Introduction
Colorectal cancer (CRC) is one of the most commonly
diagnosed cancers in both males and females. Accordingly, this
disease is a public health issue and accounts for 8.5% of all
cancer-related deaths worldwide (1). CRC progression occurs through a series
of well-defined clinical and histopathological features, ranging
from single precursor lesions through benign tumors (serrated or
tubular adenoma) to malignant disease (2).
CRC is a heterogeneous disease represented by
biologically diverse subtypes. One of the most recent gene
expression-based subtypings of CRC has proposed four consensus
molecular subtypes (CMSs), namely CMS1-microsatellite instability
immune, CMS2-canonical, CMS3-metabolic, and CMS4-mesenchymal. Each
of these CMSs shows distinguishing molecular disorders related to
different clinical outcomes (3,4);
however, consensus molecular classification has not yet been used
as a tool to guide clinical decisions. The constant development of
molecular stratification strategies is also still necessary to
reveal clinical potentials (5).
Regardless of the subtype, the disruption of the
apical junctional complex (AJC), consisting of tight junctions
(TJs) and adherens junctions, is frequently observed during CRC
progression (6). The functionality
of TJs, which constitute the barrier to the paracellular flow of
macromolecules and ions, is regulated in part by the levels of its
proteins (7,8), and claudins in particular. Claudins
are the main proteins in the regulation of TJs, and they also
affect the stability of TJs via a fine-tuned mechanism (9,10)
involving changes in the subcellular localization and an imbalance
(both overexpression and downregulation) in claudin levels. For
example, dysregulation of claudin-3 is often observed in CRC
(11). Claudin-3 overexpression
destabilizes the TJs, causing an increase in transepithelial
resistance and the paracellular flux of macromolecules leading to
induction of cell migration and colony formation that can be either
dependent or independent of anchorage (12).
Previous studies have also demonstrated that the
stability of TJs is regulated by glycoproteins, such as receptor
tyrosine kinases (RTKs) and E-cadherin (13–15),
but the regulatory role of N-linked glycans in this process
is poorly understood. Pioneering work using siRNA to DPAGT1
(the gene that encodes the enzyme that initiates the synthesis of
the dolichol lipid-linked oligosaccharide precursor for protein
N-glycosylation) showed that inhibition of
N-glycosylation promotes the assembly of TJs through the
recruitment of the PP2A protein (a negative regulator of TJ
biogenesis) to adherens junctions (16), thereby suggesting a role for
N-glycans in TJ stability. Indeed, the functionality of RTKs
is finely tuned by the N-glycans attached to its
extracellular domain.
Several studies have demonstrated that
N-glycosylation contributes to ligand binding, kinase
activity, and the determination of the proper conformation of the
RTKs (17–20). For example, β1,6-GlcNAc-branching
N-glycans are synthesized by MGAT5 (also known as GnT-V;
Fig. S1) and are inserted onto
RTKs. Their presence promotes the binding of this branched
structure to galectins to form molecular ‘lattices’ that prevent
glycoprotein receptor endocytosis and lead to the persistence of
cancer-related signaling (21).
However, a mechanism that would integrate RTK signaling and
N-glycosylation with the regulation of claudin-3 levels in
CRC remains to be established.
In the present study, we demonstrated that the high
expression levels of CLDN3 in CRC worsened the patients'
long-term survival. We also showed that inhibition of
N-glycan biosynthesis in CRC cells led to a decrease in the
levels of claudin-3 and, concomitantly, to a reduction in
phosphorylation levels of epidermal growth factor receptor (EGFR)
and insulin-like growth factor 1 receptor (IGF1R). We report that
specific inactivation of RTKs also leads to a decrease in claudin-3
levels, and we provide evidence that the regulation of claudin-3
levels by changes in RTK signaling can be mediated by phospholipase
C (PLC) and signal transducer and activator of transcription 3
(STAT3) in CRC cells. We therefore evaluated the correlation
between N-glycogenes and CLDN3 expression levels in
each of the CRC molecular subtypes, since N-glycans are
known to regulate RTKs. CMS1 (MSI immune) was the subtype that
concomitantly exhibited low expression levels of CLDN3 and
N-glycogenes (MGAT5, ST6GAL1, and B3GNT8),
whereas CMS2 (canonical) exhibited high gene expression levels of
CLDN3 and N-glycogenes (ST6GAL1 and
B3GNT8). A robust positive correlation was also detected
between CLDN3 and B3GNT8 expression levels in all
four CMS. Taken together, our results support the hypothesis that
N-glycans play a role in the regulation of RTKs, claudin-3,
and TJs, thereby providing a better understanding of CRC
biology.
Materials and methods
Chemicals and antibodies
The rabbit monoclonal antibodies used here included
anti-pEGFR (Tyr 1068, cat. no. 2234), anti-EGFR (cat. no. 2646),
anti-p-IR/p-IGF1R (Tyr 1150, 1151/Tyr 1135, 1136, cat. no. 3024),
anti-IGF1R (cat. no. 3027), anti-pAKT (Ser 473, cat. no. 9271),
anti-AKT (cat. no. 4691) and anti-α-tubulin (cat. no. 2144) and
were purchased from Cell Signaling Technology, Inc. Rabbit
polyclonal antibodies anti-claudin-3 (cat. no. 34-1700),
anti-occludin (cat. no. 33-1500) and anti-ZO1 (cat. no. 33-9100)
were obtained from Thermo Fisher Scientific, Inc. Anti-mouse GAPDH
monoclonal antibody (cat. no. 32233) were obtained from Santa Cruz
Biotechnology, Inc. Secondary peroxidase-conjugated anti-mouse
(cat. no. 2304) and anti-rabbit (cat. no. 9169) were purchased from
Sigma-Aldrich (Merck KGaA). Fluorescein-conjugated Phaseolus
vulgaris lectin (L-PHA lectin) was purchased from Vector
Laboratories. Alexa Fluor 488-conjugated anti-rabbit secondary
antibody (cat. no. 11008) was obtained from Molecular Probes
(Thermo Fisher Scientific, Inc.). The tunicamycin A1 homolog (Tun),
H-89 (a PKA inhibitor), PD 153035 (an EGFR inhibitor), forskolin (a
PKA activator), and Ly294002 (a PI3K inhibitor) were purchased from
Sigma-Aldrich (Merck KGaA). OSI906 (an IGF1R inhibitor) was
purchased from Selleck Chemicals. PD98059 (a MEK1 inhibitor) was
purchased from Cell Signaling Technology, Inc. U73122 (an inhibitor
of PLC-dependent processes) was obtained from Cayman Chemical Co.,
while STA-21 (a STAT3 inhibitor) was obtained from Santa Cruz
Biotechnology, Inc. Ruthenium red was purchased from Ted Pella
Inc.
Cell culture and treatments
CRC cell lines (Caco-2, HCT-116, and HT-29) were
obtained from the American Type Culture Collection (ATCC) and were
authenticated using their short tandem repeat (STR) profiles. Cells
were cultured at 37°C in a humidified atmosphere of 5%
CO2/air in Dulbecco's modified Eagle's medium (DMEM,
Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS) (Thermo Fisher
Scientific, Inc.), penicillin G (60 mg/l), and streptomycin (100
mg/l). For experimental purposes, the cells were seeded in culture
flasks or on plates, glass coverslips, or Transwell polycarbonate
filters with a 0.4-µm pore size (Corning, Inc.). After reaching 80%
confluence, the cells were treated with the appropriate drugs for
24 h. The drugs used for the different assays were tunicamycin at
0.25, 0.50, 0.75, and 1 µg/ml; PD153035 at 1, 10, and 20 µg/ml;
OSI906 at 2, 4, and 8 µg/ml; U73122 at 7, 14, and 21 µg/ml; PD98059
at 7, 14, and 21 µg/ml; LY294002 at 2, 4, and 6 µg/ml; STA-21 at 3,
4, 5, 6, and 7.5 µg/ml; H-89 at 9 µg/ml; and forskolin at 4
µg/ml.
Tissue samples
Well-differentiated or moderately differentiated
human colorectal specimens and mucinous adenocarcinomas were
obtained from the surgical resections of 14 Brazilian patients (5
males and 9 females, aged 64±10 years) from the Brazilian National
Cancer Institute after patient consent. These samples were
collected from February 2013 to June 2016. In all cases, control
specimens were collected from the accompanying normal mucosa, 5–10
cm away from the carcinoma. All samples were evaluated by a
board-certified pathologist. The cancer tissue and normal
epithelium samples destined for use in immunoblotting were
immediately frozen at −80°C. The study was carried out with the
approval of the National Cancer Institute Ethics Committee (nº
84/04). Clinicopathological features are listed in Table SI.
Western blotting
Cell cultures and homogenized tissue samples were
washed with phosphate-buffered saline (PBS) and then lysed in a
solution containing 1% Triton X-100, 1% NP40, a protease inhibitor
cocktail (1 tablet/50 ml buffer; Roche), and a phosphatase
inhibitor cocktail (Sigma-Aldrich; Merck KGaA, 1:100 dilution).
Total protein was quantified using a bicinchoninic acid (BCA)
protein assay kit (Bio-Rad Laboratories, Inc.). For western
blotting, the samples (30 µg of protein per lane) were subjected to
SDS-PAGE (ranging from 7.5 to 13%) and the separated proteins were
transferred to a nitrocellulose membrane. The blots were then
probed with primary (dilute solutions 1:1,000) and
peroxidase-conjugated secondary (dilute solutions 1:40,000)
antibodies or biotinylated lectins (Vector Laboratories). The
proteins were visualized using an ECL chemiluminescence kit (GE
Healthcare). Immunoreactive bands from lectin blots were then
visualized using the Vector stain ABC kit (Vector Laboratories).
The protein or carbohydrate levels were quantified by densitometry
using LabWorks 4.6 software (Bio-Rad Laboratories, Inc.). The
measurements were obtained from sub-exposed photographic films
after the chemiluminescence reaction, and the values were
normalized to the amount of a housekeeping protein (GAPDH or
tubulin). Differences in protein levels were evaluated using ANOVA
followed by the Dunnett post hoc test.
Lectin labeling by flow cytometry
Upon reaching 70% confluence, the cells were washed,
collected from the plates, and fixed with paraformaldehyde (4%) for
8 min at room temperature (RT). The cells were then washed, blocked
with bovine serum albumin (4%) (Sigma-Aldrich; Merck KGaA) for 30
min, and centrifuged at 1,500 × g for 3 min. Fluorescein-conjugated
L-PHA lectin was then added at a concentration of 5 or 2.5 µg/ml.
After incubation for 20 min at RT, the cells were collected by
centrifugation and washed three times with PBS. Finally,
1×104 cells were analyzed by flow cytometry (FASCalibur;
BD Biosciences). Unstained cells were used as negative controls for
lectin recognition. Fluorescence histograms and median fluorescence
data were created and analyzed with CellQuest Pro software (version
5.1.1; BD Biosciences).
Immunofluorescence and confocal
microscopy
Cells were grown on glass coverslips until they
reached 70% confluence. The cell monolayers were washed with PBS,
fixed with methanol for 20 min at −20°C, and then rehydrated with
PBS/CM (PBS containing 100 mM CaCl2 and 100 mM
MgCl2, pH 8.0) and blocked with 0.2% bovine serum
albumin (BSA) for 60 min. The cells were then incubated overnight
with anti-claudin-3 (1:40 dilution), washed with PBS, and incubated
for 1 h with Alexa Fluor 488-conjugated rabbit secondary antibody
(1:500 dilution). Finally, all coverslips were incubated with DAPI
and then washed and assembled using n-propyl gallate
(Sigma-Aldrich; Merck KGaA). Images were acquired using an FV10i-O
laser confocal microscope (Olympus) with constant laser intensity.
The fluorescence intensity in both the cytoplasm and cell membrane
was quantified using ICY Bioimage Analysis software (version
2.0.3.0; Institut Pasteur-France Bioimaging). For this purpose, an
intensity-based line was used to measure the fluorescence in an
area that included the junction between two cells.
Transmission electron microscopy
Cells were cultured on Transwell polycarbonate
filters (Corning, Inc.) and fixed for 60 min on the apical side of
the monolayer with a solution containing 2.5% glutaraldehyde, 1%
freshly prepared paraformaldehyde, 8% sucrose, 2 mM
CaCl2, and 6 mg/ml ruthenium red in 0.1 M cacodylate
buffer, pH 7.4. After washing with cacodylate buffer containing
ruthenium red for 10 min, the cells were postfixed with 1%
OsO4 and 6 mg/ml ruthenium red in cacodylate buffer for
45 min. The cell monolayers were then washed with cacodylate
buffer, dehydrated with an acetone series, and embedded in Epon
resin. Ultrathin sections (70 nm) were stained with lead citrate
and observed with a Zeiss CEM-900 transmission electron microscope
(Carl Zeiss).
CMS subtyping and gene expression
level analysis
The CRC molecular subtype classifier (4) was downloaded and applied to 644
primary human colorectal cancer samples using the RNA-Seq data from
The Cancer Genome Atlas (TCGA) project database.
Clinical-pathological and RNA-Seq data were obtained using the R
package TCGABiolinks (22). The
expression levels of CLDN3 and related glycogenes (MGAT5,
ST6GAL1, and B3GNT8) were then analyzed in the tumor
samples previously classified according to their specific CMS, as
well as in 51 normal samples, also from the TCGA database.
Differences in gene expression were evaluated by Kruskal-Wallis
test followed by pairwise comparisons using Dunn's test to compare
rank means between each subgroup. All analyses and plots were
performed in the R environment.
Clinical outcome analysis
The influence of the gene expression levels in
clinical outcomes was determined by classifying the tumor samples
into high or low groups according to the gene expression pattern.
The data were divided into three sections (tertiles), and the upper
and lower thirds were considered the high and low groups,
respectively. The overall survival over six years was then analyzed
within these low and high expression categories for all samples or
by molecular subtype. Survival analyses were carried out using the
‘survival’ package for R.
Statistical analysis
All statistical analyses from in vitro assays
were performed using the GraphPad Prism 5 software (GraphPad
Software). Differences were considered statistically significant at
a P-value <0.05.
Results
Identification of colorectal cancer
intra-CMS subgroups by expression analysis of CLDN3
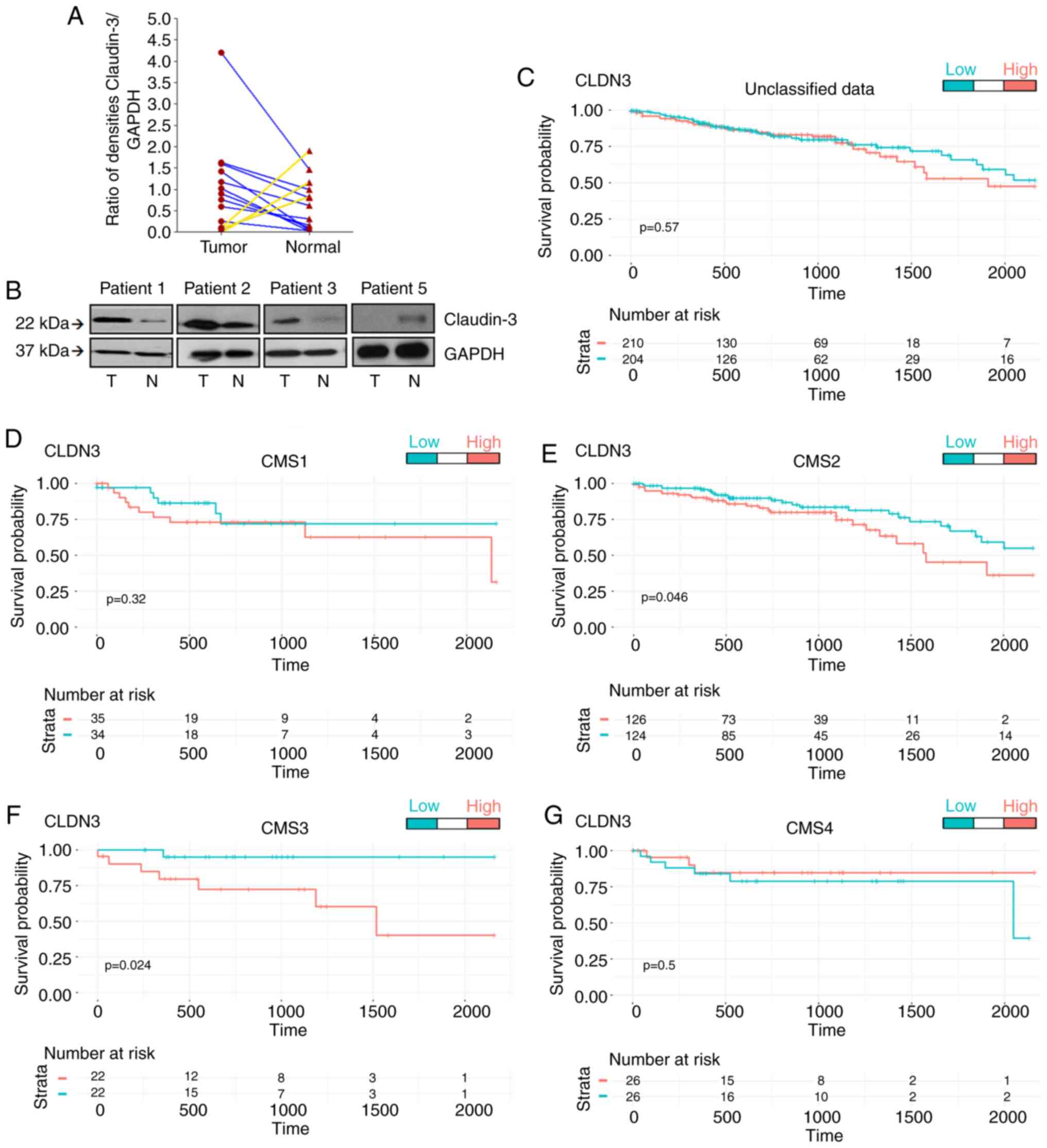
We first analyzed claudin-3 levels in CRC samples
and adjacent normal tissue and were able to identify two distinct
subgroups of tumors: Those with high levels and those with low
levels of claudin-3 protein (Fig. 1A
and B). We then performed an in silico analysis using
644 CRC samples from the TCGA database to assess the expression
levels of CLDN3. The possibility that the expression levels
of CLDN3 could affect the overall survival of CRC patients
was tested by classifying the expression values as ‘high’ or ‘low’
using the following strategy: Data were divided into tertiles, and
the values were defined as ‘high’ or ‘low’ only when they were in
the tertiles with the highest or lowest values, respectively. The
overall survival was calculated according to the expression levels
of CLDN3 in the unclassified data (Fig. 1C), as well as in data classified
into the four different molecular subtypes of CRC (Fig. 1D-G). This strategy revealed that
high expression levels of CLDN3 in CMS2 and CMS3 worsened
the patients' long-term survival (Fig.
1E and F). Taken together, these data demonstrated that
analysis of CLDN3 expression was useful for clearly
separating the CMS2/CMS3 populations into two groups with distinct
clinical outcomes. The data also showed that verification of the
expression profile of specific genes within the CRC molecular
subtypes represented an appealing strategy for identifying
intra-CMS subgroups.
Inhibition of N-glycan biosynthesis
decreases the protein levels of claudin-3 and induces its
redistribution in CRC cells
We first evaluated claudin-3 levels in different CRC
cell lines and found that HCT-116 cells had the highest level of
this protein among the cells analyzed (Fig. 2A). Interestingly, these
undifferentiated CRC cells had previously demonstrated a weaker
cell-cell adhesion phenotype than differentiated cells when the
complex N-glycan levels in HCT-116 was increased (23). We therefore chose HCT-116 cells for
subsequent investigations of the role of N-glycans in TJ
stability.
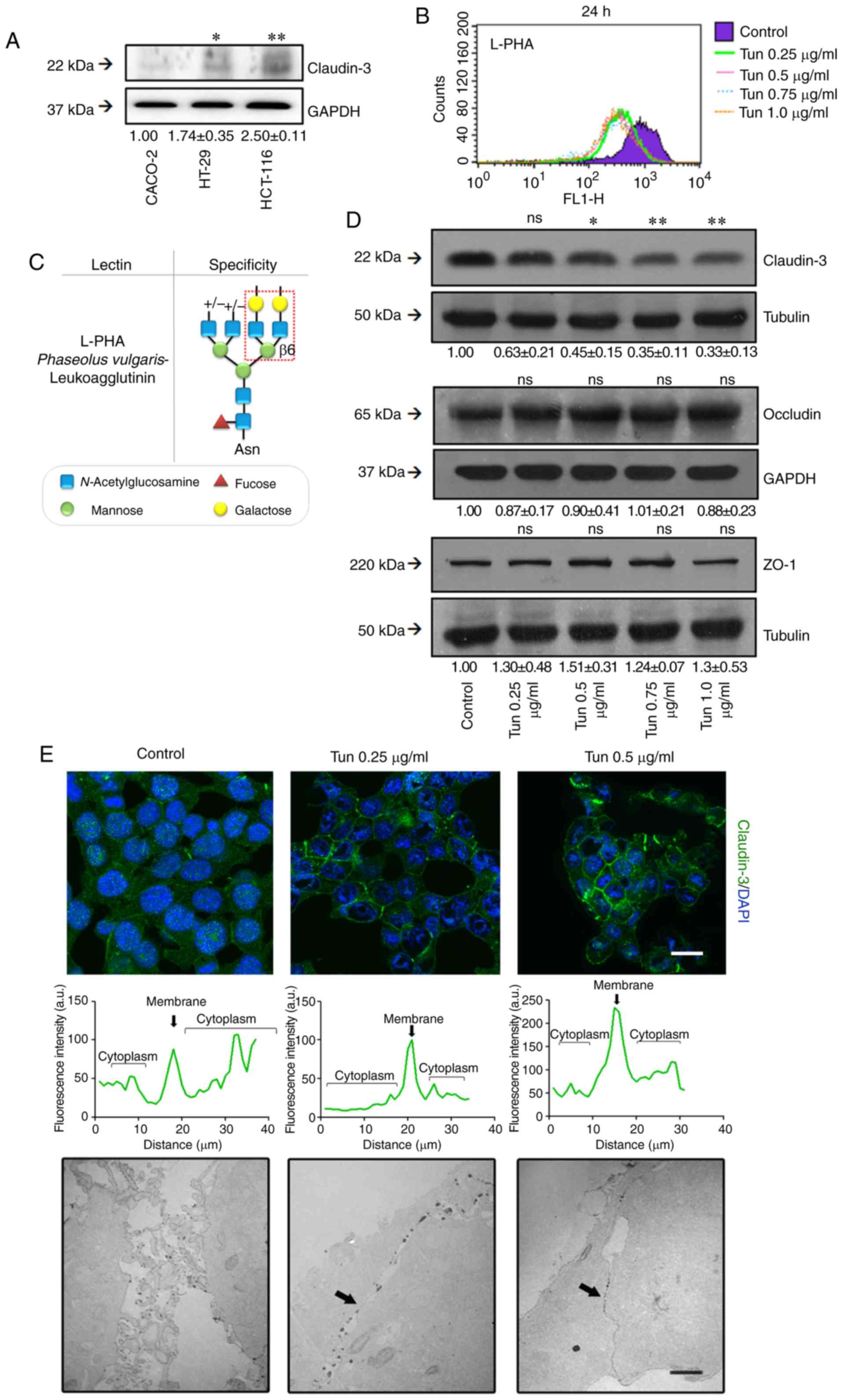 | Figure 2.Effects of N-glycan
biosynthesis inhibition on TJ stability. (A) Cell lysates from
Caco-2, HT-29, and HCT-116 cells were analyzed by western blotting
for claudin-3. (B) HCT-116 cells were treated with different
concentrations of tunicamycin for 24 h. After treatment, the cells
were incubated with FITC-conjugated lectin L-PHA and analyzed by
flow cytometry. The histograms of fluorescence were generated by
the Cell Quest software: control (purple); 0.25 µg/ml (green); 0.5
µg/ml (pink); 0.75 µg/ml (blue); and 1 µg/ml (orange). (C) Lectin
L-PHA specificity. (D) Cell lysates were obtained after 24 h
treatment with tunicamycin and analyzed by western blotting for
claudin-3, occludin, and ZO-1. Tubulin was used as an endogenous
protein control. (E, upper panel) Cell monolayers were fixed and
stained for claudin-3 (green) and nuclei (blue) (DAPI).
Representative images were obtained by confocal microscopy. The
graphs represent the fluorescence intensity in cytosolic and
membrane regions of neighboring cells. Scale bar, 10 µm. (E, lower
panel) Cells were cultured in filters of Transwell polycarbonate,
and the functionality of TJs was analyzed by transmission electron
microscopy (MET) using ruthenium red as a tracer. The images are
representative of ultrathin sections of treated and control cells.
Black arrows indicate the cell-cell contact. Scale bar, 2 µm. The
numerical values represent densitometric units ± standard error
(n=3). ns, not significant (P>0.05); *P<0.05; **P<0.01;
ANOVA. Tun, tunicamycin; TJ, tight junction; L-PHA,
phytohemagglutinin-L or Phaseolus vulgaris
leucoagglutinin. |
We determined that inhibition of
N-glycosylation with low doses of tunicamycin (Tun) for 24 h
resulted in the expected decrease in the levels of complex
N-glycans on the surfaces of the treated cells (Fig. 2B), as verified by labeling with
L-PHA lectin (Fig. 2C). We then
used western blotting to determine the effect of tunicamycin
treatment on TJ component levels [i.e., claudin-3, zonula
occludens-1 (ZO-1), and occludin levels]. We observed that
treatment with tunicamycin decreased the claudin-3 levels but did
not affect the levels of other evaluated protein constituents
(Fig. 2D). Treatment with
tunicamycin also promoted a reorganization of the subcellular
localization of claudin-3, characterized by an increase of its
levels on the cell membrane (Fig.
2E, upper panel).
We also assessed permeability to ruthenium red to
investigate whether changes in claudin-3 localization, induced by
the inhibition of N-glycosylation, could affect the
functionality of the TJs. Transmission electron microscopy
observations revealed that tunicamycin treatment did not have a
significant effect on the permeability of ruthenium red, which
permeated the paracellular region of the monolayers. However,
tunicamycin treatment did appear to promote tight cell-cell
contacts (Fig. 2E, lower panel).
Taken together, these data suggest that N-glycans are
important for regulating both the claudin-3 levels and the
stability of the TJs.
Inhibition of N-glycan biosynthesis
affects RTK phosphorylation in CRC cells
Previous studies have demonstrated that claudin
levels are regulated by signaling pathways activated through
transmembrane glycoproteins, such as RTKs (12,24).
Our evaluation of the effects of tunicamycin on the phosphorylation
levels of two RTKs (EGFR and IGF1R) revealed that inhibition of
N-glycan biosynthesis decreased the phosphorylation levels
of both receptors (Fig. 3A),
indicating that modulation of RTK function by N-glycans may
impact both receptor functionality and related signaling
pathways.
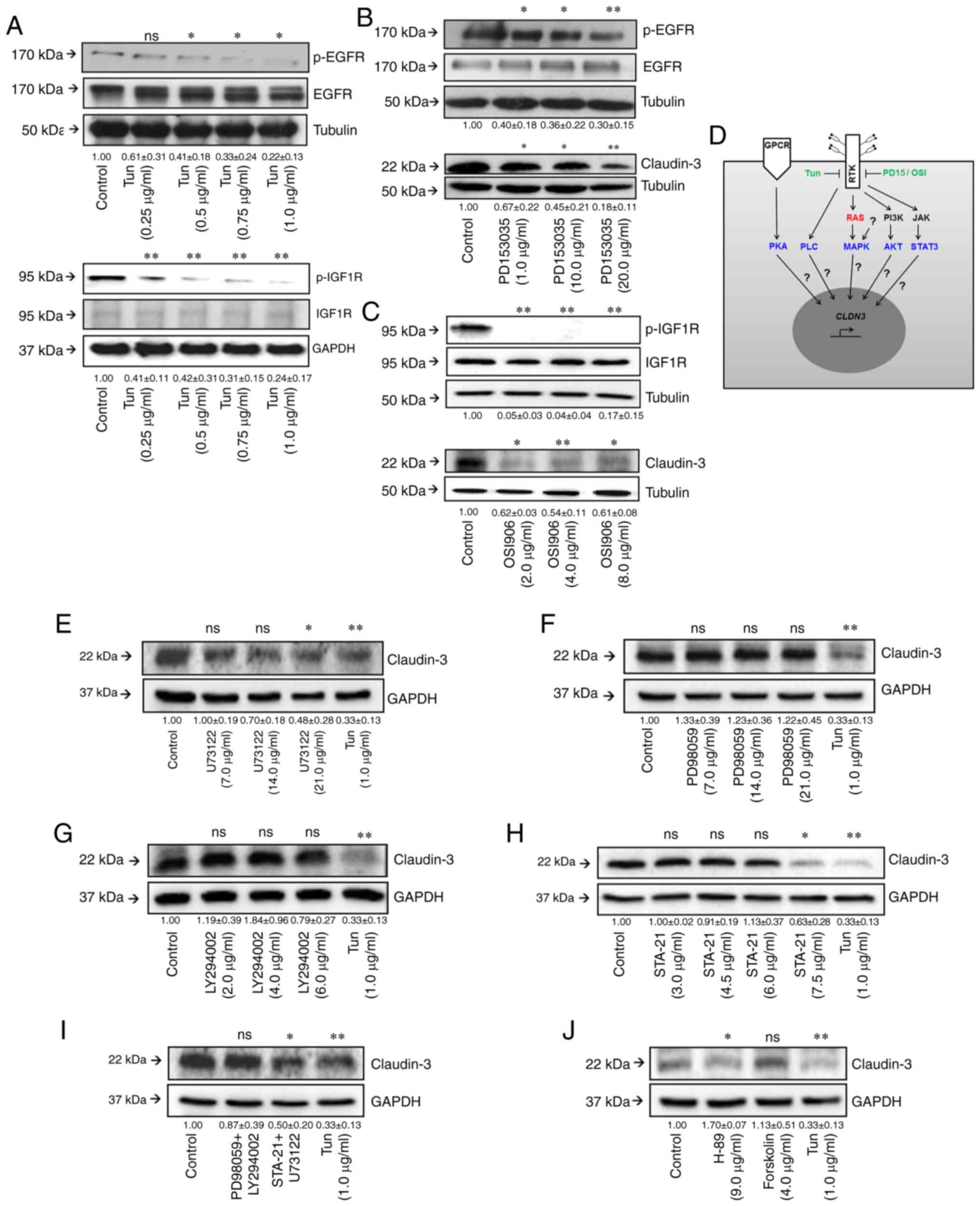 | Figure 3.Effects of inhibition of
N-glycan biosynthesis on RTK functionality. (A) After
treatment with tunicamycin, cell lysates were obtained and analyzed
by western blotting for p-EGFR, EGFR, p-IGF1R, and IGF1R. (B and C)
Cells were treated with different concentrations of PD153035 or
OSI906 for 24 h, and then levels of RTK phosphorylation and
claudin-3 were assessed by western blotting. (D) Illustration
showing RTK-related or non-RTK-related pathways, as well as on the
relationship of this regulatory network with the regulation of
CLDN3 expression. (E-H) Effects of specific inhibitors on
claudin-3 levels. Cells were treated with different concentrations
of (E) U73122 (an inhibitor of PLC-dependent processes), (F)
PD98059 (a MEK1 inhibitor), (G) Ly294002 (a PI3K inhibitor), and
(H) STA-21 (a STAT3 inhibitor) for 24 h, and then levels of
claudin-3 were assessed by western blotting. (I) Cells were treated
with paired combinations of inhibitors: PD98059 (21 µg/ml) and
Ly294002 (6 µg/ml), or STA-21 (7.5 µg/ml) and U73122 (21 µg/ml).
(J) Cells were treated with Forskolin or H-89 (a PKA inhibitor) for
24 h, and then levels of claudin-3 were assessed by western
blotting. The numerical values represent densitometric units ±
standard error (n=3). ns, not significant (P>0.05); *P<0.05;
**P<0.01; ANOVA. Tun, tunicamycin; RTKs, receptor tyrosine
kinases; EGFR, epidermal growth factor receptor; IGF1R,
insulin-like growth factor 1 receptor; p-, phosphorylated. |
Inhibition of RTK signaling affects
the levels of claudin-3
Inhibition of N-glycan biosynthesis has broad
effects on physiology irrespective of effects on RTKs; therefore,
we evaluated the effects of RTK-specific inactivation on claudin-3
levels. HCT-116 cells were treated for 24 h with different
concentrations of PD153035 (an EGFR inhibitor) or OSI906 (an IGF1R
inhibitor). These treatments decreased EGFR and IGF1R
phosphorylation levels and reduced the claudin-3 levels (Fig. 3B and C), indicating that the
specific inactivation of EGFR or IGF1R signaling decreased
claudin-3 levels. The RTK-related signaling mechanism involved was
investigated by using specific inhibitors of PLC (U73122), MAPK
(PD98059), AKT (LY294002), and STAT3 (STA-21). PLC and STAT3
inhibition, but not MAPK and AKT inhibition, significantly reduced
the claudin-3 levels (Fig. 3E-H).
Concomitant treatment with U73122 and STA-21 did not increase the
inhibitory effect on claudin-3 levels (Fig. 3I). Interestingly, inhibition of PKA
(which is not related to RTK signaling) by H-89 also led to a
significant reduction of claudin-3 levels (Fig. 3H). Collectively, these results
showed that a complex regulatory network influences the protein
levels of claudin-3 and may not be related exclusively to RTKs.
These data also suggest that the regulation of claudin-3 levels due
to modulation of RTK activity, including changes in its
N-glycosylation pattern, may be associated with disturbances
in the PLC and STAT3 pathways.
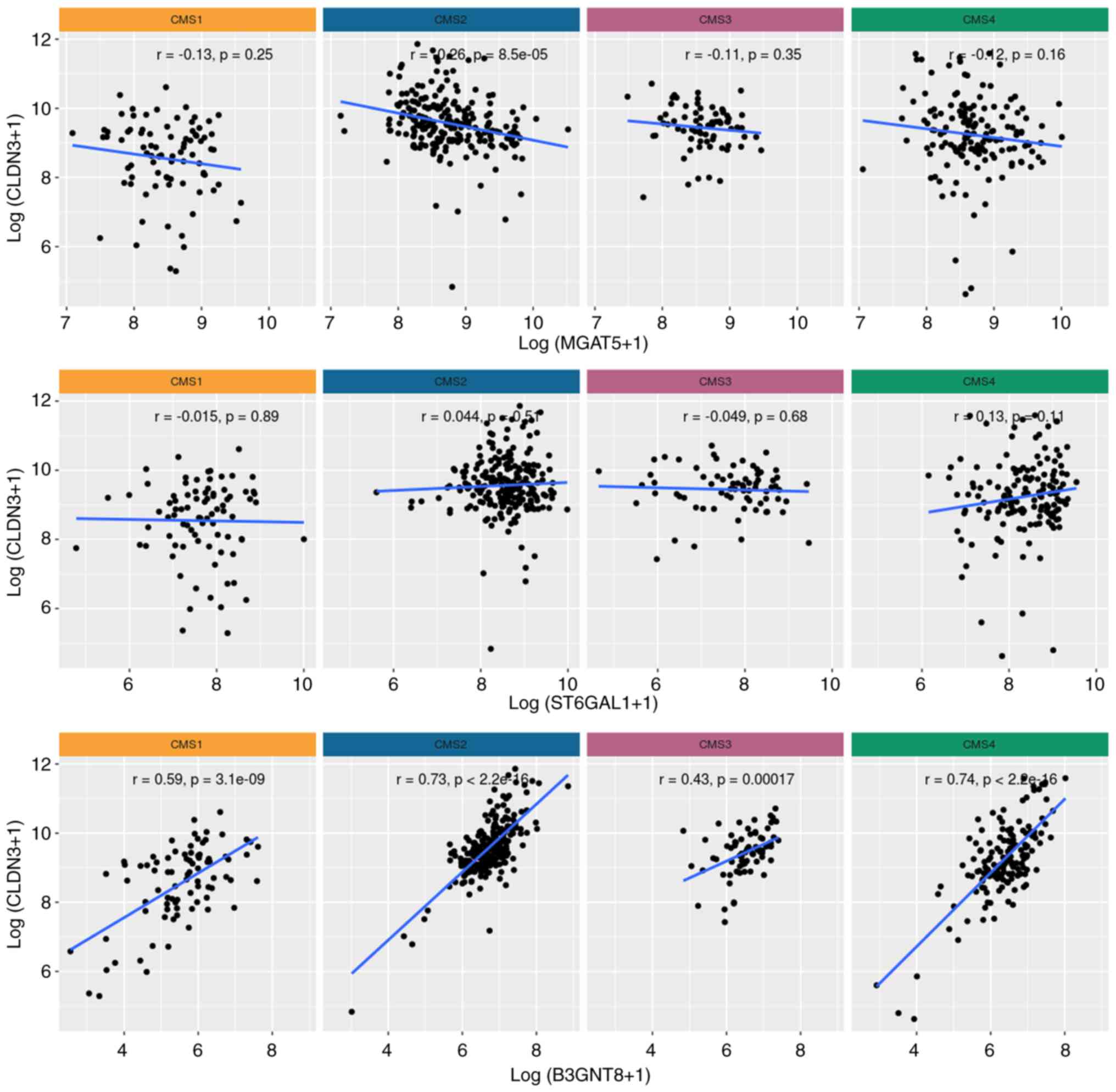
CLDN3 and B3GNT8 expression levels
correlate positively in CRC
Previous studies have shown that changes in
N-glycosylation affect the functionality of RTKs (17–19),
suggesting a possible correlation between CLDN3 expression
and transcript levels of N-glycan-related genes in CRC. We
investigated three N-glycogenes (Fig. S1). The first was MGAT5, the
gene encoding human N-acetylglucosaminyltransferase V (MGAT5
or GnT-V) that synthesizes the β1,6-GlcNAc branching
N-glycan structures widely associated with a malignant
phenotype (25). The second was
B3GNT8, the gene encoding human UDP-GlcNAc: β Gal
β-1,3-N-acetylglucosaminyltransferase 8, the enzyme involved
in the biosynthesis of poly-N-acetyllactosamine chains on
β1,6-branched N-glycan. This branching increases the
reactivity to L-PHA when the enzyme is overexpressed in CRC cells,
suggesting a potential involvement in malignancy (26). The third gene was ST6GAL1,
which encodes the human ST6 β-galactosamide α-2,6-sialyltranferase
1, a sialyltransferase that adds an α-2-6-linked sialic acid to the
N-glycan and is upregulated in CRC (27,28).
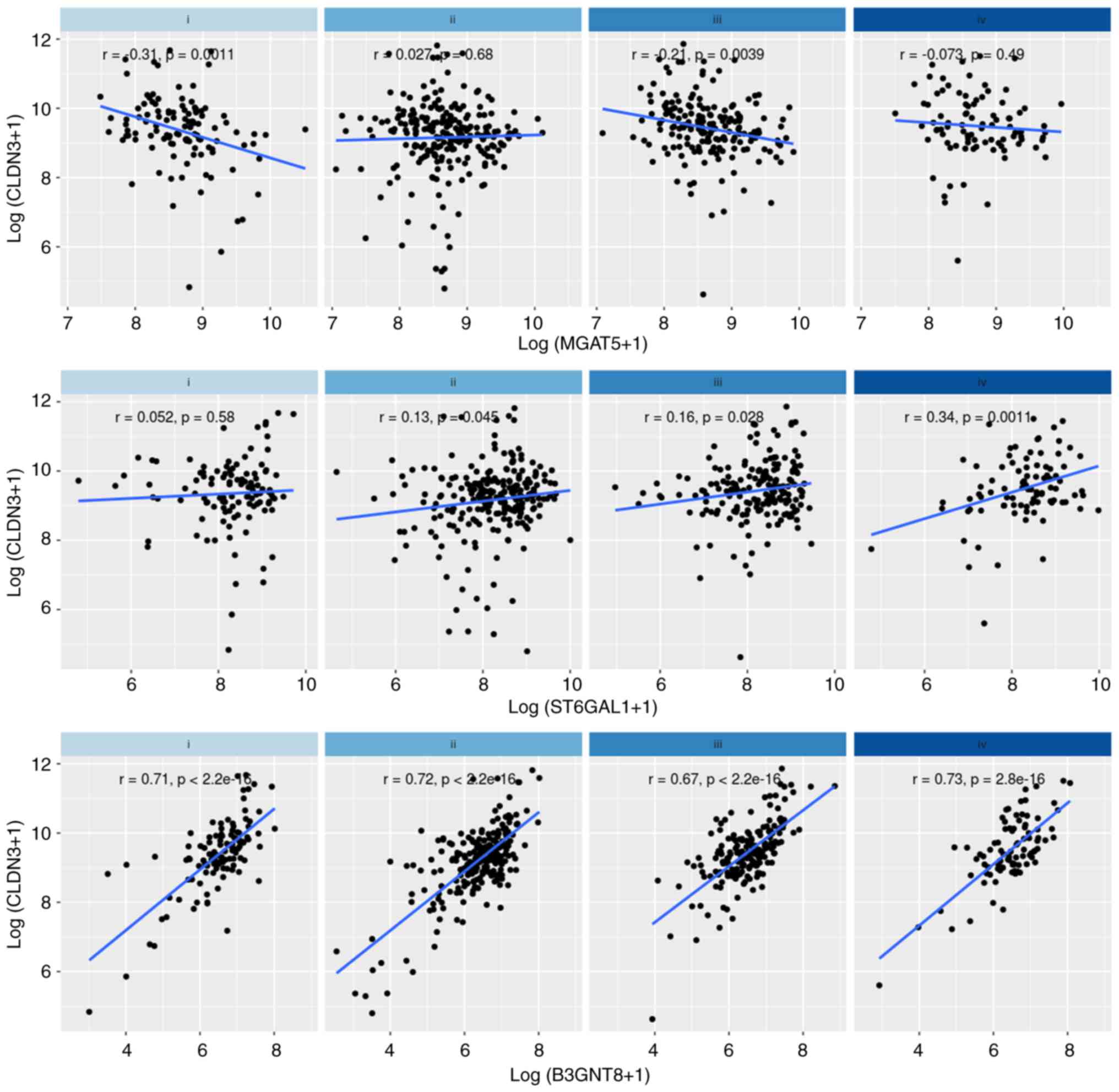
Our in silico approach revealed a robust positive
correlation between CLDN3 and B3GNT8 expression
levels in all four CMS and in all stages of CRC (Figs. 4 and 5). A weaker positive correlation was also
observed between CLDN3 and ST6GAL1 expression in
stages II, III, and IV of CRC (Fig.
5). Surprisingly, a negative correlation was observed between
CLDN3 and MGAT5 expression levels in stages I and
III, as well as in CMS2 (Figs. 4
and 5). Since N-glycans can
regulate RTK activity, these results support the possibility of a
regulatory mechanism that interconnects RTKs, CLDN3, and
N-glycan-related glycogenes.
CLDN3 and N-glycan-related glycogenes
show similar expression patterns within molecular subtypes of
colorectal cancer
We also investigated the expression profile of
CLDN3 and N-glycogenes in different CRC stages and
CMS. The in silico analysis comparing the molecular subtypes
to each other showed that CMS1 is a subtype that exhibits low
expression levels of CLDN3, MGAT5, ST6GAL1, and
B3GNT8 when compared to the other subtypes, while CMS2 is a
subtype that exhibits high expression levels of CLDN3,
ST6GAL1, and B3GNT8 when compared to the other subtypes
(Fig. 6, upper panel). In addition,
all analyzed genes had higher levels in CMS2 than in CMS1.
Upregulation of ST6GAL1 is frequently observed in CRC
samples (29,30), in agreement with significantly
higher expression levels of ST6GAL1 in CMS2, the most
frequent subtype (Fig. 6, upper
panel). Our analysis of the expression of the same genes of
interest (CLDN3, and N-glycan-related genes) in the
different stages of CRC (Fig. 6,
lower panel) revealed downregulation of CLDN3 in stage II,
and also downregulation of B3GNT8 in all stages of cancer
(Fig. 6, lower panel).
Interestingly, we observed that MGAT5 was significantly
upregulated in CRC, even in the early stages of disease (I and II)
(Fig. 6, lower panel), suggesting
that MGAT5 expression could be a potential biomarker of
CRC.
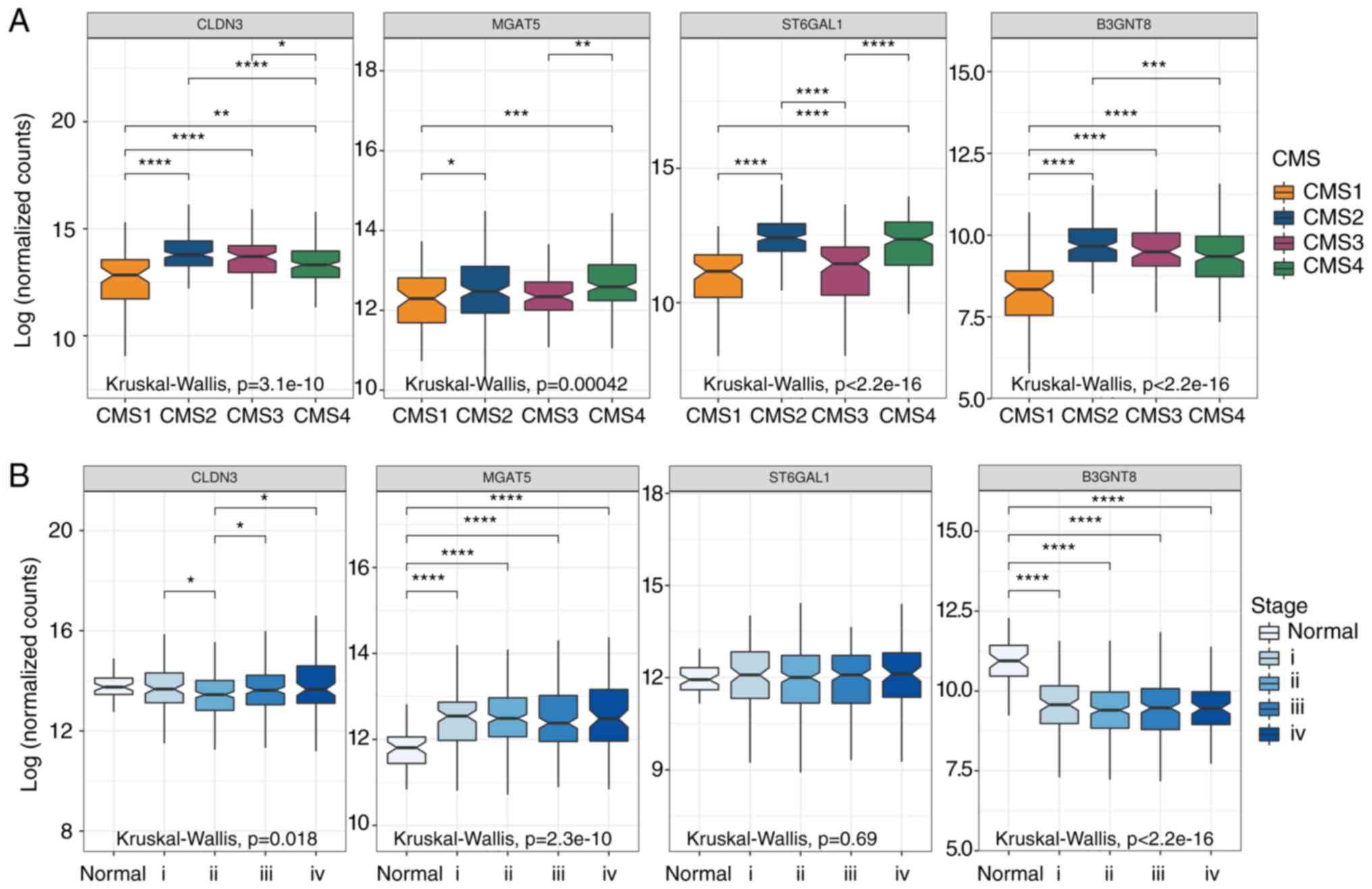 | Figure 6.Expression of CLDN3, and
N-glycan-related genes (MGAT5, ST6GAL1, and
B3GNT8) in samples from patients with colorectal cancer
(CRC). Box graphs represents absolute values of gene expression
from tumors (n=644) and normal tissue samples (n=51) accessed in
The Cancer Genome Atlas (TCGA) database. (A) Colorectal cancer
consensus molecular subtypes (CMS). CMS1 n=90; CMS2 n=242; CMS3
n=78; CMS4 n=165; and (B) colorectal cancer disease stages (i, ii,
iii, iv). Only significant differences are highlighted. The symbols
correspond to the Dunn's rank sum test P-values adjusted with
Hochberg's multiple comparisons correction *Padj <0.05; **Padj
<0.01; ***Padj <0.001; ****Padj <0.0001. CLDN3,
gene encoding claudin-3; MGAT5, α-mannoside
β-1,6-N-acetylglucosaminyltransferase; ST6GAL1, ST6
β-galactoside α-2,6-sialyltransferase; B3GNT8,
β-1,3-N-αcetylglucosaminyltransferase 8. |
Discussion
A stable apical junctional complex (AJC) has been
considered to be a suppressor of carcinoma progression due to its
role in the maintenance of apical-basolateral polarity,
intercellular adhesion, and epithelial architecture (31,32).
The dysregulation of this protein complex is correlated with a
malignant phenotype and a poor clinical outcome (12,33,34).
The regulatory role of N-glycans in the stability and
function of AJC has been demonstrated (16,35),
but few studies have been dedicated specifically to investigating
the role of N-glycosylation on TJ function. Here, we
demonstrated that the inhibition of the N-glycan
biosynthesis pathway leads to a subcellular redistribution of
claudin-3 and decreases its levels in colorectal cancer (CRC)
HCT-116 cells (undifferentiated phenotype). Changes in both protein
levels (overexpression or downregulation) and subcellular
localization of different claudins may lead to the loss of TJ
functionality (12,36,37).
We previously reported that claudin-3 overexpression in HT-29 cells
(moderately differentiated CRC cells) increases the malignancy
potential and affects the mechanisms of paracellular flux control
(12).
In the present work, we observed that inhibition of
N-glycan biosynthesis by tunicamycin led to a decrease in
claudin-3 levels in HCT-116 cells, which is a cell line that
endogenously presents high levels of this protein. However,
tunicamycin did not restore the flow of ruthenium red dye through
the TJs. We suspect that this finding may be related to the
undifferentiated phenotype of these cells, where the decrease in
claudin-3 levels was not sufficient to completely restore
TJ-mediated permeability. We did, however, observe that tunicamycin
led to the establishment of tighter cell-cell contacts, which may
contribute to a more differentiated phenotype (23) and to a decrease in invasiveness
(38). Moreover, tunicamycin also
promoted an increase in claudin-3 levels on the cellular
membrane.
The tighter cell-cell contact can be explained, at
least in part, by the protein composition of TJ strands. Changes in
claudin levels also facilitate the incorporation of other isoforms
to compose their oligomers, which may interfere in both barrier and
channel functions of the tight junctions (39). Claudin-3, for example, can interact
with claudins 1, 4, and 8 to form oligomers, besides being able to
interact with claudins 1, 2, and 5 from adjacent cells (40,41). A
previous study that analyzed the expression of several claudin
isoforms in normal and tumor tissues identified mechanisms that may
simultaneously regulate CLDN3, CLDN4, and CLDN7 and
lead to very similar expression patterns of these genes (42). Therefore, we should not disregard
the possibility of variations in the levels of other isoforms of
claudin in the context of the decreased claudin-3 that was observed
in the present study.
The influence of receptor tyrosine kinases (RTKs) on
the control of TJ stability is already known (13–15);
however, the regulatory role of N-glycans in this process
remains poorly understood. Here, we found that epidermal growth
factor receptor (EGFR) and insulin-like growth factor 1 receptor
(IGF1R) deglycosylation induced by treatment with tunicamycin led
to a decrease in the phosphorylation levels of both receptors. The
specific inhibition of EGFR or IGF1R also decreased both their
phosphorylation and the claudin-3 protein levels. These findings
show that RTK-related downstream signaling pathways regulates the
content of claudin-3 in HCT-116 cells. Although we identified
RTK-related signaling pathways (PLC and STAT3) that regulate
claudin-3 levels in CRC HCT-116 cells, the findings did not reveal
the mechanism that integrates RTKs, glycogenes, and claudin-3
levels. Our ongoing studies are addressing this issue.
The differential levels of claudins in distinct
carcinomas have been previously reported (43). A gene expression-based study
identified a molecular subtype of breast cancer characterized by
low levels of mRNA coding for claudins, referred to as the
claudin-low molecular subtype (44). Interestingly, while the low
expression of CLDN3 in this subtype was related to worse
overall survival (44), other
carcinomas, such as colorectal, breast, gastric, ovary, and
pancreas carcinomas, have shown increased levels of various
claudins (45), as well as being
related to a poor prognosis (46).
Indeed, the expression levels of cancer-related genes have been
extensively used as a parameter to determine tumor molecular
subtypes related to distinct clinical outcomes (47,48).
Regarding CRC, the recent gene profiling-based
stratification system, which has identified consensus molecular
subtypes with prognostic and predictive differences, represents a
novel classification method that can improve clinical practice
(49). In our study, we analyzed
the overall survival of CRC patients according to the expression
levels of CLDN3. Low expression levels of CLDN3 in
the CMS2 and CMS3 subtypes improved the patients' long-term
survival. Recently, similar results were reported regarding the
identification of intra-CMS subgroups using the expression levels
of claudins, thereby corroborating the use of this strategy for the
identification of molecular subtypes (50). Our findings also revealed that an
integrated analysis of functionally related genes in a particular
cellular event (e.g., TJ stability regulation) should be explored
as a useful tool to better understand the specific alterations in
each CMS.
We also found that the potential functional
relationship between CLDN3, MGAT5, ST6GAL1, and
B3GNT8 could be subtype-specific, since we observed that
these genes display higher expression levels in the CMS2 than in
the CMS1 subtype. These findings also suggest that multidimensional
analyses that considering the different stages of CRC, and
especially molecular subtypes, are crucial for the identification
of regulatory mechanisms that rely on the integrated participation
of several genes.
The differential gene expression seen among these
CMSs can be assumed to have biological significance corresponding
to their respective protein levels; however, the existence of
discrepancies cannot be disregarded between mRNA levels and protein
expressions attributable to other levels of regulation (51). Nevertheless, this correlation
appears to be quite reliable within distinct biological groups
(52).
One important issue in the classification of cancers
into molecular subtypes concerns the limitations imposed by tumor
heterogeneity. Intratumoral heterogeneity has challenged the actual
classification of CRC because the region of the tumor where the
sample is taken for molecular profiling analysis could obscure the
tumor classification (53).
Nevertheless, other authors have argued that tumor-intrinsic
subtyping captures the vast majority of biological diversity
(54). Pioneering translational
research in colorectal cancer has shown that a specific molecular
subtype of CRC, called CCS3, is resistant to anti-EGFR therapy in a
clinical setting, independent of RAS mutation status, the classical
determinant for therapy response (55).
Encouraging data have recently clarified this
issue, as no differences were observed in the survival of KRAS/BRAF
wild-type patients treated with cetuximab whose tumors had been
classified as mesenchymal-like (CMS4) (56). This confirms the importance of
discovering the molecular identities and new targets if
improvements in the efficacy of therapies against CRC are to be
achieved. Here, we identified an intra-CMS2 and intra-CMS3
subgroups that show significant differences in terms of the
patients' long-term survival based on the expression pattern of
CLDN3. We also demonstrated that the inhibition of
N-glycan biosynthesis compromises RTK activation, thereby
corroborating previous data suggesting N-glycosylation as a
promising target in cancer therapy (57–59).
Aberrant N-glycosylation in cancer cells has
been reported previously (60) and
is regulated by changes in enzyme levels that make up the
glycosylation machinery (48,61),
among other factors. Alterations in glycan structures, as well as
in the enzymes responsible for them, are accepted as possible
biomarkers in cancer (62–64). For example, MGAT5 and
β1,6-branched N-glycans are useful markers for predicting
the aggressive phenotype in CRC tumors (65,66).
Here, we observed that MGAT5 was upregulated in CRC samples,
even in those from patients with early stages of disease. This
result suggests that the overexpression of the MGAT5 gene
could be a potential CRC biomarker, once the
N-glycan-related gene expression profile has also been used
to identify CRC molecular biomarkers (48).
In conclusion, the data we have presented here
suggest a modulatory role of N-glycosylation in RTK
functionality and in the regulation of claudin-3 protein levels. We
also demonstrated that the expression analysis of CLDN3 and
N-glycan-related genes could be clinically useful for
determining relevant CRC subtypes, as well as for identifying
potential glycobiomarkers. Our findings provide insights into how
the dysregulation of claudin-3 occurs in CRC.
Supplementary Material
Supporting Data
Acknowledgements
We are grateful to all members of the laboratory,
particularly to Annie Cristhine Moraes Sousa-Squiavinato, and Bruna
dos Santos Mendonça, for assistance with relevant suggestions.
Funding
This work was funded by Ministério da Saúde,
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
(CAPES), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro
(FAPERJ), and Conselho Nacional de Desenvolvimento Científico e
Tecnológico (CNPq – grant no. 404052/2016-9).
Availability of data and materials
The data that support the findings of this study
are available from the corresponding author upon reasonable
request.
Authors' contributions
This study was designed and supervised by JCMDF Jr.
AGP performed experiments and drafted the manuscript. JADC, WFDS,
MDSF and CAFN performed experiments. MB, CADL and PTSS contributed
in collecting and analyzing data. IMDO and PVF collected patient
samples and performed experiments. JAMD co-supervised the
experiments. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the National Cancer Institute (INCA) (Rio de Janiero, Brazil).
Written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that no competing
interest exists.
Glossary
Abbreviations
Abbreviations:
|
AJC
|
apical junctional complex
|
|
B3GNT8
|
β-1,3-N-αcetylglucosaminyltransferase 8
|
|
BRAF
|
V-Raf murine sarcoma viral oncogene
homolog B
|
|
CLDN3
|
gene encoding claudin-3
|
|
CMS
|
consensus molecular subtypes
|
|
CRC
|
colorectal cancer
|
|
DPAGT1
|
dolichyl-phosphate
N-acetylglucosaminephosphotransferase 1
|
|
EGFR
|
epidermal growth factor receptor
|
|
IGF1R
|
insulin-like growth factor 1
receptor
|
|
KRAS
|
Kirsten rat sarcoma viral oncogene
homolog
|
|
L-PHA
|
phytohemagglutinin-L or Phaseolus
vulgaris leucoagglutinin
|
|
MGAT5
|
α-mannoside
β-1,6-N-acetylglucosaminyltransferase
|
|
PLC
|
phospholipase C
|
|
PP2A
|
protein phosphatase 2
|
|
RTKs
|
receptor tyrosine kinases
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
ST6GAL1
|
ST6 β-galactoside
α-2,6-sialyltransferase
|
|
TJ
|
tight junction
|
References
|
1
|
IJspeert JE, Vermeulen L, Meijer GA and
Dekker E: Serrated neoplasia-role in colorectal carcinogenesis and
clinical implications. Nat Rev Gastroenterol Hepatol. 12:401–409.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fessler E, Drost J, van Hooff SR,
Linnekamp JF, Wang X, Jansen M, De Sousa E, Melo F, Prasetyanti PR,
IJspeert JE, et al: TGFβ signaling directs serrated adenomas to the
mesenchymal colorectal cancer subtype. EMBO Mol Med. 8:745–760.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bramsen JB, Rasmussen MH, Ongen H,
Mattesen TB, Ørntoft MW, Árnadóttir SS, Sandoval J, Laguna T, Vang
S, Øster B, et al: Molecular-subtype-specific biomarkers improve
prediction of prognosis in colorectal cancer. Cell Rep.
19:1268–1280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gehren AS, Rocha MR, de Souza WF and
Morgado-Díaz JA: Alterations of the apical junctional complex and
actin cytoskeleton and their role in colorectal cancer progression.
Tissue Barriers. 3:e10176882015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lingaraju A, Long TM, Wang Y, Austin JR II
and Turner JR: Conceptual barriers to understanding physical
barriers. Semin Cell Dev Biol. 42:13–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zihni C, Mills C, Matter K and Balda MS:
Tight junctions: From simple barriers to multifunctional molecular
gates. Nat Rev Mol Cell Biol. 17:564–580. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Oliveira SS, de Oliveira IM, De Souza W
and Morgado-Díaz JA: Claudins upregulation in human colorectal
cancer. FEBS Lett. 579:6179–6185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Krug SM, Schulzke JD and Fromm M: Tight
junction, selective permeability, and related diseases. Semin Cell
Dev Biol. 36:166–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Sun T, Sun H, Yang S, Li D and
Zhou D: SCF/C-Kit/JNK/AP-1 signaling pathway promotes claudin-3
expression in colonic epithelium and colorectal carcinoma. Int J
Mol Sci. 18:7652017. View Article : Google Scholar
|
|
12
|
de Souza WF, Fortunato-Miranda N, Robbs
BK, de Araujo WM, de-Freitas-Junior JC, Bastos LG, Viola JP and
Morgado-Díaz JA: Claudin-3 overexpression increases the malignant
potential of colorectal cancer cells: Roles of ERK1/2 and PI3K-Akt
as modulators of EGFR signaling. PLoS One. 8:e749942013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh AB and Harris RC: Epidermal growth
factor receptor activation differentially regulates claudin
expression and enhances transepithelial resistance in madin-darby
canine kidney cells. J Biol Chem. 279:3543–3552. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ikari A, Sato T, Watanabe R, Yamazaki Y
and Sugatani J: Increase in claudin-2 expression by an
EGFR/MEK/ERK/c-fos pathway in lung adenocarcinoma A549 cells.
Biochim Biophys Acta. 1823:1110–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Campbell HK, Maiers JL and DeMali KA:
Interplay between tight junctions & adherens junctions. Exp
Cell Res. 358:39–44. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nita-Lazar M, Rebustini I, Walker J and
Kukuruzinska MA: Hypoglycosylated E-cadherin promotes the assembly
of tight junctions through the recruitment of PP2A to adherens
junctions. Exp Cell Res. 316:1871–1884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fernandes H, Cohen S and Bishayee S:
Glycosylation-induced conformational modification positively
regulates receptor- receptor association: A study with an aberrant
epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in
cancer cells. J Biol Chem. 276:5375–5383. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Whitson KB, Whitson SR, Red-Brewer ML,
McCoy AJ, Vitali AA, Walker F, Johns TG, Beth AH and Staros JV:
Functional effects of glycosylation at asn-579 of the epidermal
growth factor receptor. Biochemistry. 44:14920–14931. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaszuba K, Grzybek M, Orłowski A, Danne R,
Róg T, Simons K, Coskun Ü and Vattulainen I: N-Glycosylation as
determinant of epidermal growth factor receptor conformation in
membranes. Proc Natl Acad Sci USA. 112:4334–4339. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lopez Sambrooks C, Baro M, Quijano A,
Narayan A, Cui W, Greninger P, Egan R, Patel A, Benes CH, Saltzman
WM and Contessa JN: Oligosaccharyltransferase inhibition overcomes
therapeutic resistance to EGFR tyrosine kinase inhibitors. Cancer
Res. 78:5094–5106. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lau KS, Partridge EA, Grigorian A,
Silvescu CI, Reinhold VN, Demetriou M and Dennis JW: Complex
N-glycan number and degree of branching cooperate to regulate cell
proliferation and differentiation. Cell. 129:123–134. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colaprico A, Silva TC, Olsen C, Garofano
L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM,
Castiglioni I, et al: TCGAbiolinks: An R/bioconductor package for
integrative analysis of TCGA data. Nucleic Acids Res. 44:e712016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Freitas Junior JC, Silva BR, de Souza
WF, de Araújo WM, Abdelhay ES and Morgado-Díaz JA: Inhibition of
N-linked glycosylation by tunicamycin induces E-cadherin-mediated
cell-cell adhesion and inhibits cell proliferation in
undifferentiated human colon cancer cells. Cancer Chemother
Pharmacol. 68:227–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peter Y, Comellas A, Levantini E, Ingenito
EP and Shapiro SD: Epidermal growth factor receptor and claudin-2
participate in A549 permeability and remodeling: Implications for
non-small cell lung cancer tumor colonization. Mol Carcinog.
48:488–497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taniguchi N and Korekane H: Branched
N-glycans and their implications for cell adhesion, signaling and
clinical applications for cancer biomarkers and in therapeutics.
BMB Rep. 44:772–781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishida H, Togayachi A, Sakai T, Iwai T,
Hiruma T, Sato T, Okubo R, Inaba N, Kudo T, Gotoh M, et al: A novel
beta1,3-N-acetylglucosaminyltransferase (beta3Gn-T8), which
synthesizes poly-N-acetyllactosamine, is dramatically upregulated
in colon cancer. FEBS Lett. 579:71–78. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dall'Olio F, Malagolini N, di Stefano G,
Minni F, Marrano D and Serafini-Cessi F: Increased CMP-NeuAc:Gal
beta 1,4GlcNAc-R alpha 2,6 sialyltransferase activity in human
colorectal cancer tissues. Int J Cancer. 44:434–439. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Petretti T, Kemmner W, Schulze B and
Schlag PM: Altered mRNA expression of glycosyltransferases in human
colorectal carcinomas and liver metastases. Gut. 46:359–366. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dall'Olio F, Chiricolo M, Ceccarelli C,
Minni F, Marrano D and Santini D: Beta-galactoside alpha2,6
sialyltransferase in human colon cancer: Contribution of multiple
transcripts to regulation of enzyme activity and reactivity with
sambucus nigra agglutinin. Int J Cancer. 88:58–65. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dall'Olio F, Chiricolo M, Mariani E and
Facchini A: Biosynthesis of the cancer-related sialyl-alpha
2,6-lactosaminyl epitope in colon cancer cell lines expressing
beta-galactoside alpha 2,6-sialyltransferase under a constitutive
promoter. Eur J Biochem. 268:5876–5884. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Royer C and Lu X: Epithelial cell
polarity: A major gatekeeper against cancer? Cell Death Differ.
18:1470–1477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Halaoui R and McCaffrey L: Rewiring cell
polarity signaling in cancer. Oncogene. 34:939–950. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bouchagier KA, Assimakopoulos SF, Karavias
DD, Maroulis I, Tzelepi V, Kalofonos H, Karavias DD, Kardamakis D,
Scopa CD and Tsamandas AC: Expression of claudins-1, −4, −5, −7 and
occludin in hepatocellular carcinoma and their relation with
classic clinicopathological features and patients' survival. In
Vivo. 28:315–326. 2014.PubMed/NCBI
|
|
34
|
Katayama A, Handa T, Komatsu K, Togo M,
Horiguchi J, Nishiyama M and Oyama T: Expression patterns of
claudins in patients with triple-negative breast cancer are
associated with nodal metastasis and worse outcome. Pathol Int.
67:404–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pinho SS, Figueiredo J, Cabral J, Carvalho
S, Dourado J, Magalhães A, Gärtner F, Mendonfa AM, Isaji T, Gu J,
et al: E-cadherin and adherens-junctions stability in gastric
carcinoma: Functional implications of glycosyltransferases
involving N-glycan branching biosynthesis,
N-acetylglucosaminyltransferases III and V. Biochim Biophys Acta.
1830:2690–2700. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bücker R, Krug SM, Fromm A, Nielsen HL,
Fromm M, Nielsen H and Schulzke JD: Campylobacter fetus impairs
barrier function in HT-29/B6 cells through focal tight junction
alterations and leaks. Ann N Y Acad Sci. 1405:189–201. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zeissig S, Bürgel N, Günzel D, Richter J,
Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M and
Schulzke JD: Changes in expression and distribution of claudin 2, 5
and 8 lead to discontinuous tight junctions and barrier dysfunction
in active crohn's disease. Gut. 56:61–72. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nami B, Donmez H and Kocak N:
Tunicamycin-induced endoplasmic reticulum stress reduces in vitro
subpopulation and invasion of CD44+/CD24−
phenotype breast cancer stem cells. Exp Toxicol Pathol. 68:419–426.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Capaldo CT and Nusrat A: Claudin
switching: Physiological plasticity of the tight junction. Semin
Cell Dev Biol. 42:22–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Findley MK and Koval M: Regulation and
roles for claudin-family tight junction proteins. IUBMB Life.
61:431–437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koval M: Differential pathways of claudin
oligomerization and integration into tight junctions. Tissue
Barriers. 1:e245182013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hewitt KJ, Agarwal R and Morin PJ: The
claudin gene family: Expression in normal and neoplastic tissues.
BMC Cancer. 6:1862006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kwon MJ: Emerging roles of claudins in
human cancer. Int J Mol Sci. 14:18148–18180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dias K, Dvorkin-Gheva A, Hallett RM, Wu Y,
Hassell J, Pond GR, Levine M, Whelan T and Bane AL: Claudin-low
breast cancer; clinical & pathological characteristics. PLoS
One. 12:e01686692017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oliveira SS and Morgado-Díaz JA: Claudins:
Multifunctional players in epithelial tight junctions and their
role in cancer. Cell Mol Life Sci. 64:17–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Iacobuzio-Donahue CA, Maitra A, Shen-Ong
GL, van Heek T, Ashfaq R, Meyer R, Walter K, Berg K, Hollingsworth
MA, Cameron JL, et al: Discovery of novel tumor markers of
pancreatic cancer using global gene expression technology. Am J
Pathol. 160:1239–1249. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Levine EA, Votanopoulos KI, Qasem SA,
Philip J, Cummins KA, Chou JW, Ruiz J, D'Agostino R, Shen P and
Miller LD: Prognostic molecular subtypes of low-grade cancer of the
appendix. J Am Coll Surg. 222:493–503. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ashkani J and Naidoo KJ:
Glycosyltransferase gene expression profiles classify cancer types
and propose prognostic subtypes. Sci Rep. 6:264512016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Thanki K, Nicholls ME, Gajjar A, Senagore
AJ, Qiu S, Szabo C, Hellmich MR and Chao C: Consensus molecular
subtypes of colorectal cancer and their clinical implications. Int
Biol Biomed J. 3:105–111. 2017.PubMed/NCBI
|
|
50
|
Cherradi S, Martineau P, Gongora C and Del
Rio M: Claudin gene expression profiles and clinical value in
colorectal tumors classified according to their molecular subtype.
Cancer Manag Res. 11:1337–1348. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vogel C and Marcotte EM: Insights into the
regulation of protein abundance from proteomic and transcriptomic
analyses. Nat Rev Genet. 13:227–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koussounadis A, Langdon SP, Um IH,
Harrison DJ and Smith VA: Relationship between differentially
expressed mRNA and mRNA-protein correlations in a xenograft model
system. Sci Rep. 5:107752015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dunne PD, McArt DG, Bradley CA, O'Reilly
PG, Barrett HL, Cummins R, O'Grady T, Arthur K, Loughrey MB, Allen
WL, et al: Challenging the cancer molecular stratification dogma:
Intratumoral heterogeneity undermines consensus molecular subtypes
and potential diagnostic value in colorectal cancer. Clin Cancer
Res. 22:4095–4104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Prat A, Pineda E, Adamo B, Galván P,
Fernández A, Gaba L, Díez M, Viladot M, Arance A and Muñoz M:
Clinical implications of the intrinsic molecular subtypes of breast
cancer. Breast. 24 (Suppl 2):S26–S35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
De Sousa E, Melo F, Wang X, Jansen M,
Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van
Leersum R, et al: Poor-prognosis colon cancer is defined by a
molecularly distinct subtype and develops from serrated precursor
lesions. Nat Med. 19:614–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Trinh A, Trumpi K, De Sousa E, Melo F,
Wang X, de Jong JH, Fessler E, Kuppen PJ, Reimers MS, Swets M, et
al: Practical and robust identification of molecular subtypes in
colorectal cancer by immunohistochemistry. Clin Cancer Res.
23:387–398. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Carvalho S, Reis CA and Pinho SS:
Cadherins glycans in cancer: Sweet players in a bitter process.
Trends Cancer. 2:519–531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Contessa JN, Bhojani MS, Freeze HH, Ross
BD, Rehemtulla A and Lawrence TS: Molecular imaging of N-linked
glycosylation suggests glycan biosynthesis is a novel target for
cancer therapy. Clin Cancer Res. 16:3205–3214. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
de Freitas Junior JC and Morgado-Díaz JA:
The role of N-glycans in colorectal cancer progression: Potential
biomarkers and therapeutic applications. Oncotarget. 7:19395–19413.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Anugraham M, Jacob F, Nixdorf S,
Everest-Dass AV, Heinzelmann-Schwarz V and Packer NH: Specific
glycosylation of membrane proteins in epithelial ovarian cancer
cell lines: Glycan structures reflect gene expression and DNA
methylation status. Mol Cell Proteomics. 13:2213–2232. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kannagi R, Yin J, Miyazaki K and Izawa M:
Current relevance of incomplete synthesis and neo-synthesis for
cancer-associated alteration of carbohydrate
determinants-Hakomori's concepts revisited. Biochim Biophys Acta.
1780:525–531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Meany DL and Chan DW: Aberrant
glycosylation associated with enzymes as cancer biomarkers. Clin
Proteomics. 8:72011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu L, Yan B, Huang J, Gu Q, Wang L, Fang
M, Jiao J and Yue X: The identification and characterization of
novel N-glycan-based biomarkers in gastric cancer. PLoS One.
8:e778212013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Qin R, Zhao J, Qin W, Zhang Z, Zhao R, Han
J, Yang Y, Li L, Wang X, Ren S, et al: Discovery of non-invasive
glycan biomarkers for detection and surveillance of gastric cancer.
J Cancer. 8:1908–1916. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim YS, Ahn YH, Song KJ, Kang JG, Lee JH,
Jeon SK, Kim HC, Yoo JS and Ko JH: Overexpression and
β-1,6-N-acetylglucosaminylation-initiated aberrant glycosylation of
TIMP-1: A ‘double whammy’ strategy in colon cancer progression. J
Biol Chem. 287:32467–32478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Murata K, Miyoshi E, Kameyama M, Ishikawa
O, Kabuto T, Sasaki Y, Hiratsuka M, Ohigashi H, Ishiguro S, Ito S,
et al: Expression of N-acetylglucosaminyltransferase V in
colorectal cancer correlates with metastasis and poor prognosis.
Clin Cancer Res. 6:1772–1777. 2000.PubMed/NCBI
|