Introduction
Osteosarcoma is the most common primary malignancy
of bones, and accounts for ~2% of all childhood malignancies
(1–3). It is primarily diagnosed in teens and
adolescents and in the elderly population over 65 years (4,5).
Osteosarcoma is a markedly aggressive cancer with a high capacity
to form distant metastases, and there is a high rate of resistance
to the traditional therapeutic approaches (3). There have been only minimal advances
made in recent decades in the treatment of osteosarcoma (5). Although multimodal therapy has
revealed improved outcomes in patients with osteosarcoma, this is
not the case for the patients who present with metastases at the
initial diagnosis (6). Therefore,
it is necessary to study the pathogenesis and metastatic mechanisms
underlying the development of osteosarcoma, to identify novel
therapeutic targets for improving the treatment and prognosis.
Maternal embryonic leucine zipper kinase (MELK)
belongs to the Snf1/AMPK kinase family and is also referred to as
murine protein K38 (MPK38) or Eg3 protein (7,8). It is
highly expressed in the egg and in the pre-implanted embryo in
mice, and influences embryonic development (9). MELK is a highly conserved
AMP-activated Ser/Thr protein kinase (7), and its expression is upregulated in
several types of cancer, including glioblastoma, melanoma, breast
cancer, ovarian cancer, gastric cancer and colorectal cancer
(10–15). MELK performs various functions in
different types of cancer, which promote the development and
progression of cancer (13,16,17).
However, the role and mechanism of MELK in osteosarcoma has not
been studied, to the best of our knowledge.
In the present study, the effects of MELK on crucial
functions, such as proliferation, metastasis and apoptosis, which
promote the progression of osteosarcoma, was studied. OTSSP167, a
MELK inhibitor, is already in phase I clinical trials for the
treatment of solid tumors that do not respond to any other
treatment (18,19), and if successful may serve as
therapy for the treatment of several different types of cancer.
Therefore, the role of the novel molecular inhibitor of MELK,
OTSSP167, in osteosarcoma was also explored.
The aim of the present study was to determine the
roles and the underlying mechanisms of MELK in osteosarcoma. MELK
expression was determined to be upregulated in osteosarcoma
compared with the normal control samples, and the increased
expression was associated with a less favorable prognosis. Through
analysis of the clinicopathological characteristics, it was
revealed that there was a positive association between MELK
expression with metastasis and a poor response to chemotherapy. A
series of in vitro and in vivo experiments were
performed to establish the role of MELK in crucial functions such
as proliferation, metastasis, cell cycle progression and apoptosis
of osteosarcoma. Furthermore, the role and therapeutic effects of
OTSSP167 in osteosarcoma were also studied in vitro and
in vivo. The results revealed that MELK promoted
osteosarcoma proliferation and metastasis by regulating PCNA and
MMP9 expression through the PI3K/Akt/mTOR signaling pathway.
Collectively, these results indicated that MELK may be a novel
prognostic marker and therapeutic target for treatment of
osteosarcoma.
Materials and methods
Analysis of public datasets
Gene expression data including 84 cases of
osteosarcoma were acquired from the Therapeutically Applicable
Research to Generate Effective Treatments (TARGET) program, which
is accessible publicly (https://portal.gdc.cancer.gov/projects). The relative
differences in MELK expression were analyzed using X-tile software
(version 3.6.1; Yale University School of Medicine) to determine
the thresholds for producing the most significant log-rank test
P-values, which segregated the data into MELK-low and MELK-high
groups. The differences in survival between the low- and
high-expression groups were evaluated using Kaplan-Meier
analysis.
Tissue samples and cell lines
A total of 25 osteosarcoma tissue samples that had
not received any neoadjuvant chemotherapy and 5 osteoblastoma
tissue samples were collected from patients who were treated at
Qilu Hospital of Shandong University from January 2010 to June
2014. All patients signed informed consent to participate in the
present study, and the study was approved by the Ethics Committee
of Qilu Hospital of Shandong University. The small number of
samples is due the fact that most osteosarcoma patients receive
preoperative neoadjuvant chemotherapy. MNNG/HOS and hFOB1.19 cell
lines were purchased from the American Type Culture Collection.
MNNG/HOS cells were maintained in DMEM with 5% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and hFOB1.19 cells were maintained in a
1:1 mixture of Ham's F12 medium and DMEM supplemented with 10% FBS.
All the cells were incubated at 37°C with 5% CO2. MELK
inhibitor (OTSSP167; cat. no. S7159) and PI3K inhibitor (LY294002;
cat. no. S1105) were purchased from Selleck Chemicals.
Immunohistochemistry
Immunohistochemical staining was performed on tissue
sections (4-µm thick) of formalin-fixed at room temperature for 48
h, paraffin-embedded osteosarcoma tissue samples. Xylene and
ethanol were used to deparaffinize and rehydrate the tissue slides.
Antigen retrieval was performed using sodium citrate buffer
solution (pH 6.0) maintained at a sub-boiling temperature for 15
min. Endogenous peroxidase and nonspecific binding were blocked
using 3% hydrogen peroxide for 15 min and goat serum (SP-kit: cat.
no. SP9000; Beijiing Zhongshan Golden Bridge Biotechnology) for 30
min, respectively at room temperature. Tissues were incubated with
anti-MELK primary antibody (1:250; cat. no. bs-12201R; Bioss) in a
humid chamber overnight at 4°C. Expression was detected using a
I–View 3,3′-diaminobenzidine staining technique. To evaluate the
staining intensity, semiquantitative evaluation of protein levels
in tissues was performed using the H-score technique (range, 0–3)
(20). The staining intensity was
graded as: 0, none; 1, weak; 2, moderate; and 3, strong. The
H-score was calculated using the formula: H-score=ΣPi × I; where i
represents the staining intensity and Pi the percentage of cells at
each level of intensity. An H-score <1.5 was classified as low,
and a H-score ≥1.5 was classified as a high protein expression
level.
Transient transfection
Small interfering (si)RNAs and the corresponding
negative control (NC) were used to transiently knockdown expression
of MELK. The siRNAs were purchased from (Shanghai GenePharma, Co.,
Ltd.). RNAi-mediated knockdown was performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
sequences of the siRNA used were: si-MELK,
5′-GACAUCCUAUCUAGCUGCA-3′ and si-NC,
5′-UUCUCCGAACGUGUCACGUTT-3′.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from osteosarcoma cells
using TRIzol® reagent (Ambion; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The cDNA was
synthesized using a One Step PrimeScript miRNA cDNA Synthesis kit
(Takara Bio, Inc.). qPCR was performed using a SYBR Green Premix Ex
Taq II (Takara Bio, Inc.) with a StepOne Plus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The reaction
conditions were as follows: Denaturation at 95°C for 5 sec,
followed by 40 cycles of annealing at 60°C for 10 sec and an
extension at 72°C for 30 sec. GAPDH expression was used as the
endogenous control for the detection of miRNA expression levels.
The 2−ΔΔCq was used to calculate the relative gene
expression level (21). The
sequences of the primers used were: GAPDH forward,
5′-CAGGAGGCATTGCTGATGAT-3′ and reverse, 5′-GAAGGCTGGGGCTCATTT3′;
and MELK, forward 5′-TCTCCCAGTAGCATTCTGCTT3′ and reverse
5′-TGATCCAGGGATGGTTCAATAGA3′. Primers were purchased from Sangon
Biotech, Co., Ltd.
Western blotting
Cells and tissues were harvested and lysed using
RIPA Lysis Buffer (Beyotime Institute of Biotechnology) with PMSF
(1%). Protein samples were incubated for 30 min on ice and
sonicated, after which the cell debris were removed by
centrifugation at 12,000 × g. The protein concentration was
determined using a bicinchoninic acid assay kit (Thermo Fisher
Scientific, Inc.). Protein samples (30 µg) were resolved by
SDS-PAGE (5% stacking gel and 10% separating gel) and transferred
to a PVDF membrane (EMD Millipore). The membrane was blocked with
5% non-fat milk at room temperature for 1 h, incubated overnight at
4°C with the primary antibody, and subsequently incubated with
horseradish peroxidase-conjugated anti-rabbit (dilution 1:5,000;
cat. no. TA130023; OriGene Technologies, Inc.) secondary antibody
for 2 h. Signals were visualized using enhanced chemiluminescence
reagent and ImageQuant LAS 4000 (GE Healthcare Life Sciences).
ImageJ software version 1.47v (National Institutes of Health) was
used for densitometric measurement.
The antibodies used were: Phosphorylated (p)-PI3K
p85 (1:1,000; product no. 4228; Cell Signaling Technology, Inc.),
total PI3K p85 (1:1,000; product code ab40755; Abcam), p-Akt(S473)
(1:1,000; product no. 9271S; CST Biological Reagents Co., Ltd.),
total Akt (1:1,000; product no. 9272S; CST Biological Reagents,
Co., Ltd.), GAPDH (1:5,000; cat. no. AF7021; Affinity Biosciences),
MELK (1:1,000; product code ab108529; Abcam), mTOR (1:1,000;
product no. 2972S; CST Biological Reagents, Co., Ltd.), p-mTOR
(1:1,000; product no. 5536S; CST Biological Reagents, Co., Ltd.),
PCNA (1:750, cat. no. AF0239) and MMP9 (1:750; cat. no. AF5228;
both from Affinity Biosciences).
Cell proliferation assay
The proliferative capacity of cells was assessed
using a Cell Counting Kit-8 (CCK-8) assay (BestBio). Each cell line
was seeded in quintuplicate into 96-well plates (1-2×103
cells/well) for 1–5 days. At the specified time-points, 10 µl CCK-8
reagent was added to each well, and the cells were incubated for an
additional 4 h at 37°C. Subsequently, the absorbance values at 450
nm were measured using a Varioskan Flash microplate reader (Thermo
Fisher Scientific, Inc.). Cell proliferation was measured in
si-MELK and si-NC transfected cells, and in cells treated with 40
nM OTSSP167. For the latter, cells were initially treated for 48 h
with OTSSP167.
Cell viability assay
A total of 5×103 cells/well were seeded
in quintuplicate into 96-well plates. The plate was incubated at
37°C for 24 h, and subsequently treated with various concentrations
of OTSSP167 (0, 2, 4, 8, 16, 32 and 64 nM) for 48 h. For evaluation
of cell viability, 10 µl CCK-8 reagent was added to each well and
incubated at 37°C for 4 h. The cell viability was determined by
measuring the absorbance at 450 nm using Varioskan microplate
reader.
Migration and invasion assay
Cellular invasion and migration were analyzed using
Boyden chamber-style cell culture inserts with and without
Matrigel, respectively (BD Falcon; BD Biosciences). Osteosarcoma
cells (1-2×105 cells) were seeded in the upper chambers
of the Transwell inserts (24-wells, 8 µm pores; BD Falcon) with 200
µl serum-free media. The lower chambers were filled with 500 µl
culture media supplemented with 10% FBS as a chemoattractant. After
12–48 h, cells on the lower surface of the membrane were fixed at
room temperature in methanol for 20 min, stained at room
temperature with 0.1% crystal violet for 30 min, and counted using
a light microscope (magnification, ×200).
Wound healing assay
Cell migration was measured using a wound healing
assay. MNNG/HOS cells were used for the assay at a density of
5×105/well. The scratch was generated using 200-µl
pipette tip. The culture medium was replaced with serum-free DMEM
after the generation of a scratch wound, and wound closure was
imaged 48 h later under a light microscope (magnification, ×100).
Wound closure was analyzed and is represented as percent closure.
The experiments were performed in triplicate. Wound healing assays
were performed on si-MELK and si-NC transfected cells, and cells
treated with 10 nM OTSSP167.
Cell cycle distribution and apoptosis
assay
Cell cycle synchronization was achieved by serum
starvation and thymidine double blocking (22). Subsequently, the cells were
harvested and stained with 5 µl propidium iodide at 4°C for 30 min
in the dark according to the manufacturer's protocol (BestBio).
Flow cytometry was performed using a FACScan flow cytometer (BD
Biosciences).
Apoptosis was detected using an Annexin V-APC and
7-AAD kit (BioGems International, Inc.) according to the
manufacturer's protocol, and flow cytometry was performed on the
cells. MNNG/HOS cells were harvested 48 h after transfection or
treated with OTSSP167 (10 nM) and subsequently harvested using
EDTA-free trypsin, centrifuged at 800 × g for 5 min; and, after
washing twice with cold PBS, the cells were resuspended at a
concentration of 1×106/ml and stained with 5 µl Annexin
V-APC and 5 µl 7-AAD at room temperature for 15 min in the dark.
The cells were analyzed by flow cytometry (FACSCalibur) and the
results were analyzed using FlowJo software (version 10; TreeStar,
Inc.).
Animal experiments
Eight pathogen-free female Balb/c nude mice
(weighing 10–14 g; 4 weeks old) were purchased from Beijing Vital
River Laboratory Animal Technology Co., Ltd and housed in standard
sterilized conditions in individually ventilated cages (IVCs) with
high efficiency particulate air (HEPA) filters at an ambient
temperature of 30–31°C, humidity of 50–60% with 12-h light/dark
cycle and adequate access to food and water. The health and
behavior of the animals were monitored every 3 days. All animal
experiments were approved by the Ethics Committee of Qilu Hospital,
Shandong University.
For tumorigenesis assays, 1×107 MNNG/HOS
cells, resuspended in 200 µl PBS, were injected subcutaneously into
the armpits of the mice. The tumor volume was measured daily using
a Vernier caliper, with the following formula: ½ × a ×
b2 where ‘a’ and ‘b’ represent the length and width of
the tumor, respectively. After 7 days, when the tumor volumes
reached ~100 mm3, the tumor-bearing mice were orally fed
Vehicle or OTSSP167 (10 mg/kg, daily). The tumor volumes were
measured daily using Vernier calipers. After 7 days, when the
maximum tumor volumes reached ~1,000 mm3 the mice were
euthanized using cervical dislocation method and then the growth of
the tumors was examined. Following excision, the tumors were
preserved at −80°C for western blot analysis.
Statistical analysis
Data were analyzed using independent samples t-test,
log-rank test, or Fisher's exact test. Fisher's exact test was used
to analyze the relationship between MELK expression and
clinicopathological data of osteosarcoma patients. Overall survival
(OS) curves were plotted using the Kaplan-Meier method and analyzed
using a log-rank test. Quantitative data were analyzed using a
Student's t-test. Data are expressed as the mean ± the standard
deviation of three independent measurements. Statistical analysis
was performed using SPSS version 18.0 (SPSS Inc.) and GraphPad
Prism version 5.0 (GraphPad Software Inc.). Two-tailed P<0.05
was considered to indicate a statistically significant
difference.
Results
MELK expression is upregulated in
osteosarcoma and its overexpression is associated with a less
favorable prognosis
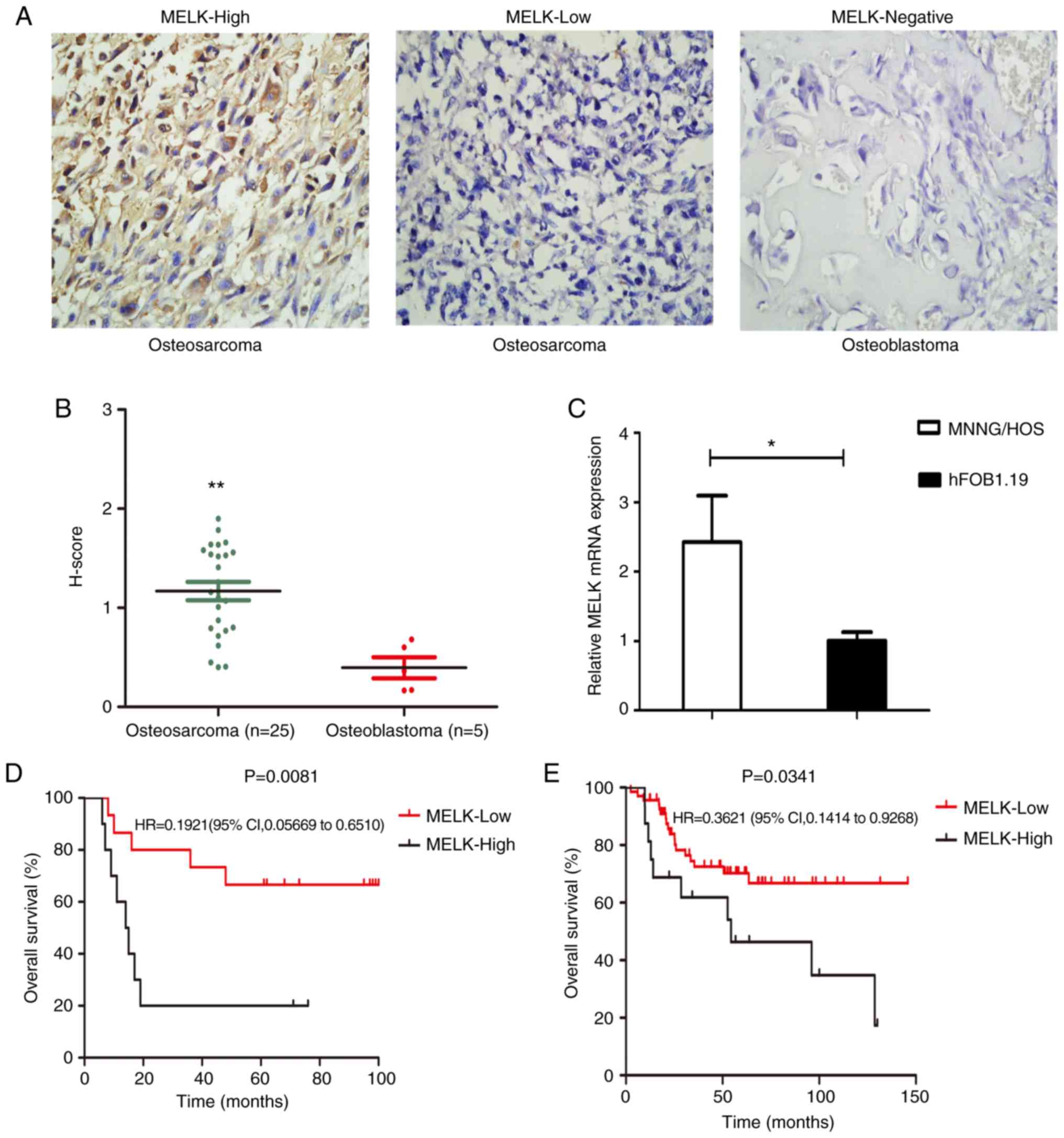
The expression levels of MELK were assessed in 25
osteosarcoma tissue samples and 5 osteoblastoma samples using
immunohistochemistry (Fig. 1A).
H-score evaluation revealed significantly higher protein expression
levels in osteosarcoma tissues compared with the osteoblastoma
tissues (Fig. 1B). Of the 25
samples, 40% exhibited high staining and 60% exhibited low
staining, whereas in the osteoblastoma samples, none of the tissues
showed MELK expression (Fig. 1A).
MELK mRNA expression levels were also compared between MNNG/HOS and
hFOB1.19 cells. MELK expression was significantly higher in the
MNNG/HOS cells compared with the hFOB1.9 cells (Fig. 1C). Subsequently, the association
between MELK expression and various clinicopathological
characteristics of the patients were assessed. As revealed in
Table I higher expression levels of
MELK were associated with metastasis and the response to
chemotherapy. Kaplan-Meier survival curves revealed that OS was
shorter in patients with high MELK expression levels, based on the
data obtained from TARGET (Fig.
1E), in agreement with the present analysis. Thus, high
expression levels of MELK were associated with less favorable OS in
patients (Fig. 1D).
 | Table I.Association between MELK expression
and clinicopathological data of osteosarcoma patients. |
Table I.
Association between MELK expression
and clinicopathological data of osteosarcoma patients.
|
| MELK
expression |
|
|
|---|
|
|
|
|
|
|---|
| Parameters | N 25 | High n=10
(40%) | Low n=15 (60%) | P-value |
|---|
| Age (years) |
|
|
| 0.6968 |
|
<18 | 14 | 5 | 9 |
|
|
≥18 | 11 | 5 | 6 |
|
| Sex |
|
|
| 0.2262 |
|
Male | 13 | 7 | 6 |
|
|
Female | 12 | 3 | 9 |
|
| Location |
|
|
| 0.1500 |
|
Extremity | 23 | 8 | 15 |
|
|
Trunk | 2 | 2 | 0 |
|
| Histological
subtype |
|
|
| 0.8889 |
|
Osteoblastic | 8 | 2 | 6 |
|
|
Fibroblastic | 4 | 3 | 1 |
|
|
Chondroblastic | 7 | 4 | 3 |
|
|
Othersa | 6 | 1 | 5 |
|
| Metastasis |
|
|
| 0.0414b |
|
Yes | 13 | 8 | 5 |
|
| No | 12 | 2 | 10 |
|
| Surgery |
|
|
| 0.4139 |
|
Amputation | 14 | 7 | 7 |
|
| Limb
salvage | 11 | 3 | 8 |
|
| Response to
chemotherapy |
|
|
| 0.0154b |
|
Good | 13 | 2 | 11 |
|
|
Poor | 12 | 8 | 4 |
|
| Enneking stage |
|
|
| 0.472 |
| I | 2 | 0 | 2 |
|
| II | 20 | 8 | 12 |
|
|
III | 3 | 2 | 1 |
|
MELK knockdown reduces proliferation
in vitro
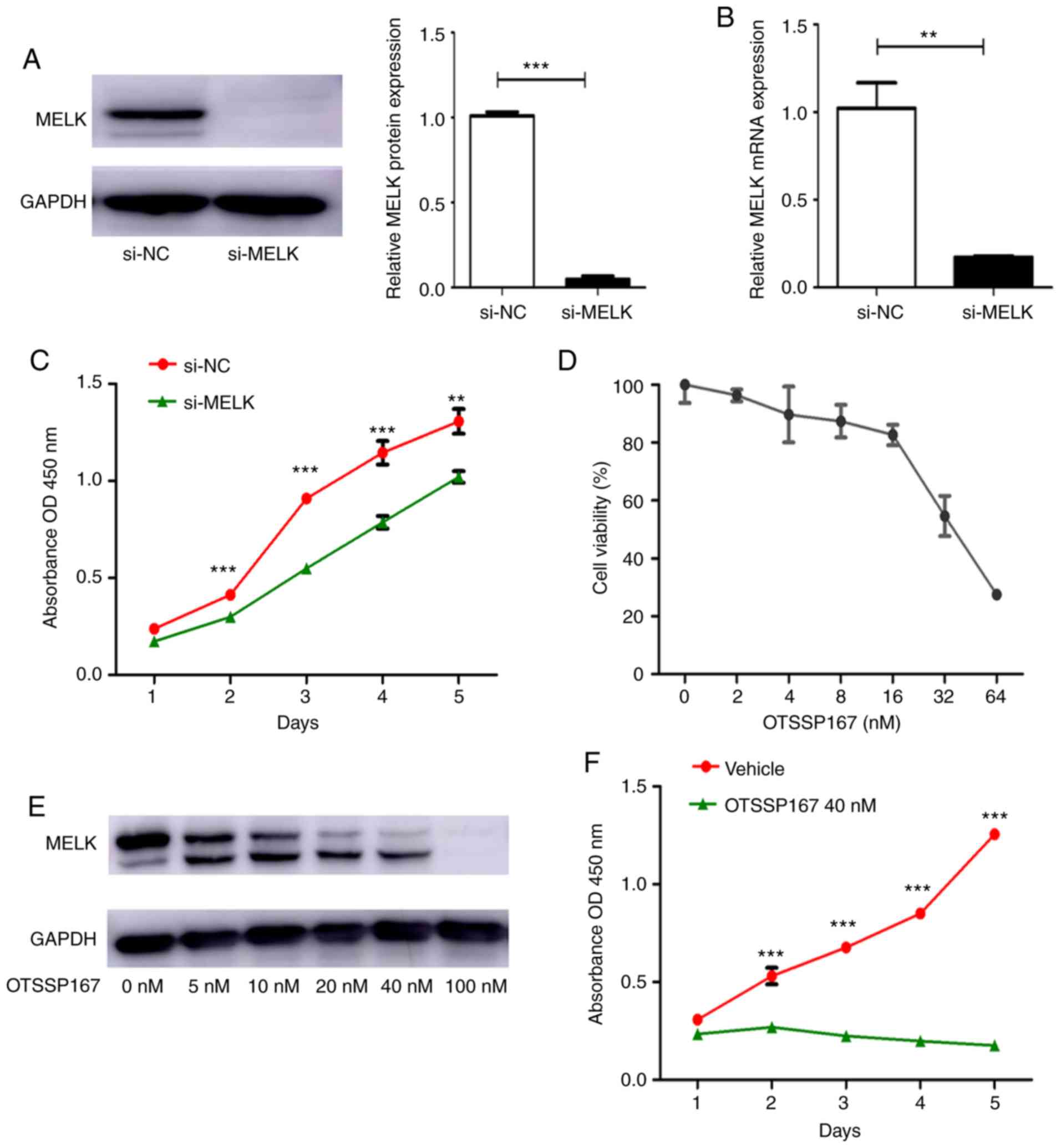
si-MELK was used to knock down the expression of
MELK in MNNG/HOS cells. The mRNA and protein expression levels of
MELK following knockdown are presented in Fig. 2A and B. To determine the effect of
MELK on the proliferation of osteosarcoma cells, a proliferation
assay was performed using MNNG/HOS cells transiently transfected
with si-NC or si-MELK. As revealed in Fig. 2C, knockdown of MELK expression
significantly decreased proliferation. OTSSP167, a MELK inhibitor,
was used to assess its effects on proliferation. First, the
IC50 of OTSSP167 was determined, which was determined to
be 40 nM/ml (Fig. 2D). There was
also a dose-dependent decrease in MELK expression levels following
treatment with OTSSP167 (Fig. 2E).
Using 40 nM OTSSP167, the proliferation of cells treated with
vehicle or OTSSP167 was assessed. As revealed in Fig. 2F, there was a significant decrease
in the proliferation of cells treated with OTSSP167. These in
vitro experiments demonstrated that MELK serves a substantial
role in the proliferation of osteosarcoma cells.
OTSSP167 inhibits tumorigenesis in
vivo
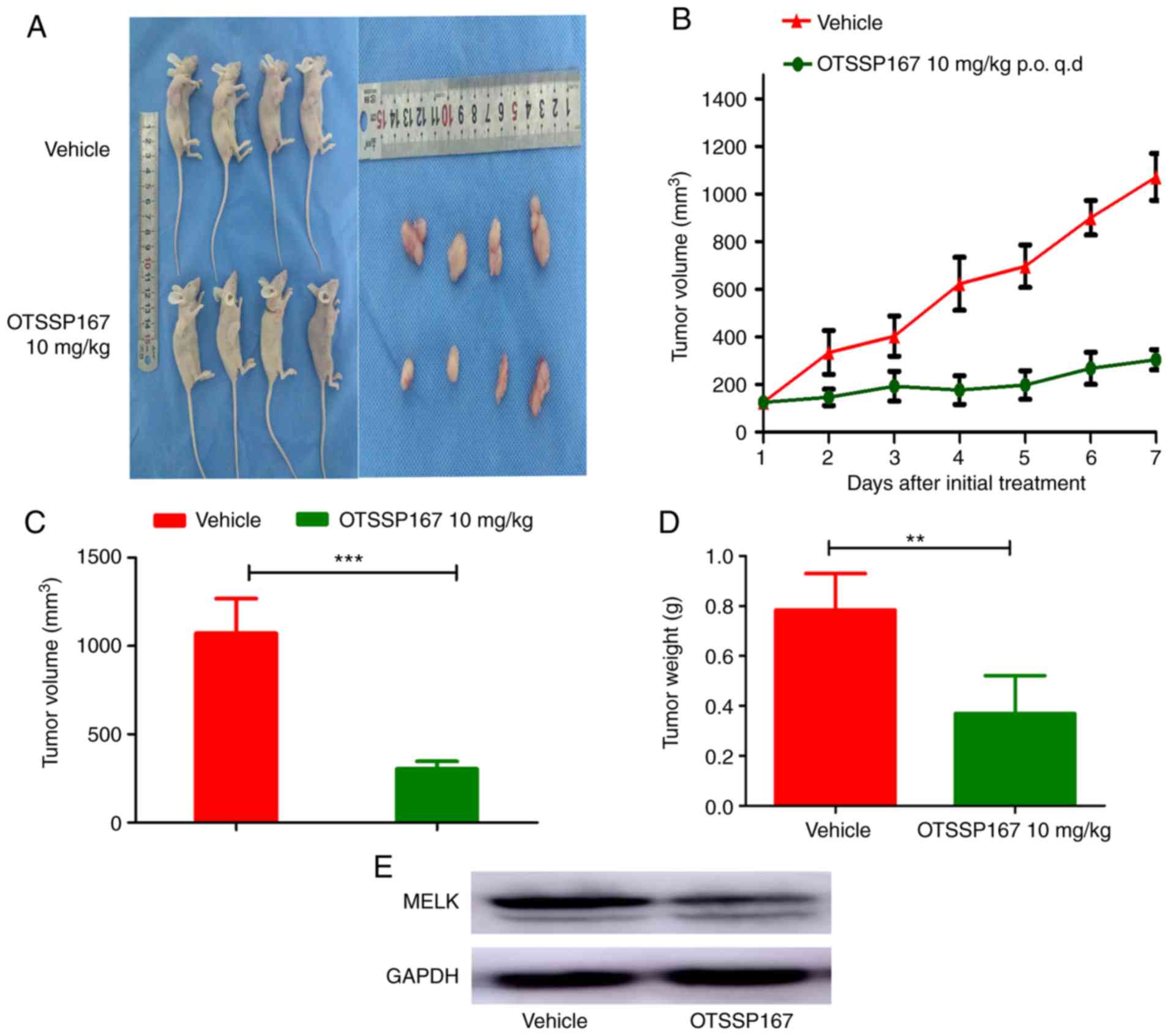
To determine the role of MELK in tumorigenesis,
MNNG/HOS cells were subcutaneously injected into the armpits of
nude mice. Tumor growth was closely monitored and tumor volume was
measured daily using a Vernier caliper, using the aforementioned
formula. When the tumor volume reached ~100 mm3, the
mice were randomly divided into two groups; one group was orally
fed OTSSP167 (10 mg/kg) daily whilst the other group was fed
vehicle. After 7 days, the mice were euthanized, and the tumors
were excised and analyzed. The tumor volume exhibited a gradual
decrease in the group fed OTSSP167 compared with the control group
fed vehicle. There was a significant difference in the tumor weight
between the two groups (Fig. 3A-D).
To evaluate the results, the protein expression levels were
assessed between the groups, and the expression of MELK was
revealed to be lower in the mice fed OTSSP167 (Fig. 3E).
MELK reduces apoptosis and promotes
cell cycle progression in osteosarcoma cells
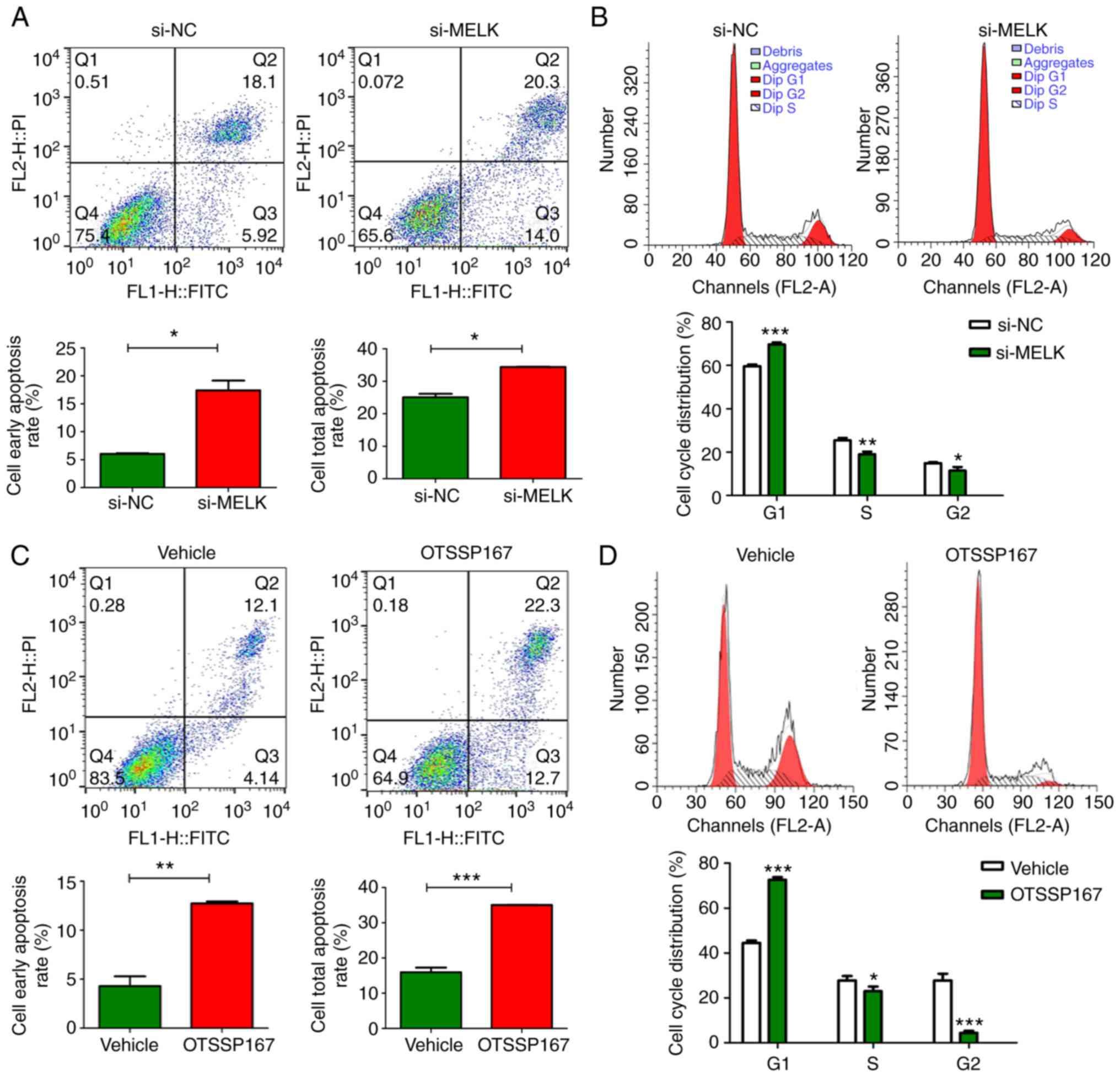
The proportion of apoptotic MNNG/HOS cells following
MELK knockdown was assessed and compared with the control cells.
The results revealed that there was an increase in the total
proportion of apoptotic cells and the portion of cells in early
stage apoptosis when MELK expression was knocked down (Fig. 4A). Additionally, MELK knockdown
resulted in inhibition of cell cycle progression in osteosarcoma
cells (Fig. 4B). There was an
increased proportion of cells in the G1 phase and a lower
proportion of cells in the G2 and S phases in the MELK-knockdown
cells compared with the control. It was hypothesized that MELK
exerted its function on osteosarcoma proliferation by inhibiting
apoptosis and potentiating the cell cycle. Such effects were also
demonstrated when MELK was inhibited by OTSSP167. Notably, the
magnitude of increase in apoptosis and cell cycle inhibition were
more pronounced with OTSSP167 (Fig. 4C
and D).
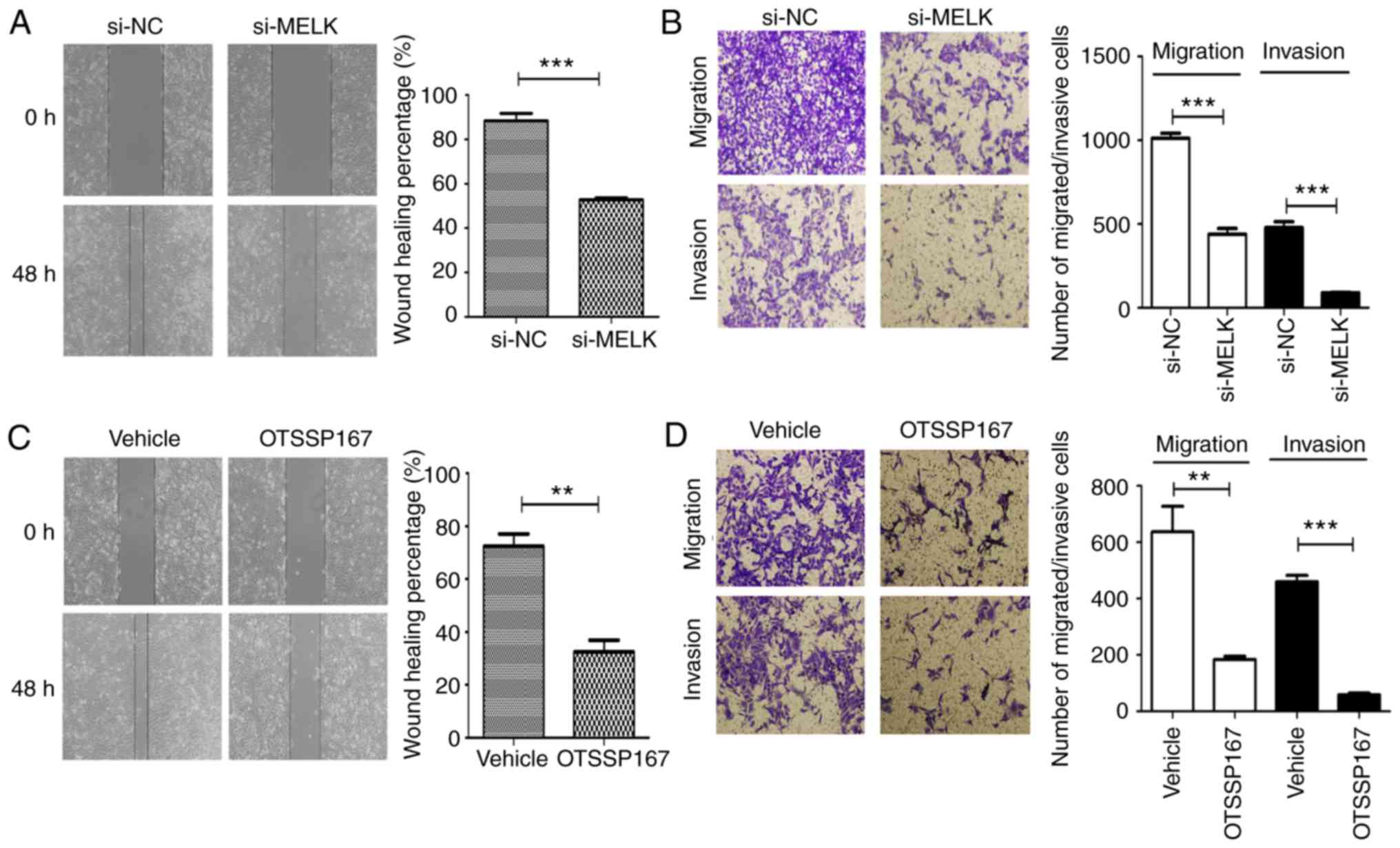
MELK knockdown decreases migration and
invasion in vitro
The role of MELK on the migration and invasion of
osteosarcoma cells was assessed using Transwell assays with or
without Matrigel, and wound healing assays. As revealed in Fig. 5A and B, MNNG/HOS cells exhibited
reduced migratory and invasive capacities following MELK knockdown.
When MELK was inhibited using the inhibitor OTSSP167, similar
inhibitory effects were observed on the migration and invasion
abilities (Fig. 5C and D). Thus, it
was hypothesized that MELK serves a role in potentiating the
migratory and invasive capacities of osteosarcoma cells. The
clinicopathological analysis from immunohistochemistry also
revealed a significant association between upregulated MELK
expression and metastasis of osteosarcoma, in agreement with the
results of the in vitro experiments.
MELK promotes proliferation and
metastasis of osteosarcoma through the PI3K/AKT/mTOR pathway
The PI3K/AKT and the mTOR signaling pathways are
considered principal pathways involved in several oncogenic
mechanisms. In the present study, it was revealed that knocking
down MELK expression resulted in reduced phosphorylation of PI3K,
AKT and mTOR, and reduced protein expression levels of MMP9 and
PCNA (Fig. 6A). Similar results
were also observed with the use of OTSSP167 (Fig. 6B). Furthermore, in MNNG/HOS cell
treated with LY294002 (20 µM), the expression of p-AKT, p-mTOR,
PCNA and MMP9 were decreased (Fig.
6C). Collectively, these results indicated that MELK promoted
proliferation and metastasis of osteosarcoma cells by modulating
PCNA and MMP9 expression via the PI3K/AKT/mTOR pathway.
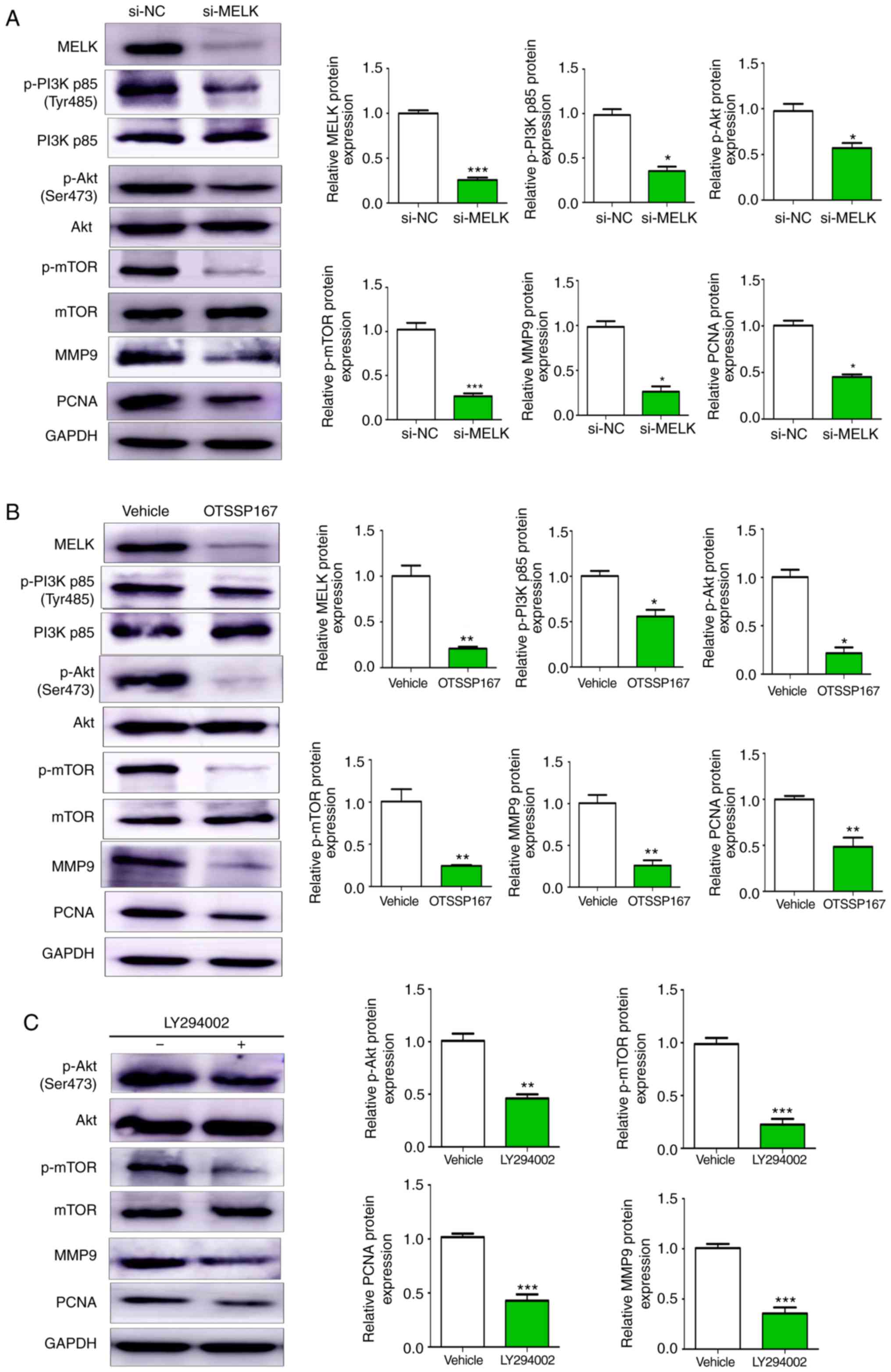 | Figure 6.MELK promotes osteosarcoma
proliferation and metastasis through the PI3K/AKT/mTOR pathway. (A)
Western blot analysis of changes in protein expression following
knockdown of MELK. Cells transfected with si-MELK exhibited lower
expression levels of p-PI3K p85 (Tyr485), p-AKT (Ser473), p-mTOR,
MMP9 and PCNA. (B) Western blotting revealed lower protein
expression levels of p-PI3K, p-AKT, p-mTOR, MMP9 and PCNA in the
cells treated with OTSSP167 compared with the vehicle control. (C)
PI3K inhibitor LY294002 decreased the expression levels of p-AKT,
p-mTOR, PCNA and MMP9. GAPDH was used as the internal control.
*P<0.05, **P<0.01, ***P<0.001. MELK, maternal embryonic
leucine zipper kinase; si, small interfering; p-, phospho-. |
Discussion
There is an increasing body of literature showing
that MELK expression is upregulated in several types of cancer
(10–15), and its upregulated expression is
associated with a less favorable prognosis in cancer (13,16,17)
Similarly, MELK has been demonstrated to exert its oncogenic
functions by affecting cellular proliferation, cell cycle,
apoptosis and metastasis (23,24).
The present study is the first study, to the best of our knowledge,
to reveal the role of MELK in the progression and metastasis of
osteosarcoma.
In the present study, MELK expression was revealed
to be significantly upregulated in osteosarcoma tissues compared
with normal tissues. Compared with the hFOB1.19 cell line, MELK
expression levels were also upregulated in the MNNG/HOS
osteosarcoma cell line. Analysis of the clinicopathological
characteristics revealed that upregulated expression of MELK was
closely associated with metastasis and chemotherapy response in
patients with osteosarcoma. Furthermore, MELK expression was
associated with the OS of the patients; patients with high MELK
expression levels had a worse prognosis than patients with low
expression.
MELK expression was knocked down in the MNNG/HOS
osteosarcoma cell line using si-MELK or the MELK inhibitor,
OTSSP167. Knockdown or inhibition of MELK activity resulted in a
significant decrease in cellular proliferation and metastatic
behavior. MELK has previously been revealed to regulate
proliferation, migration and invasion (11–17).
In agreement with the previous studies, suppressing MELK in the
MNNG/HOS osteosarcoma cell line reduced proliferation, migration
and invasion. There was also a significant decrease in cell cycle
progression and potentiation of apoptosis due to knockdown of
MELK.
In addition to identifying the role of MELK in
osteosarcoma, the underlying mechanism was also identified. PCNA
has been revealed to be an indicator of cell proliferation, and to
serve a principal role in the regulation of cellular proliferation
and cell cycle progression (25,26).
Furthermore, PCNA is a known oncogene and its upregulation has been
revealed to be associated with a poorer prognosis in patients with
osteosarcoma (27).
In the present study, the expression levels of PCNA
were revealed to be decreased following knockdown or inhibition of
MELK. Thus, it was hypothesized that MELK may promote the growth of
osteosarcoma by regulating PCNA expression.
MMPs are proteolytic enzymes that serve a role in
extracellular matrix remodeling, affecting cell growth,
differentiation, migration, invasion and angiogenesis (28). As an important member of the MMP
family, MMP-9 has been revealed to affect tumor metastasis in
several tumors, including osteosarcoma (29–31).
The results of the present study revealed that there was a
significant decrease in migration and invasion based on the
Transwell and wound healing assays. Western blot analysis also
revealed that MMP9 protein expression levels were lower in the
cells when MELK expression was knocked down, indicating reduced
invasive capacity.
The PI3K/AKT/mTOR pathway is associated with a
variety of cellular functions and is known to serve a distinct role
in cancer progression (32). In the
present study, silencing of MELK resulted in marked suppression of
p-PI3K, p-AKT, p-mTOR, PCNA and MMP9 expression. It was further
revealed that a PI3K inhibitor decreased the expression of p-AKT,
p-mTOR, PCNA and MMP9. Collectively, it was hypothesized that MELK
affected the proliferation and migration of osteosarcoma cells by
regulating PCNA and MMP9 expression via the PI3K/Akt/mTOR signaling
pathway. Studies have previously revealed a link between PCNA and
the PI3K/Akt/mTOR signaling pathway (33,34),
and this pathway is known to be involved in regulation of migration
and invasion in various types of cancer, possibly through
regulation of crucial MMPs (35–37).
OTSSP167 is a potent MELK inhibitor, which has been
demonstrated to possess antitumor properties in several types of
cancer (15,16). OTSSP167 is a protein kinase
inhibitor that prevents MELK phosphorylation and abrogates its
downstream substrates (38).
OTSSP167 is being developed as an anticancer drug for patients with
solid tumors, which have not responded to established modes of
treatment, and is emerging as a promising therapeutic for treatment
of several types of solid tumors (20). A phase I clinical trial studying the
efficiency of OTSSP167 was started in August of 2013 (18). In the present study, OTSSP167 served
an inhibitory role in osteosarcoma progression through suppression
of MELK. When xenografted tumors were produced in nude mice and
were orally fed OTSSP167, there was a gradual decrease in the size
of tumors over time, whereas the control group, which was fed the
vehicle alone, possessed tumors which grew over time. This is in
agreement with the in vitro proliferation assays and
strongly supports the hypothesis that MELK is responsible for tumor
progression and its knockdown or inhibition by RNAi or OTSSP167,
respectively, could reduce tumor progression. In recent decades,
interest in studying inhibitors of PI3K, AKT and mTOR has
increased. Several of these inhibitors are in various phases of
clinical trials (39). Since the
MELK inhibitor OTSSP167 has been revealed to downregulate the
phosphorylation of these molecules, it is hypothesized that it may
be used as a substitute for these inhibitors. However, additional
studies are required for verification.
In summary, MELK expression was upregulated in
osteosarcoma, and this was associated with a poor prognosis. MELK
increased proliferation and metastasis of osteosarcoma cells by
modulating PCNA and MMP9 expression via the PI3K/AKT/mTOR pathway.
The present study is the first to examine the role of MELK in
osteosarcoma, the first to determine the underlying mechanism of
MELK-mediated effects, and the first to determine the effect of
OTSSP167 in suppressing osteosarcoma progression through inhibition
of MELK. These results suggest that MELK may be used as a
prognostic biomarker, and highlights its potential as a therapeutic
target for the treatment of osteosarcoma.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81672655) and the
Natural Science Foundation of Shandong Province of China (grant no.
ZR2017MH047).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable request.
The results published here are in whole or in part are based on
data generated by the Therapeutically Applicable Research to
Generate Effective Treatments (https://ocg.cancer.gov/programs/target) initiative,
phs000468. The data used for this analysis are available at
https://portal.gdc.cancer.gov/projects.
Authors' contributions
SFAJ and XW provided substantial contributions to
the conception and design of the work. SFAJ and XW were responsible
for the experimental procedure and clinical studies. KL and XL
conducted the literature search. QY and JL provided the data
acquisition and carried out the statistical analysis. SD drafted
the work, edited the manuscript and revised it critically for
important intellectual content. All authors critically revised and
approved the final manuscript and agree to be responsible for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All patients signed informed consent to participate
in the present study, and the study was approved by the Ethics
Committee of Qilu Hospital of Shandong University. All animal
experiments were approved by the Ethics Committee of Qilu Hospital,
Shandong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Biermann JS, Adkins DR, Agulnik M,
Benjamin RS, Brigman B, Butrynski JE, Cheong D, Chow W, Curry WT,
Frassica DA, et al: Bone cancer. J Natl Compr Canc Netw.
11:688–723. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lindsey BA, Markel JE and Kleinerman ES:
Osteosarcoma overview. Rheumatol Ther. 4:25–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Pediatric and adolescent osteosarcoma (US).
Springer. 152:3–13. 2009. View Article : Google Scholar
|
|
6
|
Bacci G, Longhi A, Bertoni F, Briccoli A,
Versari M, Pignotti E and Picci P: Bone metastases in osteosarcoma
patients treated with neoadjuvant or adjuvant chemotherapy. The
Rizzoli experience in 52 patients. Acta Orthopaed. 77:938–943.
2006. View Article : Google Scholar
|
|
7
|
Heyer BS, Warsowe J, Solter D, Knowles BB
and Ackerman SL: New member of the SNF1/AMPK kinase family, Melk,
is expressed in the mouse egg and preimplantation embryo. Mol
Reprod Dev. 47:148–156. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Zhou X, Li Y, Xu Y, Lu K, Li P
and Wang X: Inhibition of maternal embryonic leucine zipper kinase
with OTSSP167 displays potent anti-leukemic effects in chronic
lymphocytic leukemia. Oncogene. 37:5520–5533. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang P and Zhang D: Maternal embryonic
leucine zipper kinase (MELK): A novel regulator in cell cycle
control, embryonic development, and cancer. Int J Mol Sci.
14:21551–21560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ganguly R, Hong CS, Smith LG, Kornblum HI
and Nakano I: Maternal embryonic leucine zipper kinase: Key kinase
for stem cell phenotype in glioma and other cancers. Mol Cancer
Ther. 13:1393–1398. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Janostiak R, Rauniyar N, Lam TT, Ou J, Zhu
LJ, Green MR and Wajapeyee N: MELK promotes melanoma growth by
stimulating the NF-κB pathway. Cell Rep. 21:2829–2841. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li G, Yang M, Zuo L and Wang MX: MELK as a
potential target to control cell proliferation in triple-negative
breast cancer MDA-MB-231 cells. Oncol Lett. 15:9934–9940.
2018.PubMed/NCBI
|
|
13
|
Kohler RS, Kettelhack H,
Knipprath-Mészaros AM, Fedier A, Schoetzau A, Jacob F and
Heinzelmann-Schwarz V: MELK expression in ovarian cancer correlates
with poor outcome and its inhibition by OTSSP167 abrogates
proliferation and viability of ovarian cancer cells. Gynecol Oncol.
145:159–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li S, Li Z, Guo T, Xing XF, Cheng X, Du H,
Wen XZ and Ji JF: Maternal embryonic leucine zipper kinase serves
as a poor prognosis marker and therapeutic target in gastric
cancer. Oncotarget. 7:6266–6280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gray D, Jubb AM, Hogue D, Dowd P, Kljavin
N, Yi S, Bai W, Frantz G, Zhang Z, Koeppen H, et al: Maternal
embryonic leucine Zipper Kinase/Murine protein serine-threonine
kinase 38 is a promising therapeutic target for multiple cancers.
Cancer Res. 65:9751–9761. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu S, Chen X, Hu C, Wang J, Shen Y and
Zhong Z: Up-regulated maternal embryonic leucine zipper kinase
predicts poor prognosis of hepatocellular carcinoma patients in a
Chinese Han population. Med Sci Monit. 23:5705–5713. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pickard MR, Green AR, Ellis IO, Caldas C,
Hedge VL, Mourtada-Maarabouni M and Williams GT: Dysregulated
expression of Fau and MELK is associated with poor prognosis in
breast cancer. Breast Cancer Res. 11:R602009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung S and Nakamura Y: MELK inhibitor,
novel molecular targeted therapeutics for human cancer stem cells.
Cell Cycle. 12:1655–1656. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chung S, Suzuki H, Miyamoto T, Takamatsu
N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y and Matsuo Y:
Development of an orally-administrative MELK-targeting inhibitor
that suppresses the growth of various types of human cancer.
Oncotarget. 3:1629–1640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu D, Zhang XX, Li MC, Cao CH, Wan DY, Xi
BX, Tan JH, Wang J, Yang ZY, Feng XX, et al: C/EBPβ enhances
platinum resistance of ovarian cancer cells by reprogramming H3K79
methylation. Nat Commun. 9:17392018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen G and Deng X: Cell synchronization by
double thymidine block. Bio Protoc. 8:e29942018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ganguly R, Mohyeldin A, Thiel J, Kornblum
HI, Beullens M and Nakano I: MELK-a conserved kinase: Functions,
signaling, cancer, and controversy. Clin Transl Med. 4:112015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin ML, Park JH, Nishidate T, Nakamura Y
and Katagiri T: Involvement of maternal embryonic leucine zipper
kinase (MELK) in mammary carcinogenesis through interaction with
Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer
Res. 9:R172007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Strzalka W and Ziemienowicz A:
Proliferating cell nuclear antigen (PCNA): A key factor in DNA
replication and cell cycle regulation. Ann Bot. 107:1127–1140.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Müller R, Misund K, Holien T, Bachke S,
Gilljam KM, Våtsveen TK, Rø TB, Bellacchio E, Sundan A and Otterlei
M: Targeting proliferating cell nuclear antigen and its protein
interactions induces apoptosis in multiple myeloma cells. PLoS One.
8:e704302013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang X, Wang D, Yuan N, Liu F, Wang F,
Wang B and Zhou D: The prognostic value of PCNA expression in
patients with osteosarcoma: A meta-analysis of 16 studies. Medicine
(Baltimore). 96:e82542017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jabłońska-Trypuć A, Matejczyk M and
Rosochacki S: Matrix metalloproteinases (MMPs), the main
extracellular matrix (ECM) enzymes in collagen degradation, as a
target for anticancer drugs. J Enzyme Inhib Med Chem. 31 (Suppl
1):S177–S183. 2016. View Article : Google Scholar
|
|
29
|
Viros D, Camacho M, Zarraonandia I, García
J, Quer M, Vila L and León X: Prognostic role of MMP-9 expression
in head and neck carcinoma patients treated with radiotherapy or
chemoradiotherapy. Oral Oncol. 49:322–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vilen ST, Salo T, Sorsa T and Nyberg P:
Fluctuating roles of matrix metalloproteinase-9 in oral squamous
cell carcinoma. ScientificWorldJournal. 2013:9205952013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou J, Liu T and Wang W: Prognostic
significance of matrix metalloproteinase 9 expression in
osteosarcoma: A meta-analysis of 16 studies. Medicine (Baltimore).
97:e130512018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou HY, Yao XM, Chen XD, Tang JM, Qiao ZG
and Wu XY: Mechanism of metformin enhancing the sensitivity of
human pancreatic cancer cells to gem-citabine by regulating the
PI3K/Akt/mTOR signaling pathway. Eur Rev Med Pharmacol Sci.
23:10283–10289. 2019.PubMed/NCBI
|
|
33
|
Ou JM, Qiu MK, Dai YX, Dong Q, Shen J,
Dong P and Fei ZW: Combined Blockade and Akt/mTOR pathway inhibits
growth of human hemangioma via downregulation of proliferating cell
nuclear antigen. Int J Immunopathol Pharmacol. 25:945–953. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olaisen C, Muller R, Nedal A and Otterlei
M: PCNA-interacting peptides reduce Akt phosphorylation and
TLR-mediated cytokine secretion suggesting a role of PCNA in
cellular signaling. Cell Signal. 27:1478–1487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dobbin ZC and Landen CN: The importance of
the PI3K/AKT/mTOR pathway in the progression of ovarian cancer. Int
J Mol Sci. 14:8213–8227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levine DA, Bogomolniy F, Yee CJ, Lash A,
Barakat RR, Borgen PI and Boyd J: Frequent mutation of the PIK3CA
gene in ovarian and breast cancers. Clin Cancer Res. 11:2875–2878.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsuoka T and Yashiro M: The role of
PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers (Basel).
6:1441–1463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ji W, Arnst C, Tipton AR, Bekier ME II,
Taylor WR, Yen TJ and Liu ST: OTSSP167 abrogates mitotic checkpoint
through inhibiting multiple mitotic kinases. PLoS One.
11:e01535182016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee JJ, Loh K and Yap YS: PI3K/Akt/mTOR
inhibitors in breast cancer. Cancer Biol Med. 12:342–354.
2015.PubMed/NCBI
|