Introduction
Thyroid hormones (TH) are multifunctional mediators
that fine-tune several physiological processes, including metabolic
rate, digestive function and tissue development (1,2).
T4, the major TH secreted into the bloodstream by the
thyroid gland, is converted to its active form, T3, by
type I and II deiodinases (D1 and D2) that vary among tissues,
resulting in tissue-specific distribution of circulating
T3 with the capability to bind nuclear thyroid hormone
receptors (TR) (3). Upon binding of
TH, TRs interact specifically with thyroid hormone response
elements (TRE) of target gene promoter regions to regulate their
transcription (4,5).
TRs are type II nuclear receptors encoded by two
separate genes, THRA (NR1A1) and THRB (NR1A2). THRA encodes one
functional T3-binding TRα1 while TRα2 and TRα3 have been
identified as splice variants with no T3 binding
ability. TRΔα1 and TRΔα2 are truncated variants without DNA binding
domains but retain T3 binding ability and compete with
other TRs. THRB genes encode three functional T3-binding
TRβ isoforms, specifically, β1, β2 and β3 (3). Truncated TRΔβ3 lacks the DNA binding
domain but retains T3 binding activity and acts as a
dominant-negative antagonist, similar to TRΔα1 and TRΔα2 (6–9).
Several studies to date have reported aberrant expression and/or
somatic mutations of TR in human cancers (3,10–18),
including hepatocellular carcinoma (HCC) (19–28).
HCC is the major histological subtype of primary
liver malignancy and one of the major causes of tumor-related
mortalities worldwide (29). Liver
cirrhosis is the premonitory symptom of HCC, with the majority of
cases developing from cirrhotic livers (30).
In 1986, v-erbA, a mutant form of TR devoid of
ligand binding ability borne by the avian erythroblastosis virus
causing erythroleukemia was identified and subsequently shown to
play a role in HCC in transgenic mice (4,31).
Mutated or truncated forms of TRα and TRβ are expressed at high
frequencies in human HCC (32–36).
These mutant TRs display loss of transcriptional activity, along
with defects in release and binding of ligand-driven corepressor,
and further act as dominant-negative forms, highlighting an
association between aberrant TR regulation and HCC (1).
In rat models of HCC generated by a combination of
diethylnitrosamine (DEN) and partial hepatectomy,
2-acetylaminofluorene or a High-fat choline-methionine-deficient
diet, T3/TR signaling was shown to suppress
carcinogenesis via induction of a preneoplastic hepatocyte
differentiation program (2).
Disruption of TH signaling promoted tumorigenesis, particularly in
HCC (2). Analysis of the genes
modulated by T3/TR in HCC should therefore facilitate
elucidation of the mechanisms underlying HCC progression. Previous
cDNA microarrays conducted by our group to evaluate target gene
expression following T3 treatment of wild-type
TR-expressing hepatoma cells led to the identification of forkhead
box M1 (FOXM1), also known as hepatocyte nuclear factor (HNF)-3,
HNF-3/fork head homologue-11, membrane palmitoylated protein 2,
winged-helix transcription factor or Trident (37,38),
as a molecule negatively regulated by T3.
FOXM1 is characterized by the presence of the
DNA-binding domain Forkhead/Winged-helix domain (FKH) (39). Earlier cDNA microarray data
suggested that FOXM1 is negatively regulated by T3 in a
TRα1-overexpressing hepatoma cell line (40). Similar to TR, FOXM1 participated in
the cellular developmental pathway and maintenance of homoeostasis
by activating multiple target genes regulating DNA damage repair,
cell proliferation, cell cycle progression, renewal,
differentiation, migration, angiogenesis and survival (41). Deregulation of FOXM1 signaling was
shown to trigger malignant transformation (42). The mechanisms underlying signal
transduction of FOXM1 contributing to tumor growth have been
partially elucidated and appear to involve interplay with PI3K,
ERK, epidermal growth factor receptor, estrogen receptor, vascular
endothelial growth factor and reactive oxygen species (37,41,43),
but require further clarification. Data from the current study
indicated that TH/TR-modulated FOXM1 participates in HCC
progression via affecting downstream gene expression, further
highlighting the biological importance of TH/TR homeostasis.
Materials and methods
Cell culture and T3
depletion
Human hepatoma cell lines J7 (provided by Dr CS
Yang, National Taiwan University, Taiwan) (44), Mahlavu, SK-Hep1, HepG2 (American
Type Culture Collection) and Huh7 (provided by Dr TY Hsieh,
Tri-Service General Hospital, Taiwan) (45) were routinely grown in DMEM (Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (EMD Millipore),
L-glutamine, 1% penicillin/streptomycin (PS) and non-essential
amino acids (NEAAs). HepG2 cell lines stably transfected with TRα1
(HepG2-TRα1), TRβ1 (HepG2-TRβ1) or vector control (HepG2-neo) have
been previously established in our laboratory (46). To generate T3-depleted
FBS (Td-FBS), AG 1-X8 resin (Bio-Rad Laboratories, Inc.) was washed
three times with distilled, deionized water (15 min each time),
pelleted by brief centrifugation (6,000 × g, 5 min, room
temperature) and sterilized by autoclaving. T3 of FBS
(50 ml) was depleted by incubationwith 2.2 g AG 1-X8 resin three
times each for 5 h and filtered. Cells were cultured in DMEM with
1% PS, L-glutamine, NEAAs and 10% Td-FBS and treated with 0 nM
T3 (control group, Neo T3 0 nM, FOXM1
T3 0 nM) or 10 nM T3 (experimental group, Neo
T3 10 nM, FOXM1 T3 10 nM) for 24 and 48 h at
37°C in a humidified atmosphere of 95% air and 5% CO2.
The cell lines were authenticated using the Promega StemElite ID
system (Promega Corporation), a short tandem repeat-based assay
(25).
Establishment of FOXM1 knockdown and
overexpression cell lines
FOXM1 short hairpin RNA (shRNA) TRCN0000015544
(shFOXM1#1;
5′-CCGGGCCCAACAGGAGTCTAATCAACTCGAGTTGATTAGACTCCTGTTGGGCTTTTT-3′)
and TRCN0000015547 (shFOXM1#2;
5′-CCGGCGCCGGAACATGACCATCAAACTCGAGTTTGATGGTCATGTTCCGGCGTTTTT-3′)
and pLKO.1-shLuc (control group) were purchased from the RNA
interference core laboratory (Academia Sinica, Taipei, Taiwan). A
total of 5 µg shRNA and 5 µg lentiviral package plasmids were
co-transfected in 293TN cells (System Biosciences, LLC) cultured in
DMEM with 1% PS, L-glutamine, NEAAs and 10% FBS using a TurboFect
reagent kit (Thermo Fisher Scientific, Inc.) to produce viruses.
After 24 h, viral supernatant was collected by centrifugation
(4,000 × g, 5 min, room temperature) for infection of J7 and
Mahlavu cell lines as these cell lines expressed lower levels of
FOXM1 following viral infection for FOXM1 depletion. 48 h after
infection, cells were transferred to DMEM with 1% PS, L-glutamine,
NEAAs, 10% FBS and puromycin for selection. After 48 h of
incubation, FOXM1 expression was confirmed via western blotting.
The control groups for J7 and Mahlavu (shluc#1 and shluc#2) were
established by two independent viral infections of pLKO.1-shLuc.
The FOXM1 coding sequence (NM_021953) was cloned into a pcDNA3.1
expression vector (Addgene, Inc.). A total of 5 µg FOXM1-pcDNA3.1
(Neo-FOXM1) and 5 µg control plamid pcDNA3.1 (Neo) were transfected
into SK-Hep1 and Huh7 cells with TurboFect reagent for 24 h. The
sequences of primer pairs used for FOXM1 were as follows: Forward,
5′-CGCGGATCCGCGATGAAAACTAGCCCCCGTCGGC-3′ and reverse,
5′-CCGGAATTCCGGCTACTGTAGCTCAGGAATAAAC-3′.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from HepG2-TRα1, HepG2-TRβ1 and HepG2-neo
cells was purified using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the supplier's
protocol and reverse-transcribed into cDNA using a Superscript II
kit according to the manufacturer's protocol (Thermo Fisher
Scientific, Inc.). The reaction was performed in a 25-µl reaction
mixture containing 50 nM forward and reverse primers, 1X SYBR Green
reaction mix (Applied Biosystems; Thermo Fisher Scientific, Inc.)
and 50 ng cDNA template. The following thermocycling conditions
were used for the qPCR: Initial denaturation for 10 min at 95°C,
followed by 40 cycles of 95°C for 15 sec and final extension at
60°C for 1 min. Fluorescence emitted by SYBR Green was detected on
line using a ABI PRISM 7500 sequencer (Applied Biosystems; Thermo
Fisher Scientiic, Inc.). Studies have shown that the initial copy
number can be quantified during RT-qPCR analysis based on the
threshold cycle (Ct). The Ct is defined as the cycle at which
fluorescence is determined to be statistically significant above
background. All PCRs were perfomed in duplicate on the same 96-well
plate. For quantification of gene expression changes, the
2−ΔΔCq method (47) was
used to calculate relative-fold changes normalized to 18S ribosomal
RNA expression (18S rRNA) as described by the manufacturer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following primer
pairs were used for the qPCR: FOXMI forward,
5′-TCCTCAGCTAGCAGCACCTTG-3′ and reverse, 5′-CCAGGTGTTTAAGCAGCAGA-3′
and 18S rRNA forward, 5′-CGAGCCGCCTGGATACC-3′ and reverse,
5′-CCTCAGTTCCGAAAACCAACAA-3′.
Western blot analysis
Total cell lysates of HepG2, J7, Mahlavu, SK-Hep-1
and Huh7-cells were prepared using cell lysis buffer (cat. no.
9803; Cell Signaling Technology, Inc.) and protein concentrations
were determined using a Bradford assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Western blot experiments were performed as
previously described (46). Equal
amounts of protein (60 µg/lane) were separated by 8–10% SDS-PAGE.
Separated proteins were transferred to polyvinylidene difluoride
membranes and blocked in 5% milk for 1 h at room temperature.
Following blocking, incubation with the following primary
antibodies was performed overnight at 4°C: Rabbit anti-human FOXM1
polyclonal antibody (cat. no. sc-502; 1:1,000; Santa Cruz
Biotechnology, Inc.) mouse anti-human Cdk2 monoclonal antibody
(cat. no. sc-6248; 1:1,000; Santa Cruz Biotechnology, Inc.), rabbit
anti-human cyclin E polyclonal antibody (cat. no. 07-687; 1:1,000;
Sigma-Aldrich; Merck KGaA), mouse anti-human monoclonal antibodies
against β-actin (cat. no. MAB1501; 1:8,000; Sigma-Aldrich; Merck
KGaA) and GAPDH (cat. no. MAB374; 1:8,000; Sigma-Aldrich; Merck
KGaA) and rabbit anti-human cyclin D1 monoclonal antibody (cat. no.
EPR2241; 1:1,000; Epitomics; Abcam). Following primary antibody
incubation, membranes were incubated with the following secondary
antibodies for 1 h at room temperature: Peroxidase-conjugated goat
anti-mouse immunoglobulin G (IgG; cat. no. AP124P; 1:5,000;
Sigma-Aldrich; Merck KGaA) and peroxidase-conjugated goat
anti-rabbit IgG (cat. no. AP132P; 1:5,000; Sigma-Aldrich; Merck
KGaA). Protein bands were visualized using Immobilon Western
Chemiluminescent HRP substrate (cat. no. WBKLS0500; Millipore;
Merck KGaA). Quantification was performed using Image Gauge v3.46
(Fujifilm Holdings Corporation) and relative expression was
normalized to b-actin or GAPDH expression.
Dual-luciferase reporter assay
To clone the FOXM1 5′-flanking region for the
promoter activity assay, fragments of the FOXM1 promoter (positions
−1560 to +22) were amplified with the respective forward
(5′-CCGGGTACCTCTCTTCCTCTCTCTCTCTCC-3′) and reverse
(5′-CCGCTCGAGCAGTTTGTTCCGCTGTTTG-3′) primers based on the published
nucleotide sequence (GenBank accession no. NT009759) and cloned
into a pGL3-Basic vector (Promega Corporation). Two negative TRE
(nTRE) motifs, NM23H1 and hTSHβ-N, were applied to Vector NTI
advance 11 software (Invitrogen; Thermo Fisher Scientific, Inc.)
for the prediction of potential nTREs of the FOXM1 promoter
sequence. Serially-deleted FOXM1 promoter fragments were amplified
using the following primers: Forward,
5′-CCGGGTACCCCAACTGTTCTGCCCTAATCCA-3′ (−1480 to +22); forward,
5′-CCGGGTACCCACACCACACTCCATTCAGGTC-3′ (−1300 to +22); forward
5′-CCGGGTACCCTCCAGAGTAGCTGGGACTACAGGCA-3′ (−914 to +22); forward,
5′-CCGGGTACCGATTAAAATGTCTGTGCCCCTCTTCC-3′ (−728 to +22) and
reverse, 5′-CCGCTCGAGCAGTTTGTTCCGCTGTTTG-3′. To determine the
transactivation activity of the TREs on the FOXM1 promoter,
HepG2-TRα1 cells (5×105 cells/24-well dish) were
cultured in DMEM with 1% PS, L-glutamine, NEAAs and 10% Td-FBS and
transfected with 0.2 µg pGL3-Basic vector containing FOXM1 promoter
sequences using the TurboFect reagent kit. Cells were also
transfected with 0.05 µg β-galactosidase expression vector, pSVβ
plasmid (Clontech Laboratories, Inc.). At 24 h after transfection,
cells were treated with 0 or 10 nM T3 for an additional
24 h, and were then lysed to measure luciferase and β-galactosidase
activities using a Luciferase Assay System (cat. no. E1500; Promega
Corporation). Luciferase activity was normalized to β-galactosidase
activity.
Chromatin immunoprecipitation (ChIP)
assay
HepG2-TRa1 cells (4×106) were cultured in
DMEM with 1% PS, L-glutamine, NEAAs and 10% Td-FBS and treated with
0 or 10 nM T3 24 h, then cross-linked by adding 1% (v/v)
formaldehyde for 10 min at room temperature. The reaction was
terminated by treatment with 0.125 M glycine. Following washing
with ice-cold PBS, cells were resuspended in RIPA buffer [50 mM
Tris, pH 8.0, with 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS,
150 mM NaCl and 5 mM EDTA] containing protease inhibitors (1 mM
each of PMSF, aprotinin and leupeptin). Solutions were sonicated
using a Misonix Sonicator 3000 homogenizer (Mandel Scientific
Company Inc.). All samples were precleared by the addition of 60 ml
protein A/G-agarose (Sigma-Aldrich; Merck KGaA) for 1 h at 4°C.
Next, an anti-TR antibody (3 µg/ml) (provided by Dr SY Cheng,
National Cancer Institute, National Institutes of Health, Bethesda,
Maryland, USA) or anti-IgG antibody (3 µg/ml; cat. no. MAB1101;
R&D Systems, Inc.) was added and incubation proceeded at 4°C
overnight before the addition of 80 ml protein A/G-agarose
suspension for pulldown of relevant DNA/protein complexes. A
fragment containing the predicted negative TRE (nTRE) of the FOXM1
promoter was amplified using the forward primer,
5′-CAACATTTGTTTGTTTTGGAGACGG-3′ and reverse primer,
5′-AAAAATTAGCCGGGCGTGGT-3′. A fragment lacking the nTRE of GAPDH
was detected using the forward primer, 5′-CAAGGCTGAGAACGGGAAGC-3′
and reverse primer 5′-AGGGGGCAGAGATGATGACC-3′, which served as a
negative control.
Cell proliferation assay
The proliferative capacity of the aforementioned
FOXM1 knockdown and overexpression cell lines was assessed by
proliferation assay. Cells were seeded at a density of
3×104 cells/six-well plate routinely grown in DMEM with
1% PS, L-glutamine, NEAAs and 10% FBS. Trypsin was used to isolate
cells from culture plates after 1, 3 and 5 days. Cells were then
resuspended in 1 ml DMEM with 1% PS, L-glutamine, NEAAs and 10%
FBS. A total of 10 µl cell suspension were stained with equal parts
of 0.4% trypan blue, and live cells were counted using a LUNA-II™
Automated Cell Counter (Logos Biosystems). To assess the
proliferatve capacity of T3-mediated FOXM1 expression, 5
µg FOXM1-pcDNA3.1 (Neo-FOXM1) and 5 µg control plamid pcDNA3.1
(Neo) were transfected into SK-Hep1 and Huh7 cells with TurboFect
reagent for 24 h, and cells were seeded at a density of
3×104 cells/six-well plate routinely grown in DMEM with
1% PS, L-glutamine, NEAAs and 10% Td-FBS and treated with 0 or 10
nM T3 for 24, 72 and 120 h (days 1, 3 and 5). Trypsin
was used to isolate cells from culture plates at indicated
timepoints. Cells were resuspended in 1 ml DMEM with 1% PS,
L-glutamine, NEAAs and 10% Td-FBS. A total of 10 µl cell suspension
were stained with equal parts of 0.4% trypan blue, and live cells
were counted using a LUNA-II™ Automated Cell Counter (Logos
Biosystems).
Bioinformatics
Public microarray data from the Gene Expression
Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) with accession
numbers GSE14520 (48) (cohort 1),
GSE6764 (49) (cohort 2) and
GSE14323 (50) (data not shown)
were analyzed. Raw gene expression data were normalized using the
Robust Multi-array Average method and global median centering
(51). Statistical analysis was
performed using SPSS 20 (IBM Corp.).
Statistical analysis
Statistical analysis was performed using SPSS 20
(IBM Corp.). One-way ANOVA was used to compare the results obtained
for more than one treatment. Data were analyzed using medians,
standard deviations, one-way ANOVA and Tukey's Honest Significant
Difference post hoc test. Differences between grpups with two
independent variables were analyzed using two-way ANOVA and
Bonferroni's post hoc test. Differences between groups were
analyzed with Student's t-test. Data are presented the mean ± SD
from at least three independent experiments. Expression of FOXM1
and TRα1 in GSE14520 and GSE6764 were analyzed by Spearman's rank
correlation. Quartile (Q3) expression levels of FOXM1
were used as the cutoff, overall survival and recurrence survival
were analyzed using the Kaplan-Meier survival analysis curve for
high- or low-FOXM1 expression in 242-paired HCC patients of
GSE14520. P-values were determined by the log-rank test. P<0.05
was considered to indicate a statistically significant
difference.
Results
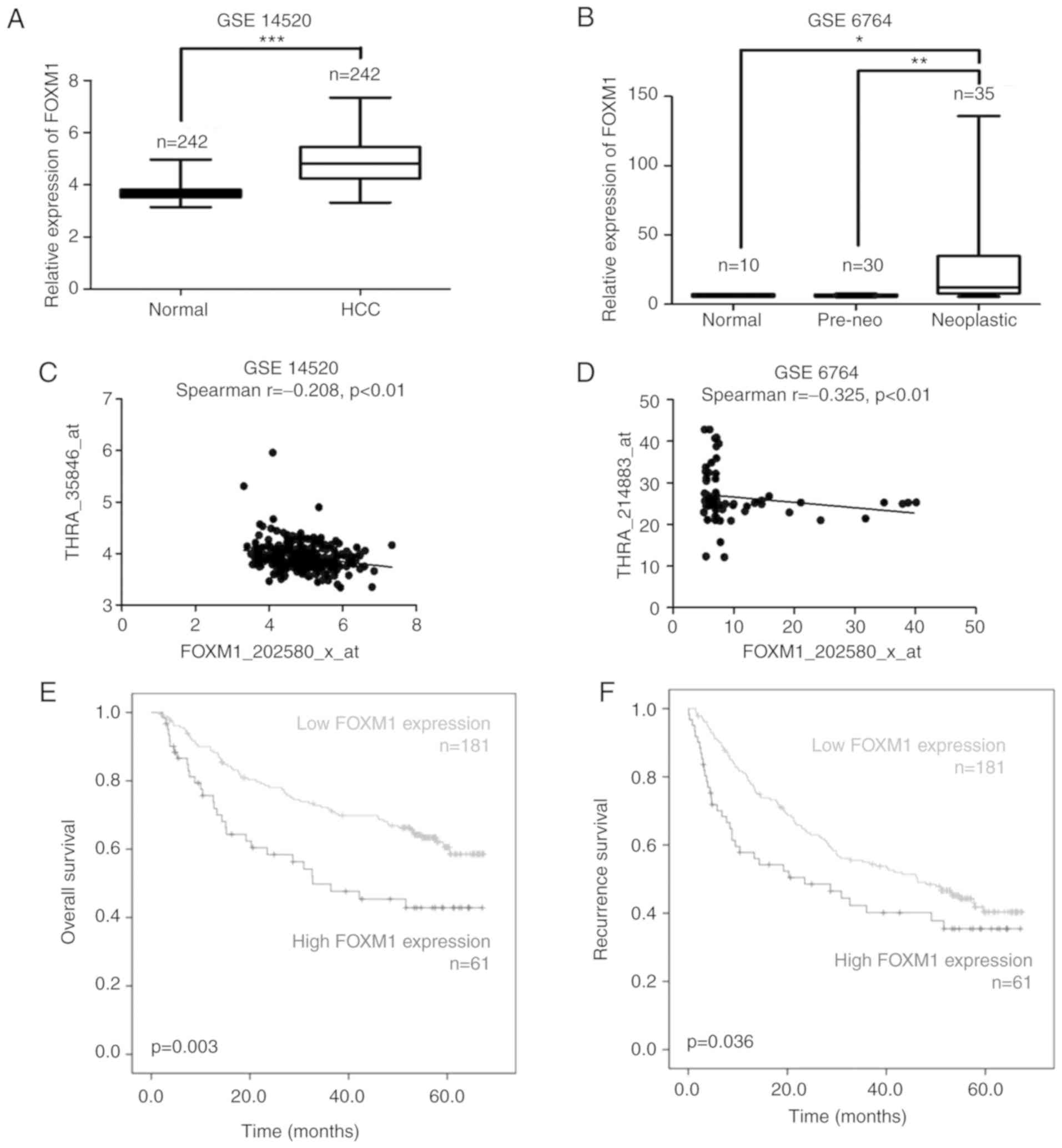
High expression of FOXM1 is associated
with poor survival in HCC
FOXM1 expression was analyzed using GEO datasets, a
web-based microarray database and data mining platform. Analysis of
three independent cohorts revealed significantly higher expression
of FOXM1 mRNA in tumor regions of patients compared with normal
tissues (Fig. 1A and B).
Signifciantly decreased FOXM1 mRNA was accompanied by a concomitant
increase in THRA (Fig. 1C and D) in
cohort 1 and cohort 2. Although FOXM1 mRNA expression is inversely
correlated with THRA mRNA expression, there was no significant
difference of FOXM1 mRNA expression between tumor regions and
normal tissues of patients in GSE14323 (data not shown). Further
analysis of GSE14520 indicated that FOXM1 expression is negatively
associated with overall survival (Fig.
1E) and recurrence survival (Fig.
1F). Significantly increased FOXM1 expression was detected at
the late stages of HCC (Fig. 1gG
and positively associated with specific clinicopathological
parameters, including tumor size (Fig.
1H), hepatitis V viral status (Fig.
1I), alpha fetoprotein (Fig.
1J) and predicted metastasis risk signatures (Fig. 1K).
TH inhibits FOXM1 mRNA and protein
levels in HCC cells
To determine the potential significance of
T3/TR in modulating FOXM1, HepG2-TRα1, HepG2-TRβ1 and
HepG2-Neo (vector-control) cell lines previously established in our
laboratory were used for experiments (46). FOXM1 mRNA was quantified after
T3 treatment of cells via RT-qPCR. In the presence of
T3, compared with controls, FOXM1 mRNA expression was
significantly decreased in HepG2-TRα1 (Fig. 2A) and HepG2-TRβ1 (Fig. 2B) cells but less reduced in
HepG2-Neo cells (Fig. 2C). Western
blot analysis further demonstrated a significant decrease in FOXM1
protein expression in HepG2-TRα1 (Fig.
2D) and HepG2-TRβ1 (Fig. 2E)
cells following treatment with T3 for 48 h but not in
HepG2-Neo cells (Fig. 2F). To
ascertain whether T3-induced repression of FOXM1 mRNA in
HepG2 cells is mediated by direct effects of TR on transcription,
the promoter region of FOXM1 with two nTRE motifs, NM23H1 and
hTSHβ-N, was analyzed. Several potential nTREs were identified in
FOXM1 promoter regions from positions −1560 to +22 (Fig. 2G). Subsequently, serially-deleted
FOXM1 promoter fragments were cloned into a pGL3-Basic vector. The
promoter activity of FOXM1 was significantly repressed in fragments
encompassing positions −1560 to +22, −1480 to +22 and −1300 to +22
with T3 treatment compared with controls. However, the
effects of T3 were reduced in fragments −914 to +22 and
−728 to +22 (Fig. 2H). Data from
ChIP performed to validate the potential binding site of TR
suggested that T3 mediated suppression of FOXM1 promoter
activity, with the promoter binding site of TR potentially located
between positions −1033 and −862 (Fig.
2I). This indicated that T3 suppressed both FOXM1
mRNA and protein expression in a concentration- and time-dependent
manner, which is positively associated with TR expression.
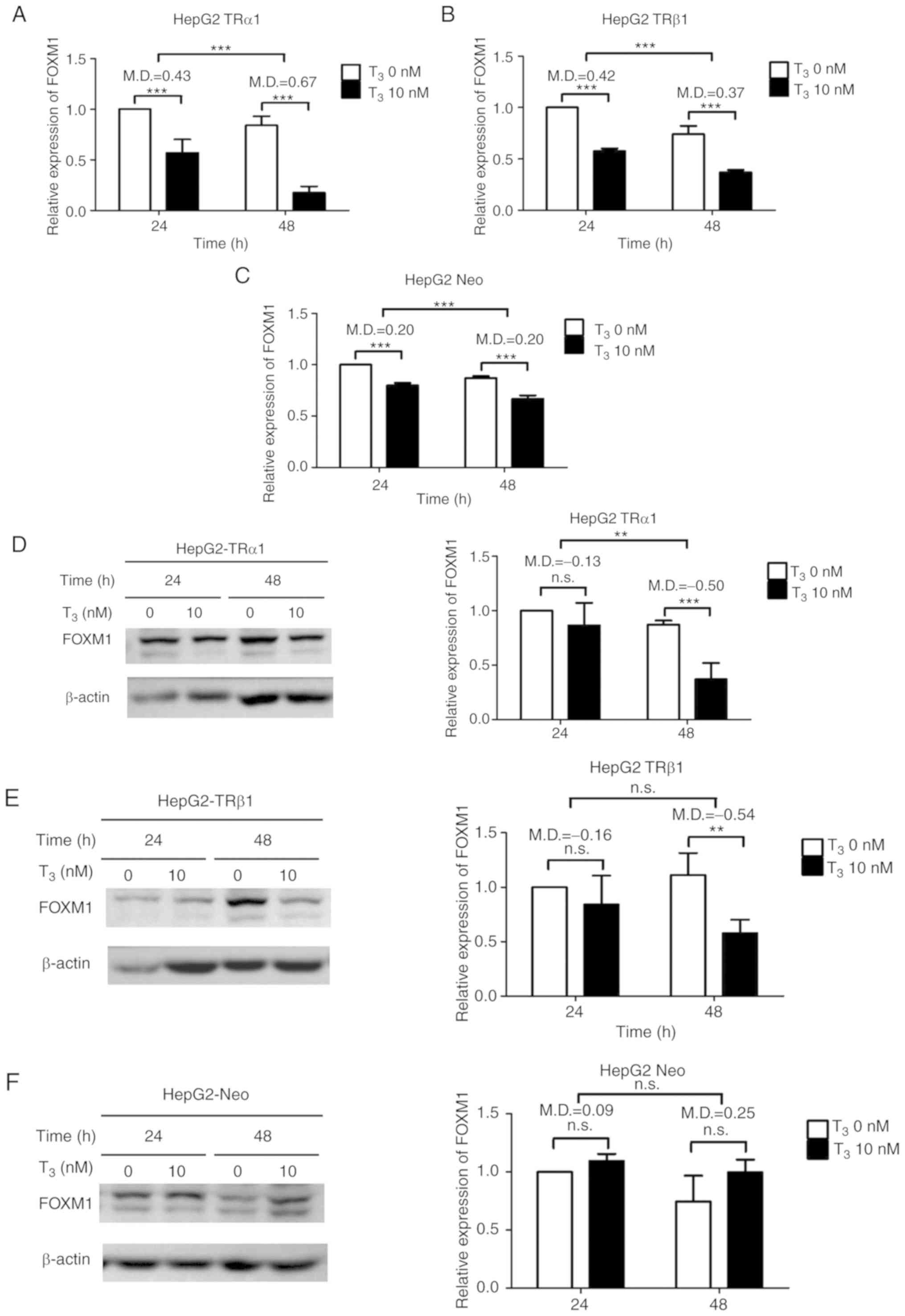 | Figure 2.T3/TR suppresses FOXM1
expression in HepG2 cells. Analysis of FOXM1 mRNA expression in (A)
HepG2-TRα1, (B) HepG2-TRβ1 and (C) HepG2-Neo cells after treatment
with 0 and 10 nM T3 for 24 and 48 h. Protein expression
of FOXM1 was analyzed in (D) HepG2-TRα1, (E) HepG2-TRβ1 and (F)
HepG2-Neo cells after treatment with 0 and 10 nM T3 for
24 and 48 h. Quantitative results are shown at the right panel.
T3/TR suppresses FOXM1 expression in HepG2 cells. (G)
Illustration of predicted nTREs within the FOXM1 promoter region.
(H) Reporter constructs containing serially deleted fragments of
the FOXM1 5′-flanking region in a pGL3-luc vector were transfected
into HepG2-TRα1 cells treated with or without 10 nM T3.
(I) For the chromatin immunoprecipitation assay, HepG2-TRα1 cell
lysates were immunoprecipitated with rabbit nonspecific IgG or
antibodies against TR. The promoter region of GAPDH acted as the
negative control. Data are presented as the mean ± standard
deviation. n=3. **P<0.01 and ***P<0.001. MD, mean of
difference; FOXM1, forkhead box protein M1; TR, thyroid hormone
receptors; nTRE, negative thyroid hormone response elements; ns,
not significant; IgG, immunoglobulin G; C1, −1560~+22; C2,
−1480~+22; C3, −1300~+22); C4, −914~+22); C5, −728~+22, Luc,
pGL3-Basic vector containing serially deleted FOXM1 promoter
fragments. |
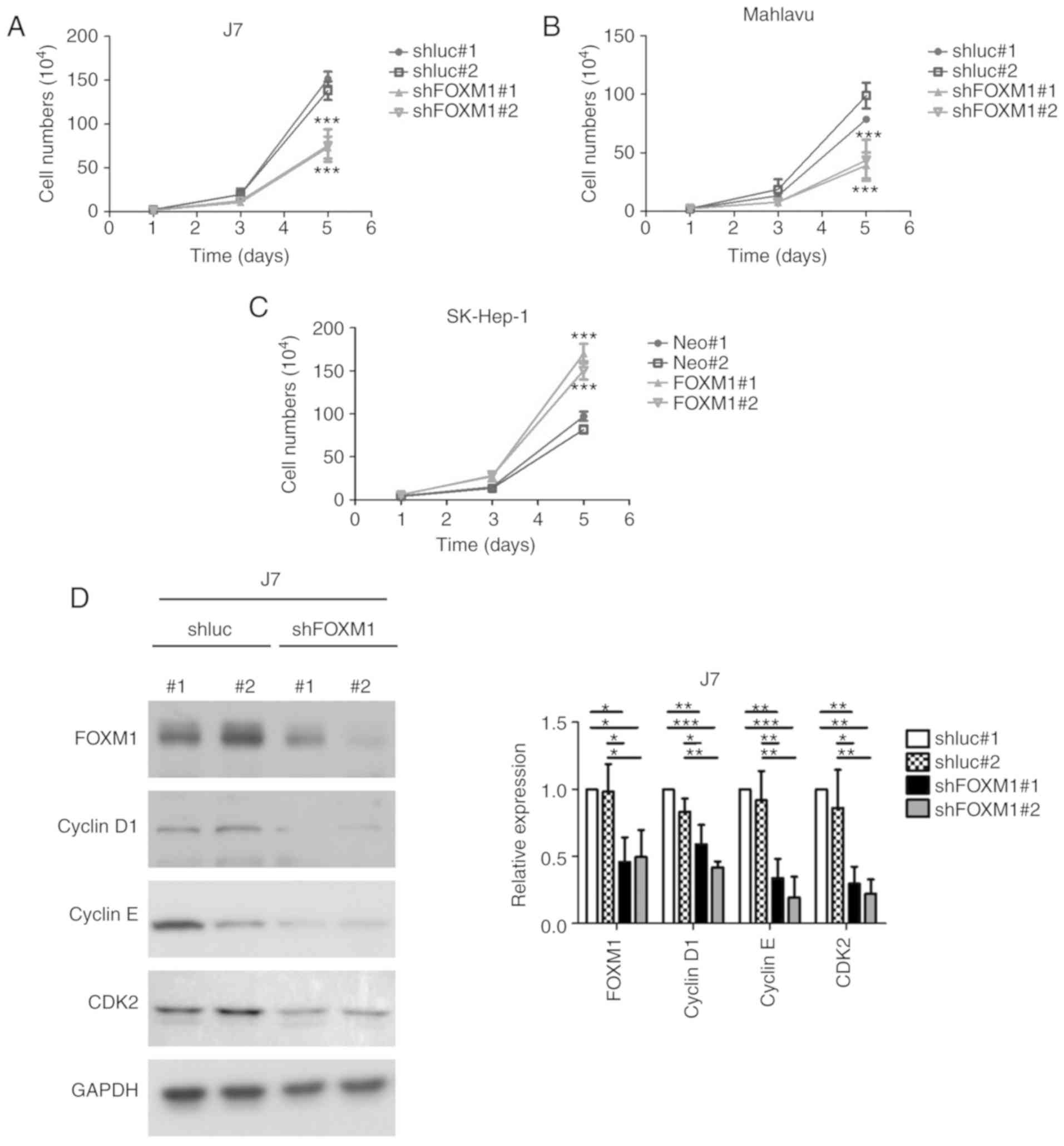
Manipulation of FOXM1 expression in
HCC cell lines affects cell proliferation
To ascertain whether FOXM1 participates in liver
cancer progression, the present study established stable J7 and
Mahlavu cells with shRNA-mediated FOXM1 knockdown (shFOXM1#1 and
shFOXM1#2) and overexpression in SK-Hep1 cells via transient
transfection of control pcDNA3.1 vector (Neo#1 and Neo#2) and
pcDNA3.1-FOXM1 (FOXM1#1 and FOXM1#2). Depletion of FOXM1 led to
significantly reduced growth of both cell lines at day 5 (Fig. 3A and B) compared with their
respective controls (shluc#1 and shluc#2). Conversely,
overexpression of FOXM1 induced a significant increase in the cell
growth rate (Fig. 3C) compared with
the vector-transfected control group. The signal transduction
pathway associated with alterations in FOXM1 expression were
additionally investigated. Depletion of FOXM1 led to significantly
reduced expression of positive regulators of cell cycle progression
(cyclin D1, cyclin E and CDK2) in both J7 (Fig. 3D) and Mahlavu cells (Fig. 3E) compared with their respective
controls. Consistent with these findings, enhanced expression of
FOXM1 significantly increased the levels of cell cycle-promoting
cyclin D1, cyclin E and CDK2 (Fig.
3F). These results collectively demonstrated a tumor-promoting
role of FOXM1 in HCC cells, highlighting the role of
T3-mediated regulation of FOXM1 in liver cancer.
Overexpression of FOXM1 in HCC cells
leads to partial recovery of response to T3
treatment
The suppressive effect of T3 on cell
proliferation rate was demonstrated in a previous study by our
group (46). To determine whether
T3-mediated FOXM1 decline regulates cancer progression,
FOXM1 was overexpressed in Huh7 and SK-Hep-1 cells subjected to
T3 treatment. Expression of FOXM1 was marginally
decreased upon T3 treatment due to low expression of
endogenous TR (Fig. 4A and B). The
cell proliferation rate significantly decreased in Huh7 and SK-Hep1
cells (Neo) after T3 treatment compared with controls.
Re-expression of FOXM1 in Huh7 and SK-Hep1 cells (Neo-FOXM1)
partially rescued suppression of cell proliferation induced by
T3 (Fig. 4C and D),
supporting its role in T3-mediated regulation of cell
proliferation (Fig. 4E).
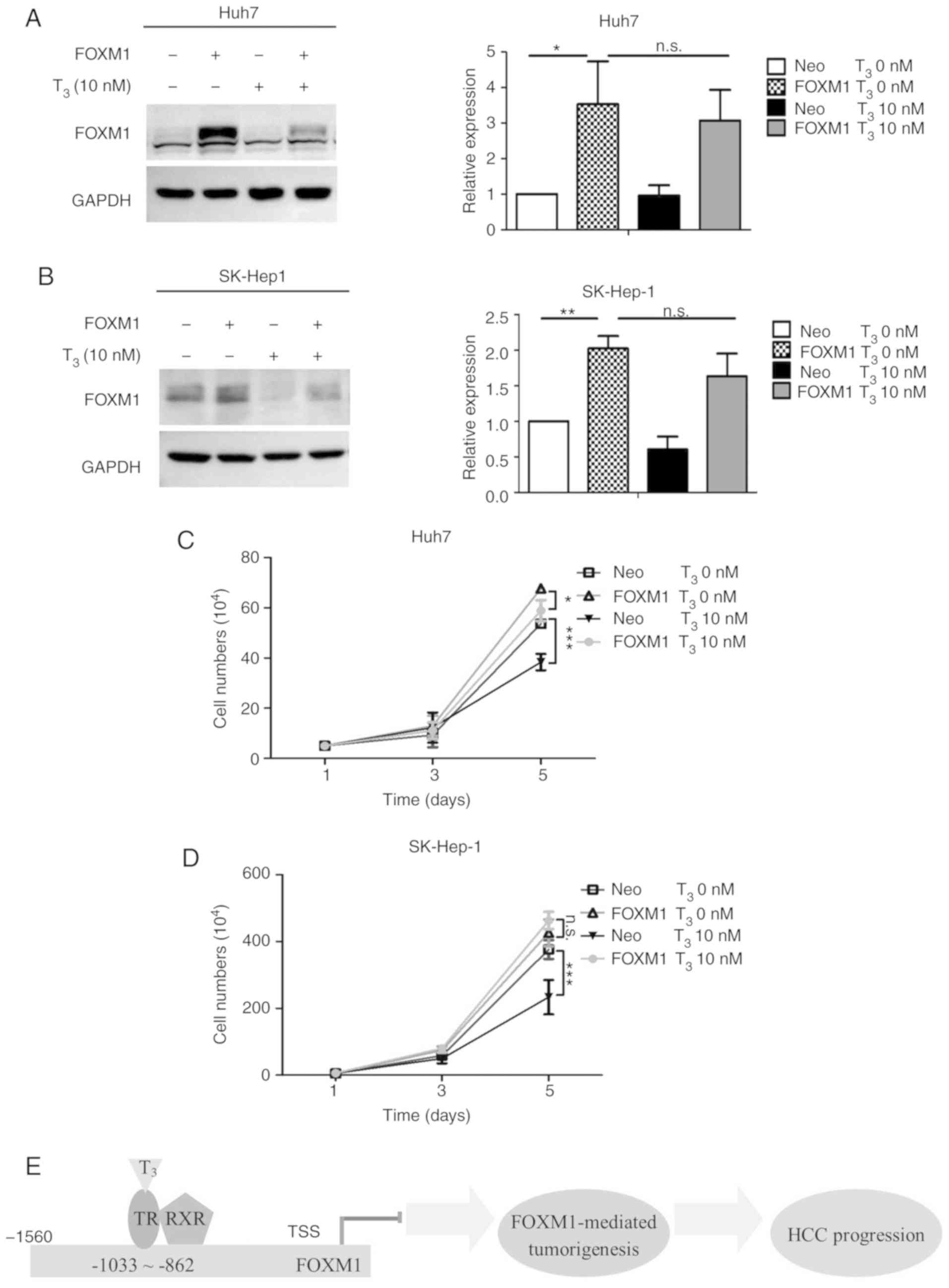 | Figure 4.Overexpression of FOXM1 in HCC cells
partially rescues suppression of proliferation induced by
T3. FOXM1 expression in (A) Huh7 and (B) SK-Hep-1
control cells (Neo) or FOXM1-overexpressing cells (FOXM1) with
T3 depletion (Neo T3 0 nM, FOXM1
T3 0 nM) or 10 nM T3 (Neo T3 10
nM, FOXM1 T3 10 nM) for 24 h. Quantitative results are
presented in the right panel. (C) Huh7 and (D) SK-Hep-1 Neo-Td,
FOXM1-Td, Neo-T3 and FOXM1-T3 cells were
seeded in six-well plates and counted at the indicated times (days
1, 3 and 5). (E) Schematic diagram of the mechanism underlying
thyroid hormone/TR signaling-mediated suppression of FOXM1 to
reduce cancer progression. The red line indicates repression.
Yellow arrows indicate promotion. Data are presented as the mean ±
standard deviation. n=3. *P<0.05, **P<0.01 and ***P<0.001.
FOXM1, forkhead box protein M1; ns, not significant; HCC,
hepatocellular carcinoma; TR, thyroid hormone receptors. RXR,
Retinoid X receptor; TSS, translation start site. |
Discussion
Homeostasis of TH/TR signaling is critical for life
processes via regulation of various downstream genes, and
disruption of this fine-tuning can lead to malignancies (8). The liver has been identified as one of
the targets of TR, with alterations in TH levels shown to trigger
HCC (3). However, the underlying
mechanisms remain to be elucidated.
FOXM1 is highly expressed in several cancer types,
including lung, ovarian, gastric and liver cancer (52). Data from the present study showed
that TH/TR inhibited FOXM1 expression in HCC cell lines and FOXM1
and TR are negatively correlated in HCC, providing evidence
supporting a tumor suppressor role of thyroid hormone signaling. A
promoting role of FOXM1 in HCC development was reported by
Kalinichenko et al (39),
where depletion of FOXM1 had little effect in normal hepatocytes
but suppressed HCC progression in a diethylnitrosamine
(DEN)/phenobarbital (PB)-induced HCC mouse model, and depletion of
FOXM1 following HCC establishment led to a significant decrease in
tumor size. Park et al (42)
further reported FOXM1 overexpression in the absence of p19Arf, a
potent inhibitor of FOXM1. The researchers generated a
bi-transgenic strain in which FOXM1 was expressed from the Rosa26
promoter in an Arf-/- (FOXM1bTg; Arf-/-) background. Mice developed
highly metastatic HCC (>70%) following DEN/PB treatment.
Metastasis significantly declined in the presence of one copy of
Arf (FOXM1bTg; Arf+/- mice), supporting a critical role of FOXM1 in
HCC development and its potential application as a therapeutic
target.
CDKs and cyclins are involved in cell cycle
progression. For instance, phosphorylation of retinoblastoma
mediated by cyclin D/CDK4/CDK6 and cyclin E/CDK2 complexes
contribute to cell cycle progression (53). Moreover, the cyclin E/CDK2 complex
is reported to activate E2F transcription factors and other cell
cycle-related genes, facilitating S-phase entry (54). Development of HCC is usually based
on a background of chronic hepatitis and occurs as a multistep
process involving dysregulation of multiple cell cycle-related
genes, including cyclin E (55).
Investigation of the role of CDK2 in hepatocarcinogenesis in
Cdk2Δhepa mice revealed significant reduction of tumor load
following DEN treatment (56). In
liver regeneration studies, Foxm1-/- hepatocytes of Alb-Cre Foxm1
fl/fl mice displayed reduced expression of the cyclin E/CDK2
complex (57), similar to
FOXM1-depleted U2OS cells. Several studies reported that the cyclin
E/CDK2 complex is critical for early tumorigenesis but dispensable
for advanced tumor progression, highlighting the complexity of
carcinogenesis and diverse roles of FOXM1 in tumor progression.
Drug resistance is a major challenge in cancer
therapy. Several reports indicated that FOXM1 contributes to
chemoresistance of various human carcinomas (52). Docetaxel (DTX) is a second-line
chemotherapeutic drug for non-small-cell lung carcinoma. Compared
to parental A549 cells, FOXM1 expression was significantly elevated
in a DTX-resistant A549 cell line, while depletion of FOXM1
promoted DTX sensitivity of these cells (58). Increasing levels of FOXM1
upregulated stathmin to mediate microtubule dynamics, leading to
tumor cell escape from DTX-induced apoptosis (59).
N6-methyladenosine has been identified as the most
common internal modification of eukaryotic mRNAs, although its
specific functions are yet to be elucidated (60). Recently, Zhang et al
(61) showed that overexpression of
the m6A demethylase, α-ketoglutarate-dependent dioxygenase alkB
homolog 5 (ALKBH5), is required for proliferation and tumorigenesis
of glioblastoma stem-like cells and FOXM1 nascent transcripts are
demethylated by ALKBH5. This step promoted the interaction between
FOXM1 pre-mRNA with HuR, thereby maintaining FOXM1 expression.
ALKBH5-dependent gene expression provided insights into the pivotal
role of RNA m6A methylation in cancer progression, supporting a
therapeutic strategy involving targeting of RNA epigenetic
modulators. In the analysis of the GSE14520 dataset (48) (data not shown), THRA mRNA expression
was negatively correlated with the m6A
methyltransferase, METTL3, but positively correlated with
m6A demethylase fat-mass and obesity-associated protein
(FTO) (data not shown). Additionally, FOXM1 mRNA expression was
positively correlated with METTL3 and negatively correlated with
FTO, suggesting the possibility that FOXM1 is regulated by TH/TR
through RNA modifications (data not shown).
In conclusion, TH/TR signaling suppressed FOXM1
expression, which participated in HCC progression via exerting its
effects on downstream gene expression, highlighting the biological
significance of thyroid hormone homeostasis in maintenance of
physiological cell functions.
Acknowledgements
We greatly appreciate the valuable work carried out
by Professor Sheng-Ming Wu at Taipei Medical University (New Taipei
City, Taiwan) for candidate selection and primary validation.
Funding
This work was supported by grants from Chang Gung
Memorial Hospital, Taoyuan, Taiwan (grant nos. CMRPD1G0421,
CMRPD1G0422, CMRPD1G0423, NMRPD1G0941, NMRPD1G0942, NMRPD1G0951 and
NMRPD1G0952) and from the Ministry of Science and Technology of the
Republic of China (grant nos. 106-2320-B-182-031-MY3 and
106-2320-B-182-032-MY3).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CHW designed the study, performed the experiments,
analyzed the data and wrote the manuscript. CTY analyzed and
interpreted the data. KHL designed the study, analyzed the data,
critically revised the manuscript and gave final approval of the
version to be published. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wu SM, Cheng WL, Lin CD and Lin KH:
Thyroid hormone actions in liver cancer. Cell Mol Life Sci.
70:1915–1936. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chi HC, Chen CY, Tsai MM, Tsai CY and Lin
KH: Molecular functions of thyroid hormones and their clinical
significance in liver-related diseases. Biomed Res Int.
2013:6013612013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu YC, Yeh CT and Lin KH: Molecular
functions of thyroid hormone signaling in regulation of cancer
progression and anti-apoptosis. Int J Mol Sci. 20:49862019.
View Article : Google Scholar
|
|
4
|
Sap J, Muñoz A, Damm K, Goldberg Y,
Ghysdael J, Leutz A, Beug H and Vennström B: The c-erb-A protein is
a high-affinity receptor for thyroid hormone. Nature. 324:635–640.
1986. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weinberger C, Thompson CC, Ong ES, Lebo R,
Gruol DJ and Evans RM: The c-erb-A gene encodes a thyroid hormone
receptor. Nature. 324:641–646. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hammes SR and Davis PJ: Overlapping
nongenomic and genomic actions of thyroid hormone and steroids.
Best Pract Res Clin Endocrinol Metab. 29:581–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davis PJ, Glinsky GV, Lin HY and Mousa SA:
Actions of thyroid hormone analogues on chemokines. J Immunol Res.
2016:31476712016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang PS, Wang CS, Yeh CT and Lin KH:
Roles of thyroid hormone-associated microRNAs affecting oxidative
stress in human hepatocellular carcinoma. Int J Mol Sci.
20:52202019. View Article : Google Scholar
|
|
9
|
Davis PJ, Leonard JL, Lin HY, Leinung M
and Mousa SA: Molecular basis of nongenomic actions of thyroid
hormone. Vitam Horm. 106:67–96. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iwasaki Y, Sunaga N, Tomizawa Y, Imai H,
Iijima H, Yanagitani N, Horiguchi K, Yamada M and Mori M:
Epigenetic inactivation of the thyroid hormone receptor beta1 gene
at 3p24.2 in lung cancer. Ann Surg Oncol. 17:2222–2228. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu R, Li Z, Bai S, Zhang H, Tang M, Lei
Y, Chen L, Liang S, Zhao YL, Wei Y and Huang C: Mechanism of cancer
cell adaptation to metabolic stress: Proteomics identification of a
novel thyroid hormone-mediated gastric carcinogenic signaling
pathway. Mol Cell Proteomics. 8:70–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brown AR, Simmen RC and Simmen FA: The
role of thyroid hormone signaling in the prevention of digestive
system cancers. Int J Mol Sci. 14:16240–16257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davis PJ, Lin HY, Hercbergs AA, Keating KA
and Mousa SA: How thyroid hormone works depends upon cell type,
receptor type, and hormone analogue: Implications in cancer growth.
Discov Med. 27:111–117. 2019.PubMed/NCBI
|
|
14
|
Davis PJ, Sudha T, Lin HY and Mousa SA:
Thyroid hormone, hormone analogs, and angiogenesis. Compr Physiol.
6:353–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cicatiello AG, Ambrosio R and Dentice M:
Thyroid hormone promotes differentiation of colon cancer stem
cells. Mol Cell Endocrinol. 459:84–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin HY, Chin YT, Yang YC, Lai HY,
Wang-Peng J, Liu LF, Tang HY and Davis PJ: Thyroid hormone, cancer,
and apoptosis. Compr Physiol. 6:1221–1237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rostkowska O, Spychalski P, Dobrzycka M,
Wilczyński M, Łachiński AJ, Obołończyk Ł, Sworczak K and Kobiela J:
Effects of thyroid hormone imbalance on colorectal cancer
carcinogenesis and risk-a systematic review. Endokrynol Pol.
70:190–197. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hercbergs A, Mousa SA, Leinung M, Lin HY
and Davis PJ: Thyroid hormone in the clinic and breast cancer. Horm
Cancer. 9:139–143. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin YH, Wu MH, Liao CJ, Huang YH, Chi HC,
Wu SM, Chen CY, Tseng YH, Tsai CY, Chung IH, et al: Repression of
microRNA-130b by thyroid hormone enhances cell motility. J Hepatol.
62:1328–1340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chi HC, Chen SL, Tsai CY, Chuang WY, Huang
YH, Tsai MM, Wu SM, Sun CP, Yeh CT and Lin KH: Thyroid hormone
suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent
selective autophagy. Autophagy. 12:2271–2285. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tseng YH, Huang YH, Lin TK, Wu SM, Chi HC,
Tsai CY, Tsai MM, Lin YH, Chang WC, Chang YT, et al: Thyroid
hormone suppresses expression of stathmin and associated tumor
growth in hepatocellular carcinoma. Sci Rep. 6:387562016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chi HC, Chen SL, Lin SL, Tsai CY, Chuang
WY, Lin YH, Huang YH, Tsai MM, Yeh CT and Lin KH: Thyroid hormone
protects hepatocytes from HBx-induced carcinogenesis by enhancing
mitochondrial turnover. Oncogene. 36:5274–5284. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Puliga E, Min Q, Tao J, Zhang R,
Pradhan-Sundd T, Poddar M, Singh S, Columbano A, Yu J and Monga SP:
Thyroid hormone receptor-β agonist GC-1 inhibits
met-β-catenin-driven hepatocellular cancer. Am J Pathol.
187:2473–2485. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang PS, Lin YH, Chi HC, Chen PY, Huang
YH, Yeh CT, Wang CS and Lin KH: Thyroid hormone inhibits growth of
hepatoma cells through induction of miR-214. Sci Rep. 7:148682017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin YH, Wu MH, Huang YH, Yeh CT, Chi HC,
Tsai CY, Chuang WY, Yu CJ, Chung IH, Chen CY and Lin KH: Thyroid
hormone negatively regulates tumorigenesis through suppression of
BC200. Endocr Relat Cancer. 25:967–979. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CY, Wu SM, Lin YH, Chi HC, Lin SL,
Yeh CT, Chuang WY and Lin KH: Induction of nuclear protein-1 by
thyroid hormone enhances platelet-derived growth factor A mediated
angiogenesis in liver cancer. Theranostics. 9:2361–2379. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kowalik MA, Puliga E, Cabras L, Sulas P,
Petrelli A, Perra A, Ledda-Columbano GM, Morandi A, Merlin S, Orrù
C, et al: Thyroid hormone inhibits hepatocellular carcinoma
progression via induction of differentiation and metabolic
reprogramming. J Hepatol. 72:1159–1169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chi HC, Tsai CY, Tsai MM, Yeh CT and Lin
KH: Molecular functions and clinical impact of thyroid
hormone-triggered autophagy in liver-related diseases. J Biomed
Sci. 26:242019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barlow C, Meister B, Lardelli M, Lendahl U
and Vennström B: Thyroid abnormalities and hepatocellular carcinoma
in mice transgenic for v-erbA. EMBO J. 13:4241–4250. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chan IH and Privalsky ML: Thyroid hormone
receptors mutated in liver cancer function as distorted antimorphs.
Oncogene. 25:3576–3588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chan IH and Privalsky ML: Thyroid hormone
receptor mutants implicated in human hepatocellular carcinoma
display an altered target gene repertoire. Oncogene. 28:4162–4174.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin KH, Shieh HY, Chen SL and Hsu HC:
Expression of mutant thyroid hormone nuclear receptors in human
hepatocellular carcinoma cells. Mol Carcinog. 26:53–61. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin KH, Wu YH and Chen SL: Impaired
interaction of mutant thyroid hormone receptors associated with
human hepatocellular carcinoma with transcriptional coregulators.
Endocrinology. 142:653–662. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin KH, Zhu XG, Shieh HY, Hsu HC, Chen ST,
McPhie P and Cheng SY: Identification of naturally occurring
dominant negative mutants of thyroid hormone alpha 1 and beta 1
receptors in a human hepatocellular carcinoma cell line.
Endocrinology. 137:4073–4081. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koo CY, Muir KW and Lam EW: FOXM1: From
cancer initiation to progression and treatment. Biochim Biophys
Acta. 1819:28–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lam EW, Brosens JJ, Gomes AR and Koo CY:
Forkhead box proteins: Tuning forks for transcriptional harmony.
Nat Rev Cancer. 13:482–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kalinichenko VV, Major ML, Wang X,
Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A,
Raychaudhuri P and Costa RH: Foxm1b transcription factor is
essential for development of hepatocellular carcinomas and is
negatively regulated by the p19ARF tumor suppressor. Genes Dev.
18:830–850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shih CH, Chen SL, Yen CC, Huang YH, Chen
CD, Lee YS and Lin KH: Thyroid hormone receptor-dependent
transcriptional regulation of fibrinogen and coagulation proteins.
Endocrinology. 145:2804–2814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bella L, Zona S, Nestal de Moraes G and
Lam EW: FOXM1: A key oncofoetal transcription factor in health and
disease. Semin Cancer Biol. 29:32–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park HJ, Gusarova G, Wang Z, Carr JR, Li
J, Kim KH, Qiu J, Park YD, Williamson PR, Hay N, et al:
Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med.
3:21–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nandi D, Cheema PS, Jaiswal N and Nag A:
FoxM1: Repurposing an oncogene as a biomarker. Semin Cancer Biol.
52:74–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shiu TY, Huang TY, Huang SM, Shih YL, Chu
HC, Chang WK and Hsieh TY: Nuclear factor κB down-regulates human
UDP-glucuronosyltransferase 1A1: A novel mechanism involved in
inflammation-associated hyperbilirubinaemia. Biochem J.
449:761–770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen CJ, Tsai NM, Liu YC, Ho LI, Hsieh HF,
Yen CY and Harn HJ: Telomerase activity in human hepatocellular
carcinoma: Parallel correlation with human telomerase reverse
transcriptase (hTERT) mRNA isoform expression but not with cell
cycle modulators or c-Myc expression. Eur J Surg Oncol. 28:225–234.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu SM, Cheng WL, Liao CJ, Chi HC, Lin YH,
Tseng YH, Tsai CY, Chen CY, Lin SL, Chen WJ, et al: Negative
modulation of the epigenetic regulator, UHRF1, by thyroid hormone
receptors suppresses liver cancer cell growth. Int J Cancer.
137:37–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX and Wang XW: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wurmbach E, Chen YB, Khitrov G, Zhang W,
Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J, et
al: Genome-wide molecular profiles of HCV-induced dysplasia and
hepatocellular carcinoma. Hepatology. 45:938–947. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mas VR, Maluf DG, Archer KJ, Yanek K, Kong
X, Kulik L, Freise CE, Olthoff KM, Ghobrial RM, McIver P and Fisher
R: Genes involved in viral carcinogenesis and tumor initiation in
hepatitis C virus-induced hepatocellular carcinoma. Mol Med.
15:85–94. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Borhani S and Gartel AL: FOXM1: A
potential therapeutic target in human solid cancers. Expert Opin
Ther Targets. 24:205–217. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Goel B, Tripathi N, Bhardwaj N and Jain
SK: Small molecule CDK inhibitors for the therapeutic management of
cancer. Curr Top Med Chem. May 16–2020.doi:
10.2174/1568026620666200516152756. Online ahead of print.
View Article : Google Scholar
|
|
54
|
Kim B, Shin HC, Heo YJ, Ha SY, Jang KT,
Kim ST, Kang WK, Lee J and Kim KM: CCNE1 amplification is
associated with liver metastasis in gastric carcinoma. Pathol Res
Pract. 215:1524342019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Aziz K, Limzerwala JF, Sturmlechner I,
Hurley E, Zhang C, Jeganathan KB, Nelson G, Bronk S, Fierro Velasco
RO, van Deursen EJ, et al: Ccne1 overexpression causes chromosome
instability in liver cells and liver tumor development in mice.
Gastroenterology. 157:210–226.e12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sonntag R, Giebeler N, Nevzorova YA,
Bangen JM, Fahrenkamp D, Lambertz D, Haas U, Hu W, Gassler N,
Cubero FJ, Müller-Newen G, et al: Cyclin E1 and cyclin-dependent
kinase 2 are critical for initiation, but not for progression of
hepatocellular carcinoma. Proc Natl Acad Sci USA. 115:9282–9287.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang IC, Chen YJ, Hughes D, Petrovic V,
Major ML, Park HJ, Tan Y, Ackerson T and Costa RH: Forkhead box M1
regulates the transcriptional network of genes essential for
mitotic progression and genes encoding the SCF (Skp2-Cks1)
ubiquitin ligase. Mol Cell Biol. 25:10875–10894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang K, Zhu X, Zhang K, Zhu L and Zhou F:
FoxM1 inhibition enhances chemosensitivity of docetaxel-resistant
A549 cells to docetaxel via activation of JNK/mitochondrial
pathway. Acta Biochim Biophys Sin (Shanghai). 48:804–809. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li X, Yao R, Yue L, Qiu W, Qi W, Liu S,
Yao Y and Liang J: FOXM1 mediates resistance to docetaxel in
gastric cancer via up-regulating Stathmin. J Cell Mol Med.
18:811–823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: m6A
demethylase ALKBH5 maintains tumorigenicity of glioblastoma
stem-like cells by sustaining FOXM1 expression and cell
proliferation program. Cancer Cell. 31:591–606.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|