxPC-3 cells and sh-NC BxPC-3 cells were injected
into two groups of mice through the tail vein. Six weeks later,
lung metastasis in the sh-Girdin group was significantly decreased
compared with that in the sh-NC group, not only in the number of
nodules but was also observed with the area of H&E staining
(Fig. 6F and G). Therefore, these
results indicated that knockdown of Girdin could exert an antitumor
effect in vivo, which further demonstrated the important
role of Girdin in PDAC growth and metastasis.
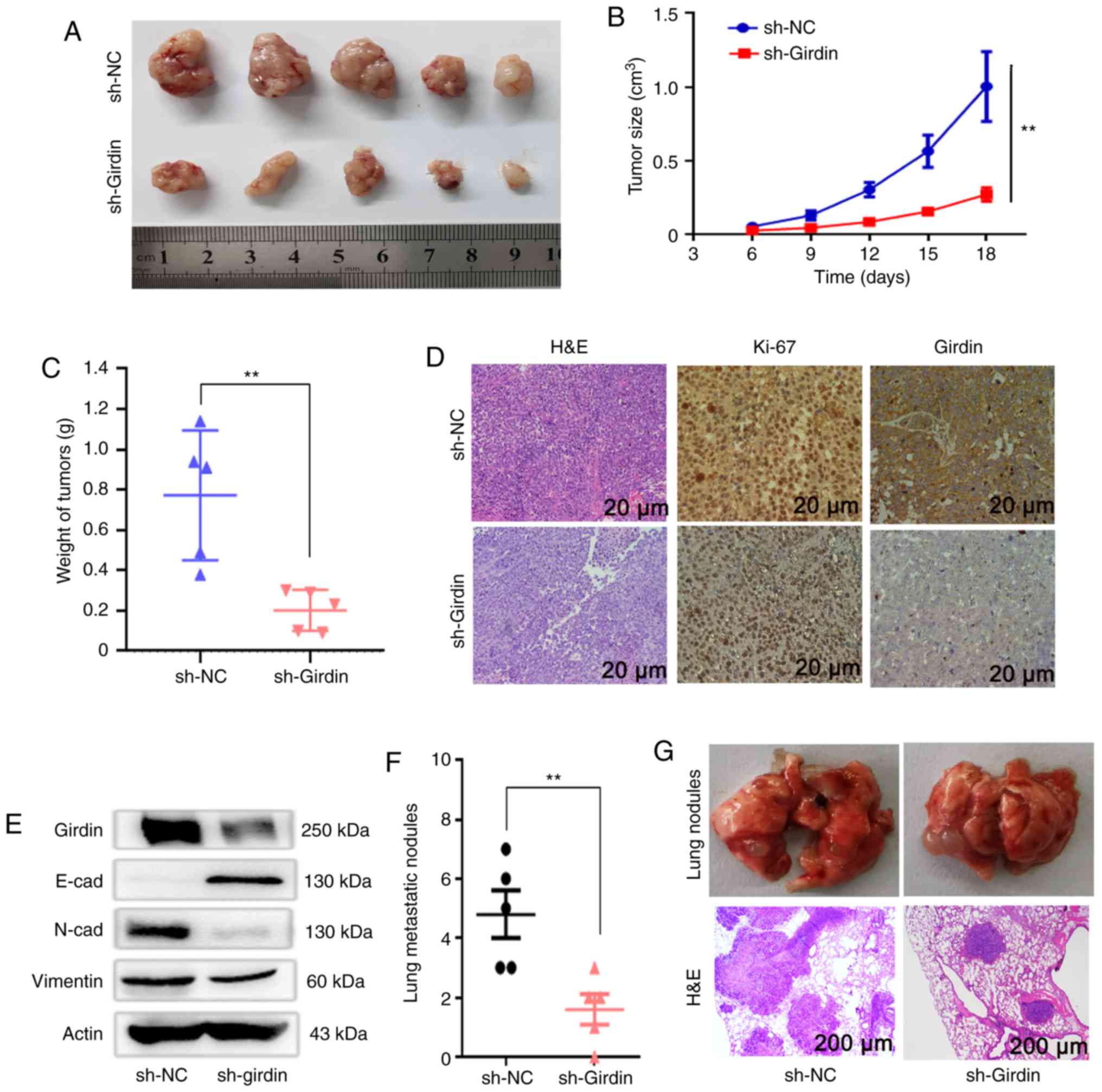 | Figure 6.Girdin regulates PDAC cell tumor
growth and metastasis in vivo. (A) Stable Girdin-knockdown
BxPC-3 cells were injected under the skin of nude mice. (B) Tumor
volumes were first calculated after injection for 6 days and
measured every three days after. (C) The tumors were dissected and
measured, and the tumor weights in the sh-Girdin group were reduced
compared to those in the sh-NC group. (D) Immunostaining revealed
H&E staining and staining using antibodies against Ki-67 and
Girdin in xenograft tumors. The levels of Ki-67 and Girdin in the
samples from the sh-Girdin group were lower than those in the
control group. Scale bar, 20 µm. (E) The protein level of Girdin
was also detected by western blotting, and the changes in EMT
markers were similar to those observed in vitro. (F) Stable
Girdin-knockdown BxPC-3 cells were injected into the mice through
the tail vein, and six weeks later, the lung metastasis in the
sh-Girdin group was significantly decreased compared with that in
the sh-NC group based on the number of nodules and the area of
H&E staining. Scale bar, 200 µm. The tumor weight and the lung
metastatic nodules are presented as the mean ± SD. **P<0.01
compared with sh-NC group. PDAC, pancreatic ductal adenocarcinoma;
H&E, hematoxylin and eosin; EMT, epithelial-mesenchymal
transition; sh-NC, non-targeting control short hairpin RNA;
sh-Girdin, a specific short hairpin RNA of Girdin. |
Discussion
Among pancreatic tumors, PDAC is the most common
type, and the poor prognosis of PDAC is mainly related to its
metastasis (33). Despite
significant attention being paid to enhancing early detection
strategies in PDAC patients, 85% of patients are diagnosed with
metastatic or locally advanced PDAC (34). However, the underlying mechanisms of
PDAC metastasis are unclear. Our previous study reported that
Girdin was highly expressed in pancreatic cancer tissues and was
associated with pathological grade (30). In the present study, it was revealed
that the level of Girdin was upregulated in PDAC cell lines and
that high Girdin expression was related to poor prognosis in PDAC
patients by analyzing GEPIA data. Therefore, the present research
mainly explored the mechanism of Girdin in pancreatic cancer
metastasis. Girdin was knocked down in PDAC cell lines, and the
results revealed that cell viability and mobility in vitro
were inhibited. In addition, knockdown of Girdin suppressed PDAC
growth and metastasis in vivo. The molecular mechanism
assays indicated that Girdin may activate the PI3K/AKT signaling
pathway, which is related to the invasion and migration of PDAC
cells. Mechanistic investigations also elucidated that Girdin may
interact with vimentin to suppress TGF-β-induced EMT in pancreatic
cancer.
To date, numerous studies have revealed that Girdin
plays an important role in tumorigenesis and progression (27–30).
Evidence that Girdin plays a key role in human PDAC development and
progression was also provided, partially by activating the PI3K/AKT
signaling pathway. It was revealed that the mRNA expression of
Girdin and the protein levels of Girdin were increased in PANC-1,
AsPC-1 and BxPC-3 (human PDAC cell lines) compared with HPNE (a
normal human pancreatic epithelial cell line).
In our previous study, IHC staining also
demonstrated higher protein expression of Girdin in PDAC tissues
compared to matched adjacent cancer tissues (30). The expression of Girdin was also
demonstrated to be higher in breast cancer and colon cancer tissues
than in adjacent normal tissues (29–30).
The functional role of Girdin revealed in the present study was
also similar to previous studies in other cancers (20,24,25).
The knockdown of Girdin in pancreatic cancer cells not only
suppressed the proliferation of PDAC in vitro but also
suppressed the growth of PDAC in vivo. Furthermore, Girdin
knockdown also decreased the metastasis of PDAC. Collectively, this
evidence indicated that Girdin may play a critical role in the
development and progression of PDAC. The present results suggest a
new potential intervention target for PDAC metastasis treatment
that may improve the prognosis of PDAC patients.
Cancer cell invasion and metastasis are primarily
responsible for most cancer-associated deaths, including those
related to PDAC (2). EMT is an
important developmental process in the distant metastasis of
pancreatic cancer (35). EMT is
also linked to invasion and dissemination in numerous types of
cancer (13–14). Epithelial cells undergo various
biochemical changes, including the loss of epithelial
characteristics and the acquisition of mesenchymal properties
(11–12). The characteristic changes in EMT are
the loss of epithelial marker E-cadherin and the increase in
mesenchymal markers vimentin and N-cadherin (34,36).
Recently, it was revealed that downregulation of Girdin suppressed
breast cancer and neuroglioma migration and invasion in
vitro and that Girdin may interact with the epithelial marker
E-cadherin to regulate cell-cell adhesion (24,26,37).
Similar to a previous study (21),
it was observed that Girdin may interact with the mesenchymal
marker vimentin to affect pancreatic cancer cell migration. The
results indicated that the Girdin knockdown-induced increase in
E-cadherin and decrease in N-cadherin may at least partly
contribute to vimentin- and MMP-mediated invasion. TGF-β is a
crucial driver of EMT and has been demonstrated to regulate cell
mobility and EMT progression in pancreatic cancer (15–16).
It was hypothesized that Girdin may also affect PDAC progression by
being involved in TGF-β-induced EMT. Therefore, it was explored
whether Girdin regulated the progression of TGF-β-induced EMT in
PDAC. Evidence was provided that with increasing concentrations and
24-h TGF-β treatment, the EMT markers in PDAC were significantly
altered, which is similar to previous studies (38,39),
and knockdown of Girdin effectively reversed TGF-β-induced EMT.
These results demonstrated the potential role of Girdin suppression
in reversing EMT in PDAC. This research, to the best of our
knowledge, is the first to highlight the potential roles of the
Girdin-vimentin complex in the EMT process. The present results
revealed that Girdin may be a novel actin protein that has a
significant effect on tumor EMT progression, which is associated
with distant metastasis and could be a potential metastatic target
for the treatment of PDAC.
The PI3K/AKT pathway is a classic signaling pathway
associated with the development and progression of numerous types
of cancers, and the PI3K/AKT pathway is aberrantly activated in the
disease progression of pancreatic cancer (40–41).
In pancreatic cancer cells, evidence has revealed that the PI3K/AKT
pathway plays a crucial role in regulating cellular metabolism,
proliferation, metastasis and survival (42–44).
It has also been revealed that the PI3K/AKT pathway is a key
regulator in pancreatic tumorigenesis and may be a treatment target
of other cancers (45). It has been
reported that knockdown of the expression of Girdin inhibits the
migration and invasion ability of glioma cancer cells by
suppressing the phosphorylation/activation of the PI3K/AKT pathway
(46). Mechanistically, Girdin is
also known as Akt phosphorylation enhancer because it can be
regarded as a substrate for the phosphorylation of AKT (17,47).
Therefore, further study of the role of Girdin in the PI3K/AKT
pathway may be beneficial to reveal the underlying molecular
mechanism of the regulatory effect of Girdin on PDAC. As revealed
in the present study, the knockdown of Girdin decreased the levels
of p-Akt and p-PI3K via the PI3K/AKT pathway in PDAC cell lines,
which was enhanced by treatment with the PI3K inhibitor LY294002.
The metastatic ability of Girdin through inhibition of PI3K/AKT was
further determined, and knockdown of Girdin and inhibition of the
PI3K/AKT pathway, significantly decreased PDAC migration. Thus, it
was concluded that the promoting effect of Girdin on PDAC cells
partly contributed to the aberrant activation of PI3K/AKT
signaling.
In the present study, a novel actin protein, Girdin,
was first identified, whose knockdown by recombinant adenoviruses
suppressed the growth and metastasis of PDAC cells. Further study
involving in vivo tumorigenicity assays and metastasis
assays demonstrated that Girdin knockdown also suppressed PDAC
growth and metastasis. In addition, it was revealed that the
Girdin-related suppression of PDAC cell invasion and metastasis may
rely on the inhibition of aberrant PI3K/Akt pathway activation.
Moreover, whether Girdin interacts with vimentin to suppress
TGF-β-induced EMT was also investigated. In summary, the present
findings revealed that Girdin may contribute to the EMT and
metastasis of PDAC. Girdin may act as a promising biological marker
and a candidate metastatic target for the treatment of pancreatic
cancer patients. Although the mechanisms by which Girdin elicits
its effects on PDAC cell apoptosis were reported in our previous
study (30), its potential effects
on autophagy should be further investigated and verified. To the
best of our knowledge, this is the first study that investigates
the metastasis and TGF-β-induced EMT effect of Girdin on PDAC. We
acknowledge several limitations in the present study. First, there
was no analysis of the correlation between Girdin and vimentin in
pancreatic cancer tissues, and there was no in-depth study on the
binding region of Girdin and vimentin. Thus, further studies are
required to reveal the underlying mechanism by which the
Girdin/vimentin axis regulates EMT in pancreatic cancer.
Acknowledgements
All of the authors gratefully thank Professors
Xiubin Liang and Dongming Su, Department of Pathophysiology, School
of Basic Medical Sciences (Nanjing Medical University, Nanjing,
China) for providing an available experimental platform and
experimental guidance and Ms. Min Li, a research assistant, for
experimental management and assistance.
Funding
Funding for the present study was mainly provided
from the National Natural Science Foundation of China (nos.
30972910 and 81172269); the Six Talent Peak Funding Projects of
Jiangsu Province (WSW-032); the Natural Science Foundation of
Jiangsu Province (no. 050104313) and the key Research and
Development Projects of Suqian (S201809).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
WW, HC, WH and CY conceived the study and designed
the experiments. WW, SW and KW performed the experiments on cell
culture, construction of the xenograft and lung metastasis model of
subcutaneous tumor formation in a nude mouse, RT-qPCR and western
blot analysis. XL, CL and LL performed analysis of data and
interpretation of data and critically reviewed the manuscript for
important intellectual content. WW, HC, WG and CY drafted the
manuscript and integrated all the results. All the authors approved
the manuscript preparation and submission.
Ethics approval and consent to
participate
This study strictly followed the guidelines issued
[Eighth Edition (2011)] by the National Institutes of Health for
the Care and Use of Laboratory Animals. The Animal Ethics and
Welfare Committee (AEWC) of Nanjing Medical College provided
protocol approval (IACUC-1601161)
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
TGF-β
|
transforming growth factor-β
|
|
BSA
|
bovine serum albumin
|
|
p-AKT
|
phosphorylated protein kinase B
|
|
AKT
|
protein kinase B, SDS-PAGE,
sulphate-polyacrylamide gel electrophoresis
|
|
PBS
|
phosphate-buffered saline
|
|
TBST
|
Tris-buffered solution containing
Tween
|
|
PVDF
|
polyvinylidene fluoride
|
|
MMP2/9
|
matrix metalloproteinase-2/9
|
|
p-PI3K
|
phosphorylated phosphatidylinositol
3-kinase
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
References
|
1
|
Owens DK, Davidson KW, Krist AH, Barry MJ,
Cabana M, Caughey AB, Curry SJ, Doubeni CA, Epling JW, Kubik M, et
al: Screening for pancreatic cancer. JAMA. 322:4382019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moffat GT, Epstein AS and O'Reilly EM:
Pancreatic cancer-A disease in need: Optimizing and integrating
supportive care. Cancer. 125:3927–3935. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rahib L, Fleshman JM, Matrisian LM and
Berlin JD: Evaluation of pancreatic cancer clinical trials and
benchmarks for clinically meaningful future trials: A systematic
review. JAMA Oncol. 2:1209–1216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi Y, Gao W, Lytle NK, Huang P, Yuan X,
Dann AM, Ridinger-Saison M, Del Giorno KE, Antal CE, Liang G, et
al: Targeting LIF-mediated paracrine in- teraction for pancreatic
cancer therapy and monitoring. Nature. 569:131–135. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fox RG, Lytle NK, Jaquish DV, Park FD, Ito
T, Bajaj J, Claire S, Koechlein CS, Zimdahl B, Yano M, et al: Image
based detection and targeting of therapy resistance in pancreatic
adenocarcinoma. Nature. 534:407–411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karmazanovsky G, Fedorov V, Kubyshkin V
and Kotchatkov A: Pancreatic head cancer: Accuracy of CT in
determination of resectability. Abdom Imaging. 30:488–500. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strobel O, Neoptolemos J, Jager D and
Buchler MW: Optimizing the outcomes of pancreatic cancer surgery.
Nat Rev Clin Oncol. 16:11–26. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Amrutkar M and Gladhaug I: Pancreatic
cancer chemoresistance to gemcita-bine. Cancers. 9:1572017.
View Article : Google Scholar
|
|
11
|
Feng H, Zhao X, Guo Q, Feng Y, Ma M, Guo
W, Dong X, Deng C, Li C, Song X, et al: Autophagy resists EMT
process to maintain retinal pigment epithelium homeostasis. Int J
Biol Sci. 15:507–521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pastushenko I and Blanpain C: EMT
transition states during tumor pro-gression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Costanza B, Rademaker G, Tiamiou A, De
Tullio P, Leenders J, Blomme A, Bellier J, Bianchi E, Turtoi A,
Delvenne P, et al: Transforming growth factor beta-induced, an
extracellular matrix interacting protein, enhances glycolysis and
promotes pancreatic cancer cell migration. Int J Cancer.
145:1570–1584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang N, Liu Y, Wang Y, Zhao M, Tu L and
Luo F: Decitabine reverses TGF-β1-induced epithelial-mesenchymal
transition in non-small-cell lung cancer by regulating miR-200/ZEB
axis. Drug Des Devel Ther. 11:969–983. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Enomoto A, Murakami H, Asai N, Morone N,
Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K and
Takahashi M: Akt/PKB regulates actin organization and cell motility
via Girdin/APE. Dev Cell. 9:389–402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weng L, Enomoto A, Ishida-Takagishi M,
Asai N and Takahashi M: Girding for migratory cues: Roles of the
Akt substrate Girdin in cancer progression and angiogenesis. Cancer
Sci. 101:836–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anai M, Shojima N, Katagiri H, Ogihara T,
Sakoda H, Onishi Y, Ono H, Fujishiro M, Fukushima Y, Horike N, et
al: A novel protein kinase B (PKB)/AKT-binding protein enhances PKB
kinase activity and regulates DNA synthesis. J Biol Chem.
280:18525–18535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang P, Enomoto A, Jijiwa M, Kato T,
Hasegawa T, Ishida M, Sato T, Asai N, Murakumo Y and Takahashi M:
An actin-binding protein Girdin regulates the motility of breast
cancer cells. Cancer Res. 68:1310–1318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Enomoto A, Weng L, Mizutani Y,
Abudureyimu S, Esaki N, Tsuyuki Y, Chen C, Mii S, Asai N, et al:
Girdin/GIV regulates collective cancer cell migration by
controlling cell adhesion and cytoskeletal organization. Cancer
Sci. 109:3643–3656. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Natsume A, Kato T, Kinjo S, Enomoto A,
Toda H, Shimato S, Ohka F, Motomura K, Kondo Y, Miyata T, et al:
Girdin maintains the stemness of glioblastoma stem cells. Oncogene.
31:2715–2724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamamura Y, Asai N, Enomoto A, Kato T, Mii
S, Kondo Y, Ushida K, Niimi K, Tsunoda N, Nagino M, et al:
Akt-Girdin signaling in cancer-associated fibroblasts contributes
to tumor progression. Cancer Res. 75:813–823. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi J, Kim KH, Oh E, Shin YK, Seo J, Kim
S, Park S and Choi Y: Girdin protein expression is associated with
poor prognosis in patients with invasive breast cancer. Pathology.
49:618–626. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghosh P, Tie J, Muranyi A, Singh S,
Brunhoeber P, Leith K, Bowermaster R, Liao Z, Zhu Y, LaFleur B, et
al: Girdin (GIV) expression as a prognostic marker of recurrence in
mismatch repair-proficient stage ii colon cancer. Clin Cancer Res.
22:3488–3498. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shibata T, Matsuo Y, Shamoto T, Hirokawa
T, Tsuboi K, Takahashi H, Ishiguro H, Kimura M, Takeyama H and
Inagaki H: Girdin, a regulator of cell motility, is a potential
prognostic marker for esophageal squamous cell carcinoma. Oncol
Rep. 29:2127–2132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gu F, Wang L, He J, Liu X, Zhang H, Li W,
Fu L and Ma Y: Girdin, an actin-binding protein, is critical for
migration, adhesion, and invasion of human glioblastoma cells. J
Neurochem. 131:457–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weng L, Han Y, Enomoto A, Kitaura Y,
Nagamori S, Kanai Y, Asai N, An J, Takagishi M, Asai M, et al:
Negative regulation of amino acid signaling by MAPK-regulated
4F2hc/Girdin complex. PLoS Biol. 16:e20050902018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia-Marcos M, Ear J, Farquhar MG and
Ghosh P: A GDI (AGS3) and a GEF (GIV) regulate autophagy by
balancing G protein activity and growth factor signals. Mol Biol
Cell. 22:673–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang S, Lei Y, Cai Z, Ye X, Li L, Luo X
and Yu C: Girdin regulates the proliferation and apoptosis of
pancreatic cancer cells via the PI3K/Akt signalling pathway. Oncol
Rep. 40:599–608. 2018.PubMed/NCBI
|
|
31
|
Livak JK and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang C, Qiao C, Wang R and Zhou W:
KiSS-1-mediated suppression of the invasive ability of human
pancreatic carcinoma cells is not dependent on the level of KiSS-1
receptor GPR54. Mol Med Rep. 13:123–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Si W, Liu X, Wei R, Zhang Y, Zhao Y, Cui L
and Hong T: MTA2-mediated inhibition of PTEN leads to pancreatic
ductal adenocarcinoma carcinogenicity. Cell Death Dis. 10:2062019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang J, Mei H, Tang Z, Li J, Zhang X, Lu
Y, Huang F, Jin Q and Wang Z: Triple-amiRNA VEGFRs inhibition in
pancreatic cancer improves the effificacy of chemotherapy through
EMT regulation. J Cont Rele. 245:1–14. 2017. View Article : Google Scholar
|
|
35
|
Aiello NM, Brabletz T, Kang Y, Nieto MA,
Weinberg RA and Stanger BZ: Upholding a role for EMT in pancreatic
cancer metastasis. Nature. 547:E7–E8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Braitsch CM, Azizoglu DB, Htike Y, Barlow
HR, Schnell U, Chaney CP, Carroll TJ, Stanger BZ and Cleaver O:
LATS1/2 suppress NFκB and aberrant EMT initiation to permit
pancreatic progenitor differentiation. PLoS Biol. 17:e30003822019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lekka K, Tzitzi E, Giakoustidis A,
Papadopoulos V and Giakoustidis D: Contemporary management of
borderline resectable pancreatic ductal adenocarcinoma. Ann
Hepatobiliary Pancreat Surg. 23:972019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bu JQ and Chen F: TGF-β1 promotes cells
invasion and migration by inducing epithelial mesenchymal
transformation in oral squamous cell carcinoma. Eur Rev Med
Pharmacol Sci. 21:2137–2144. 2017.PubMed/NCBI
|
|
39
|
Zhang Y, Li JH, Yuan QG, Cao G and Yang
WB: Upregulation of LASP2 inhibits pancreatic cancer cell migration
and invasion through suppressing TGF-β-induced EMT. J Cell Biochem.
120:13651–13657. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mann KM, Ying H, Juan J, Jenkins NA and
Copeland NG: KRAS-related proteins in pancreatic cancer. Pharmacol
Ther. 168:29–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song M, Bode AM, Dong Z and Lee MH: AKT as
a therapeutic target for cancer. Cancer Res. 79:1019–1031. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Martini M, De Santis MC, Braccini L,
Gulluni F and Hirsch E: PI3K/AKT signaling pathway and cancer: An
updated review. Ann Med. 46:372–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamamoto S, Tomita Y, Hoshida Y, Morooka
T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H,
et al: Prognostic significance of activated Akt expression in
pancreatic ductal adenocarcinoma. Clin Cancer Res. 10:2846–2850.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stoll V, Calleja V, Vassaux G, Downward J
and Lemoine NR: Dominant negative inhibitors of signalling through
the phosphoinositol 3-kinase pathway for gene therapy of pancreatic
cancer. Gut. 54:109–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ng SSW, Tsao MS, Chow S and Hedley DW:
Inhibition of phosphatidylinositide 3-kinase enhances
gemcitabine-induced apoptosis in human pancreatic cancer cells.
Cancer Res. 60:5451–5455. 2000.PubMed/NCBI
|
|
46
|
Ni W, Fang Y, Tong L, Tong Z, Yi F, Qiu J,
Wang R and Tong X: Girdin regulates the migration and invasion of
glioma cells via the PI3K-Akt signaling pathway. Mol Med Rep.
12:5086–5092. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mittal Y, Pavlova Y, Garcia-Marcos M and
Ghosh P: Src homology domain 2-containing protein-tyrosine
phosphatase-1 (SHP-1) binds and dephosphorylates
G(alpha)-interacting, vesicle-associated protein (GIV)/Girdin and
attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt
signaling pathway. J Biol Chem. 286:32404–32415. 2011. View Article : Google Scholar : PubMed/NCBI
|