Introduction
Colorectal cancer (CRC) is the third most common
cancer and the second most common cause of death worldwide
(1). The prognosis of early-stage CRC
is relatively favorable; 70–80% are eligible for curative-intent
surgery, with a 5-year survival of 72–93% for stages I–II. In
contrast, approximately 25% of CRC patients have metastases at the
time of diagnosis; nearly 10% of those have synchronous peritoneal
metastases (2,3). In addition, 8.3% of CRC patients have
peritoneal dissemination (PD) regardless of synchronous or
metachronous metastases (4). PD is a
progressive and intractable state of CRC that is related to a poor
prognosis. Despite the recent development of chemotherapeutic
agents, the median survival of CRC patients with PD treated with
systemic chemotherapy is 10–16 months after diagnosis, which is
much shorter than the median survival of those with other organ
metastases derived from CRC (5,6).
Therefore, it is essential to elucidate the underlying mechanisms
of the generation and progression of PD to identify effective
therapeutic targets.
The tumor immune microenvironment (TIME) has drawn
attention as a key contributing factor of tumor progression
(7). The TIME consists of immune,
vascular, and mesenchymal cells, as well as structural
extracellular matrix proteins such as collagen, laminin,
fibronectin, proteoglycans; it also contains soluble components
such as metabolites, growth factors, cytokines, chemokines, and
proteases (8–10). These components surround tumor cells,
interact with each other, and work as a tumor-promoting ecosystem.
Of note, TIME components in metastatic lesions differ from those in
the primary lesions of the same cancer (8). Likewise, the TIME in PD is assumed to
differ from that of primary or metastatic lesions. When PD occurs,
tumor cells are isolated from the primary lesion, migrate in the
peritoneal cavity, adhere to peritoneal mesothelial cells, and
grow. Given the confined but spacious peritoneal cavity, ascitic
flow primarily affects cell migration and the immune context of PD
progression. From this viewpoint, ascites or peritoneal lavage is
considered a feasible but important material to analyze the unique
immune cell context of the PD-relevant TIME.
Of all TIME components, myeloid-derived suppressor
cells (MDSCs) are considered to play a responsible role in the
immunosuppressive characteristics of the TIME. MDSCs consist of
immature myeloid cells in the early stages of differentiation to
mature into macrophages, dendritic cells, and granulocytes
(11). In the TIME, MDSCs
immunologically promote tumor progression by suppressing the
activation and function of T cells (12). In this regard, we previously reported
that the accumulation of MDSCs in tumor sites, lung, and spleen
correlates with tumor progression (13) and that the frequency of MDSCs in the
peripheral blood correlates with tumor recurrence after surgical
resection in mice (14). However, few
studies have demonstrated whether and how MDSCs affect the
generation and progression of PD.
MDSCs are classified into two major subsets:
Monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs)
(15). The surface antigen phenotype
of M-MDSCs is defined as
CD11b+Ly6ChighLy6G− in mice,
whereas that of PMN-MDSCs is
CD11b+Ly6ClowLy6G+ in mice, which
is identical to that of normal neutrophils. Recently, CD244 has
been highlighted as a surface antigen to distinguish PMN-MDSCs from
normal neutrophils (11,16–18). Here,
CD244 is an immunoregulatory transmembrane receptor molecule that
belongs to the signaling lymphocyte activation molecule (SLAM)
family (18). It is known to be
expressed on natural killer (NK) cells and CD8+ T cells
(19,20). Recently, PMN-MDSCs isolated from
tumor-bearing mouse have been shown to have substantially higher
expression levels of CD244 than neutrophils isolated from naïve
mouse. Also, sorted CD244+ cells have shown higher
immune-suppressive activities than CD244− cells
(21). Thereafter, CD244 has been
used as the most reliable marker to distinguish PMN-MDSCs from
normal neutrophils (17).
These findings led us to hypothesize that
CD244+ MDSCs (PMN-MDSCs) may play a key role in the
pathogenesis of PD and be an effective therapeutic target of PD. To
this end, we established a PD mouse model and extensively analyzed
cytokines as well as immune cells in the peritoneal lavage to
evaluate the PD-relevant TIME. We also conducted PMN-MDSC
elimination in vivo to confirm the therapeutic impacts on
PD.
Materials and methods
Cell lines
The C57BL/6J mouse-derived colon cancer cell line
MC38 was originally established by Dr F. James Primus at Beckman
Research Institute, Duarte, CA, USA (22); Dr Toshiyasu Ojima at Wakayama Medical
University, Wakayama, Japan kindly provided us this cell line
(23). Luciferase-tagged line MC38
(MC38-luc) was generated as follows. MC38 cells were transfected
with a CMV-GFP-T2A-luciferase pre-packaged lentivirus vector system
BLIV101VA (System Biosciences, LLC). The cell lines stably
expressing high levels of GFP and luciferase were established by
sorting cells after the lentiviral infections. The BALB/c
mouse-derived colon cancer cell line CT26 was obtained from
American Type Culture Collection (ATCC). The MC38, MC38-luc, and
CT26 cells were maintained in RPMI-1640 medium supplemented with
10% (v/v) heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich;
Merck KGaA) at 37°C in 5% CO2. Cultured cells were
confirmed negative for mycoplasma and viral contamination.
Mice
All animal experiments in this study were conducted
with the approval of the Institutional Animal Care and Use
Committee of Kobe University (approval no. P190404) in accordance
with the ARRIVE guideline (24).
Female C57BL/6J wild-type and BALB/c wild-type mice were purchased
from CLEA Japan. OT-1 C57BL/6 transgenic mice were purchased from
Charles River. All mice were maintained in pathogen-free
conditions, fed ad libitum, and had free access to water in
quiet humidified rooms on a 12:12-h light: Dark cycle. The mice
were kept in plastic cages of 25×15×17 cm with five animals per
cage. The health and behavior of mice were monitored once a day.
Mice were acclimatized for at least five days before experiments.
The administration of aspirin was used for analgesia, and
inhalation using 4% isoflurane was used for anesthesia.
In vivo tumor models
In vivo experimental models of PD and
subcutaneous inoculation (SC) were established by inoculating the
corresponding strains of mice with the MC38, MC38-luc or CT26 cell
lines. For survival analysis, female C57BL/6J mice were
intraperitoneally or subcutaneously inoculated with
2×106 MC38 cells and female BALB/c mice were
intraperitoneally inoculated with 2×106 CT26 cells. For
sample collections of PD nodules, peritoneal lavage, spleens, and
peripheral blood, 5×105 MC38 cells or 1×105
CT26 cells were transplanted as indicated above, and the
tumor-bearing mice were sacrificed on Days 6, 13, and 20 after cell
inoculation. Peritoneal lavage was conducted with 1.5 ml
phosphate-buffered saline (PBS); spleens and peripheral blood were
simultaneously collected. For an ex vivo antigen-specific
T-cell suppression assay, spleens were harvested from OT-1 C57BL/6
transgenic mice and splenocytes were purified to use. For tumor
volume analyses following Ly6G-mediated immune cell depletion,
female C57BL/6 mice were intraperitoneally inoculated with
2×106 MC38-luc cells and Ly6G mAb (clone 1A8; BioXCell)
was intraperitoneally injected at 200 µg/mouse every 3 days from
Day 7 after the tumor inoculation. Control mice were treated with
PBS with the same regimen.
From a viewpoint of animal welfare, the humane
endpoints were set as follows: Difficulty in feeding and fluid
intake, agonizing symptoms (self-injurious behavior, abnormal
posture, breathing problems, crying), and marked abdominal
distention. In addition, for the subcutaneous inoculation (SC)
model, the following conditions were also considered: Tumor
diameter >20 mm, tumor necrosis, ulceration, or tumor infection.
Mice judged to have reached the humane endpoint were euthanized by
cervical dislocation under anesthesia. Euthanasia was confirmed by
their unresponsiveness to pain stimuli, apnea, and cardiac arrest.
In this study, we used 74 C57BL/6J mice and 46 BALB/c mice in
total. Most of the mice were euthanized for analyses; 12 C57BL/6J
mice and 7 BALB/c mice were found dead of tumor development.
Histological analyses
The procedure used in this study has been published
previously (25). Briefly, PD nodules
were harvested from the MC38-based PD model and placed in 10%
formalin overnight. The tissues were dehydrated, embedded in
paraffin, and sliced into 5-µm-thick sections. The sections were
soaked in xylene, ethanol in a gradient concentration and then
stained with hematoxylin and eosin solutions. After drying, the
sections were observed and photographed by a florescence microscope
IX71 (Olympus) at ×40 and ×200 magnification for the morphology of
tumor cells and immune cells in the PD nodules.
Gross tumor burden evaluation
To evaluate the tumor burden of the PD models, the
number of PD nodules was measured grossly and scored as follows: P0
for no nodules, P1 for 1–3 nodules, P2 for 4–9 nodules, and P3 for
10 or more nodules. To evaluate the tumor burden of the SC models,
the maximum and minimum diameters of tumor masses were measured and
the tumor volume (V) was calculated as follows: V=maximum diameter
× (minimum diameter)2/2.
In vivo lumino-imaging for tumor
growth evaluation
Tumor growth of the MC38-luc derived tumors in
vivo was quantitively evaluated by total flux of luminescence
(photons/sec) every 2–3 days. Briefly, D-luciferin substrate (OZ
Biosciences) was intraperitoneally injected into the mice at 10 min
before the measurement of luciferase flux. Bioluminescent images
were captured by an In Vivo Imaging System (IVIS; Xenogen)
and normalized by Living Image software (Xenogen) with minimum and
maximum radiances of 5×106 and 1×108
photons/sec, respectively.
Immune cell isolation
To isolate splenocytes, spleens were harvested,
ground, and filtered with cell strainers consisting of a nylon mesh
with 70-µm pores (BD Biosciences). To isolate intraperitoneal
immune cells, the peritoneal cavity was washed with 1.5 ml PBS and
the lavage fluid was filtered with cell strainers consisting of a
nylon mesh with 100-µm pores. Red blood cells were lysed with lysis
buffer (Sigma-Aldrich; Merck KGaA). PD nodules were minced into
small pieces and digested in a 1 mg/ml collagenase D (Worthington)
and 60 U/ml DNase I (Roche) solution at 37°C for 30 min. The cell
suspension was passed through cell strainers consisting of a nylon
mesh with 40-µm pores and centrifuged at 500 × g for 5 min. After
discarding the supernatant, the cells were resuspended with 5 ml of
40% Percoll (GE Healthcare), centrifuged at 500 × g for 10 min, and
lysed with lysis buffer to obtain single cell suspensions. The
cells were then stained with the antibodies described below.
Flow cytometry
The procedure used in this study has been published
previously (14,26). Briefly, the following anti-mouse
monoclonal antibodies were purchased from BD Biosciences:
Ly6G-peridinin chlorophyll protein (PerCP)-Cy5.5 (1A8; 560602),
Ly6C-fluorescein isothiocyanate (FITC) (AL-21; 553104),
CD4-allophycocyanin (APC) (RM4-5; 553051), CD25-FITC (7D4; 553071),
and CD19-APC (1D3; 550992). The following antibodies were purchased
from BioLegend: CD45-FITC (30F-11;103108), CD45-PerCP-Cy5.5
(30F-11;103132), CD45-brilliant violet (BV510) (30F-11;103138),
CD11b-APC/Fire750 (M1/70;101262), CD244-Alexa Fluor 647
(2B4;133509), F4/80-phycoerythrin (PE) (BM8;123110), PD-L1-BV421
(10F.9G2;124315), CD8a-APC/Fire 750 (53-6.7;100766), CD8a-PE
(53-6.7; 100708), CD127-BV510 (A7R34;135033), CD1d-PE (1B1;123509),
and CD5-BV421 (53-7.3;100618). The isolated immune cells were
stained in a 96-round-bottom-well plate for 20 min at 4°C and
washed with PBS containing 1% bovine serum albumin. Sorting of
Ly6G+CD244+ and
Ly6G+CD244− cells was conducted on a FACSAria
instrument (BD Biosciences). Flow cytometric data were obtained by
a FACS Verse instrument (BD Biosciences) and analyzed by FlowJo
software (TreeStar, Inc.). CD45-gated cells were analyzed in this
study.
Analysis of gene expression by
quantitative real-time PCR
The procedure used in this study has been published
previously (27–29); gene expression levels of
Ly6G+CD244+ and
Ly6G+CD244− cells were assessed by
quantitative real-time (q)PCR with the 2ΔCT method for relative
quantitation of target genes to an internal control. Total RNA was
extracted from the cells with TRIzol reagent (Invitrogen) and
reverse transcribed into complementary DNA (cDNA) using a
High-Capacity cDNA Reverse Transcription kit (Applied Biosciences).
qPCR was conducted on a Takara Thermal Cycler Dice Real-time System
(Takara Bio Inc.) with specific primers (Table I) and KOD SYBR qPCR Mix (Toyobo)
according to the manufacturer's instructions. The thermal cycling
conditions consisted of 95°C for 30 sec, 40 cycles of 95°C for 5
sec, and 60°C for 30 sec. Amplification of glyceraldehyde
3-phosphate dehydrogenase (GAPDH) was used as the internal
control.
 | Table I.Gene-specific primers used in
qPCR. |
Table I.
Gene-specific primers used in
qPCR.
| Gene | Forward primer | Reverse primer |
|---|
| ARG1 |
CATGGGCAACCTGTGTCCTT |
TCCTGGTACATCTGGGAACTTTC |
| NOS2 |
GACGAGACGGATAGGCAGAG |
GTGGGGTTGTTGCTGAACTT |
| IL6 |
CCGGAGAGGAGACTTCACAG |
TCCACGATTTCCCAGAGAAC |
| IL10 |
AAGGCAGTGGAGCAGGTGAA |
CCAGCAGACTCAATACACAC |
| TGFB |
CACCGGAGAGCCCTGGATA |
TGTACAGCTGCCGCACACA |
| MPO |
CCATGGTCCAGATCATCACA |
GCCGGTACTGATTGTTCAGG |
| GAPDH |
TGTGTCCCTCGTGGATCTGA |
TTGCTGTTGAAGTCGCAGGAG |
Antigen-specific T cell suppression
assay
The procedure used in this study has been published
previously (21). Briefly,
splenocytes of OT-1 C57BL/6 transgenic mice were purified, labeled
with 125 nM carboxyfluorescein diacetate succinimidyl ester (CFSE;
Invitrogen; Thermo Fisher Scientific, Inc.), seeded at
5×105 cells/well in 48-well plates, and co-cultured with
Ly6G+CD244+ or
Ly6G+CD244− cells at indicated ratios in a
complete medium [RPMI-1640 medium with 10% (v/v) heat-inactivated
FBS, 1% penicillin/streptomycin, and 50 µM 2-mercaptoethanol] with
1 µg ovalbumin (OVA)-derived peptide SIINFEKL at 37°C in 5%
CO2 for 48 h. The CFSE fluorescence of OT-1
CD8+ T cells was measured by flow cytometry. All
experiments were performed in triplicate.
Cytometric bead array assay
The procedure used in this study has been published
previously (14). Briefly, we
purchased cytometric bead array (CBA) assay kits from BD
Biosciences. The plasma and supernatants of peritoneal lavage
samples were collected, aliquoted into polypropylene
microcentrifuge tubes, and stored at −80°C until use. According to
the manufacturer's protocol, the samples were mixed with
antibody-coated capture beads against the following cytokines:
Granulocyte colony-stimulating factor (G-CSF),
granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor
necrosis factor-α (TNF-α), interferon (IFN)-γ, interleukin (IL)-1β,
IL-2, IL-4, IL-6, IL-10, and IL-13. The samples were incubated for
1 h at room temperature, mixed with PE detection reagent, and
further incubated for 1 h at room temperature. Then, the sample
were washed, centrifuged at 500 × g for 5 min, and resuspended in
assay buffer. The cytokine concentrations were measured by the
FACSVerse instrument and FCAP Array software (BD Biosciences). All
experiments were performed in duplicate and quantitated using a
standardized curve.
Statistical analysis
Differences between two groups were analyzed using
the Student's t-test. One-way analysis of variance (ANOVA) with
Holm's post-hoc test was performed to analyze differences
among multiple groups. Continuous variables are expressed as means.
Survival curves were drawn using the Kaplan-Meier method and
analyzed using the log-rank test. Statistical analyses were
conducted using JMP software (version 10; SAS Institute). Values of
P<0.05 were considered significant.
Results
Peritoneal dissemination is associated
with poor prognosis of tumor-bearing hosts
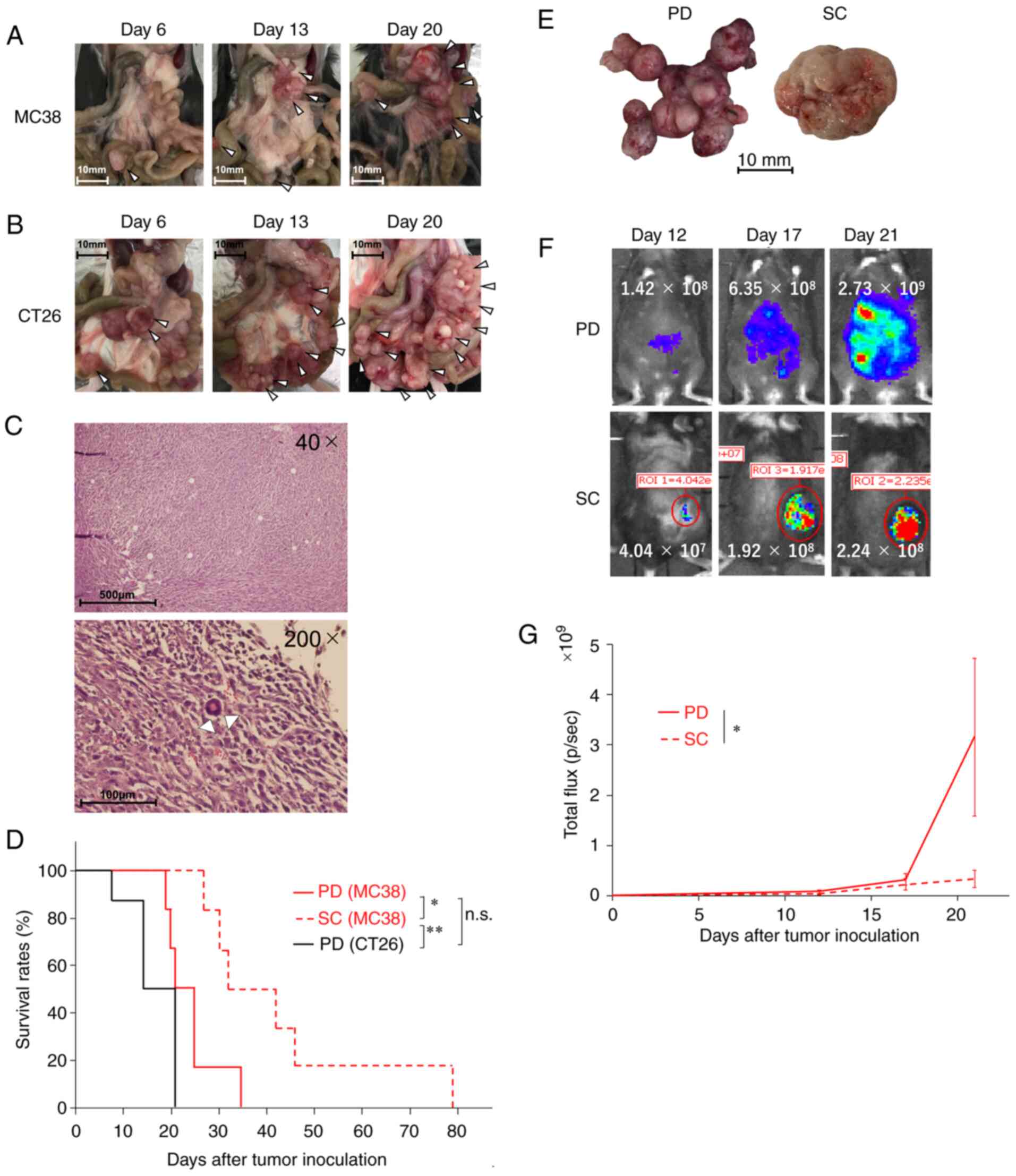
We first established PD mouse models using the
murine colon cancer cell lines MC38 (Fig.
1A) and CT26 (Fig. 1B). PD
nodules became enlarged and increased in both models over time,
with a slight predominance in the CT26-based model compared with
the MC38 model (Table SI).
Histopathological analyses of the MC38-based model revealed that
the PD nodules consisted mostly of tumor cells and scarcely of
immune cells (Fig. 1C). Consistent
with the tumor burden evaluation (Table
SI), survival analyses revealed that the prognosis of the
CT26-based PD model was poorer than that of the MC38 (P=0.12)
(Fig. 1D, red solid line vs. black
solid line). Based on these results, we decided to use the MC38
cell line primarily in this study because of its feasibility of
experimental handling. We also tested the MC38-based SC model
(Table SII) to compare with the PD
model. The prognosis of the PD model was significantly poorer than
that of the SC tumor model (P=0.015) (Fig. 1D, red solid line vs. red dotted line).
These results suggest that peritoneal dissemination is associated
with poor prognosis of tumor-bearing hosts.
Next, we sought to compare the total amount of tumor
burdens of the PD and SC models. As mentioned above (Tables SI and SII), the total tumor volume in the SC model
was grossly measurable, whereas the total tumor volume in the PD
model was extremely difficult to measure grossly due to the
multiple nature of the PD model (Fig.
1E). Therefore, to the same end, we used the bioluminescent
MC38-luc cell line and an in vivo bioluminescence detection
system (Fig. 1F). Consistent with the
survival data (Fig. 1D), total tumor
burden was significantly increased in the PD model compared with
the SC model (P=0.048) (Fig. 1G).
Taken together, these results suggest that peritoneal dissemination
is associated with poor prognosis of tumor-bearing hosts.
Intraperitoneal PMN-MDSCs
substantially increase with PD progression
Based on the finding that immune cell infiltration
was very rare in the PD nodules (Fig.
1C), we hypothesized that the peritoneal cavity through the
systemic circulation would be the main site of immune responses for
PD. To address this hypothesis, we evaluated immune cell profiles
in the peritoneal cavity and the systemic circulation in detail
using flow cytometry (Fig. 2A).
Representative flow cytometric figures of the MC38-based model are
provided (Fig. 2B). In the peritoneal
cavity of the MC38-based PD model, the numbers of M-MDSCs and
PMN-MDSCs were significantly increased with tumor progression
(M-MDSCs: P=0.027 and PMN-MDSCs: P=0.046 for Day 0 vs. Day 20)
(Fig. 2C, left panel, solid black
line and dotted black line). The numbers of intraperitoneal
PMN-MDSCs rapidly increased on Day 13 after tumor inoculation
(Fig. 2C, left panel, solid black
line). We also analyzed the MDSC subsets in the spleen and
peripheral blood in both the MC38-based PD and SC models over time
(Fig. 2D). As in the peritoneal
cavity, the number of PMN-MDSCs in the peripheral blood increased
after Day 13 in the PD model (Fig.
2D, right panel, red solid line). In comparison, M-MDSCs
consistently had a numerical advantage over PMN-MDSCs in the SC
model (Fig. 2D, dotted lines).
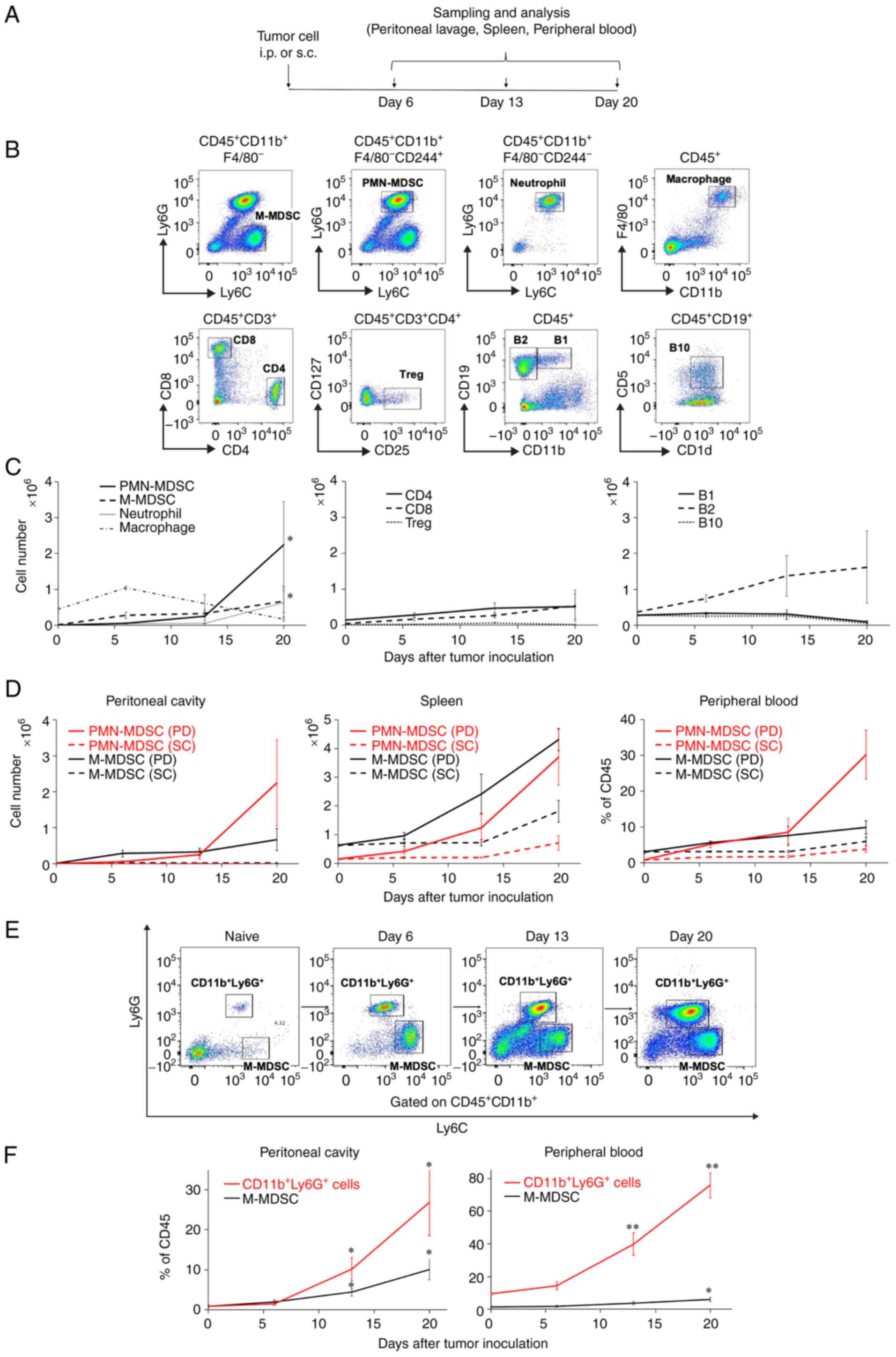 | Figure 2.Intraperitoneal PMN-MDSCs
substantially increase with PD progression. (A) Female C57BL/6J
mice (n=6) were intraperitoneally (i.p.) or subcutaneously (s.c.)
inoculated with 5×105 MC38 colon cancer cells and
sacrificed to collect peritoneal lavage, spleens, and peripheral
blood on Days 6, 13 and 20 as indicated. (B) Representative flow
cytometric data of intraperitoneal immune cell subsets. The cells
were defined as follows:
CD11b+F4/80−Ly6ChighLy6G−
for monocytic myeloid-derived suppressor cells (M-MDSCs),
CD11b+F4/80−CD244+Ly6CmidLy6G+
for polymorphonuclear (PMN)-MDSCs,
CD11b+F4/80−CD244−Ly6CmidLy6G+
for neutrophils, CD11b+F4/80+ for
macrophages, CD3+CD4+ for CD4 T cells,
CD3+CD8+ for CD8 T cells,
CD3+CD4+CD25+CD127− for
regulatory T cells (Tregs), CD19+CD11b+ for
B1 cells, CD19+CD11b− for B2 cells, and
CD19+CD1d+CD5+ for B10 cells. (C)
Time-course enumeration of the immune cell numbers identified. (D)
Time-course enumeration of the number of MDSCs in the peritoneal
cavity, spleen, and peripheral blood in the MC38-based peritoneal
dissemination (PD) and subcutaneously (SC) inoculated models.
Female BALB/c mice (n=4) were intraperitoneally inoculated with
1×105 CT26 colon cancer cells and sacrificed to collect
peritoneal lavage and peripheral blood on Days 6, 13 and 20. (E)
Representative flow cytometric data of intraperitoneal
CD11b+Ly6G+ cells and M-MDSCs. (F)
Time-course enumeration of their frequencies in the peritoneal
cavity and the peripheral blood. Values are expressed as mean
values with standard errors. One-way ANOVA with Holm's
post-hoc test was performed. *P<0.05, **P<0.01. |
To confirm the results above, we complementarily
tested the CT26-based PD models (Fig. 2E
and F). Consistent with the MC38 data (Fig. 2D), longitudinal evaluations of flow
cytometry demonstrated a rapid increase in the
Ly6G+CD11b+ cell population, which has been
shown to include CD244+ PMN-MDSCs (21), in both the peritoneal cavity and the
peripheral blood (Fig. 2F). Taken
together, these results suggest that the local and/or systemic
predominance of PMN-MDSCs is involved in the poor prognosis of the
PD-bearing hosts.
Intraperitoneal
Ly6G+CD11b+ population predominantly consists
of CD244+ cells
To confirm how the Ly6G+CD11b+
myeloid cell population consisted of CD244+ PMN-MDSCs in
this study (21), we evaluated the
ratios of CD244+ PMN-MDSCs in Ly6G+ cells
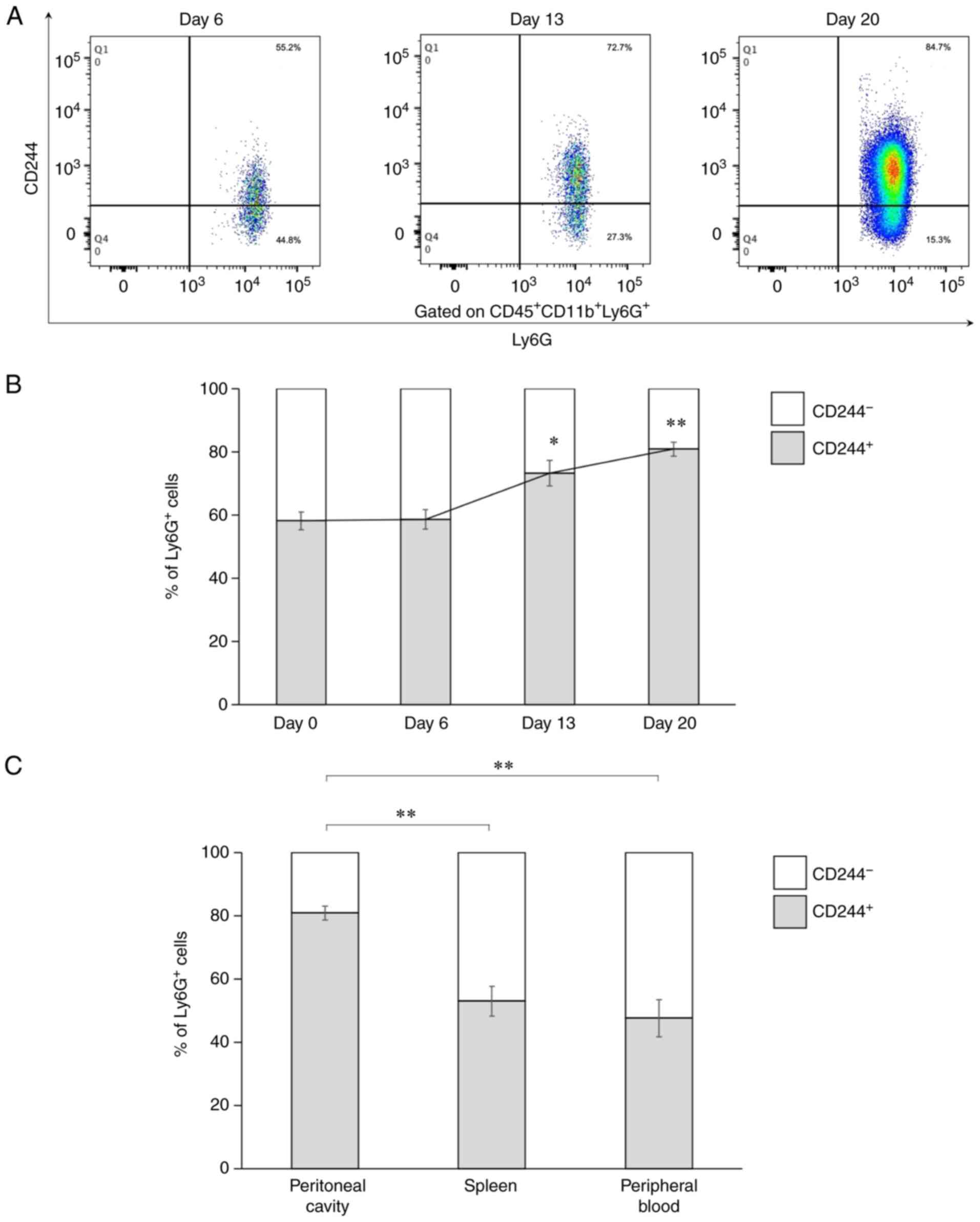
(Fig. 3). Representative flow
cytometric figures of the MC38-based model are provided (Fig. 3A). The intraperitoneal
Ly6G+CD244+ cells gradually increased over
time (Fig. 3B), and the frequencies
of the CD244+ cells in the Ly6G+ cells were
significantly high on Days 13 and 20 in the tumor-bearing mice
compared with Day 0 (P=0.011 on Day 13, P=0.0002 on Day 20). Also,
the frequencies of the CD244+ cells in the
Ly6G+ cells increased over time in the spleen and the
peripheral blood (data not shown). Among the samples tested on Day
20, the peritoneal cavity showed the highest frequency of the
CD244+ cells in the Ly6G+ cells (P=0.0007 vs.
spleen, P=0.0007 vs. peripheral blood) (Fig. 3C). These results suggest that
intraperitoneal CD244+ PMN-MDSCs contributed to tumor
progression and poor prognosis in the PD model.
Ly6G+CD244+
cells function as PMN-MDSCs
As the findings thus far suggest the functional
importance of Ly6G+CD244+ cells, we
subsequently addressed the immunological function of
Ly6G+CD244+ cells to determine whether they
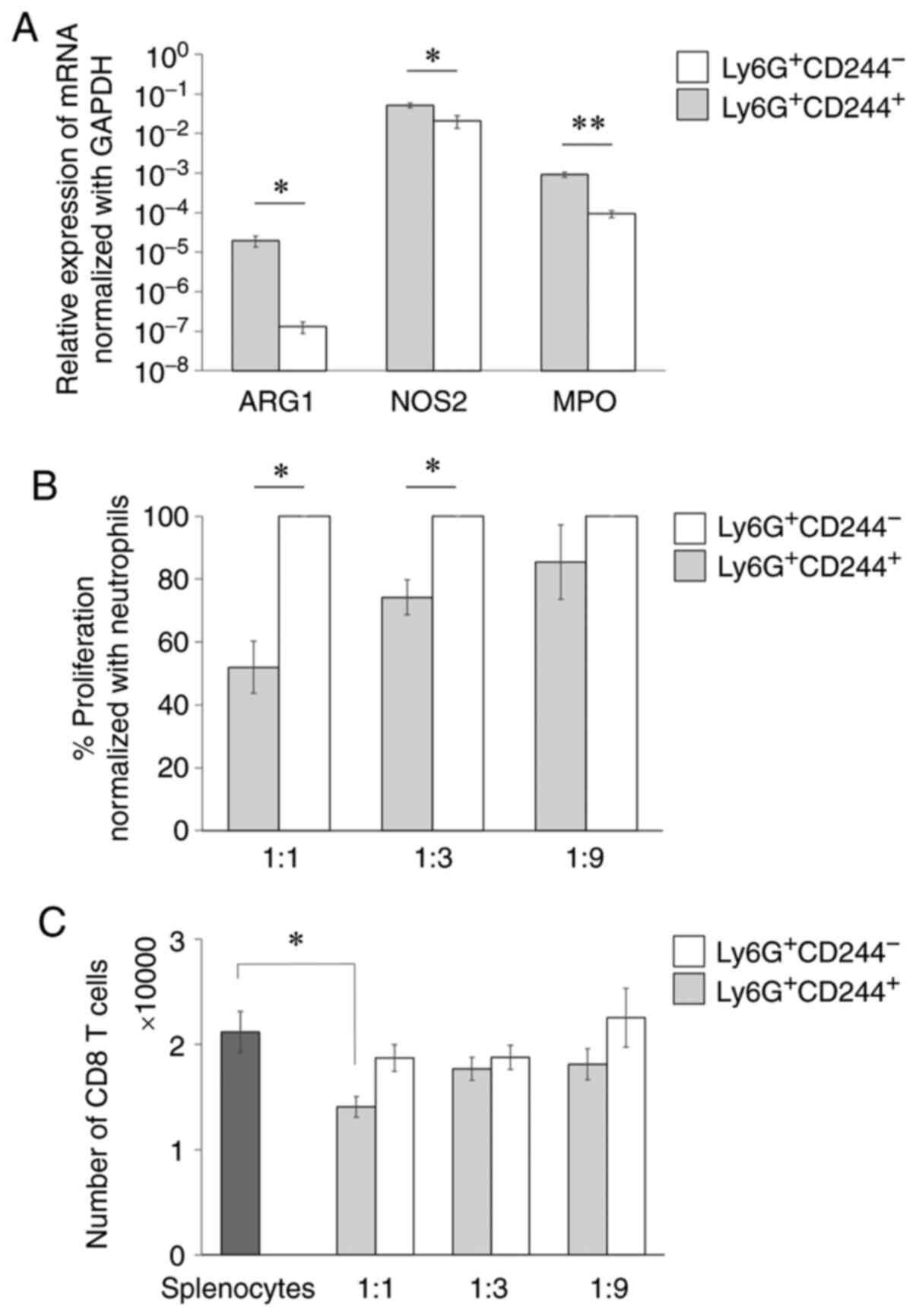
function as PMN-MDSCs. To this end, we conducted qPCR to analyze
the mRNA levels of the immunosuppressive genes in the
Ly6G+CD244+ cells (Table I). The following immunosuppressive
genes were significantly increased in the
Ly6G+CD244+ cells: Arginase-1 (ARG1:
P=0.030), nitric oxide synthase 2 (NOS2: P=0.036), and
myeloperoxidase (MPO: P=0.005) (Fig. 4A). To confirm the immunosuppressive
function of the Ly6G+CD244+ cells, we
performed an ex vivo antigen-specific T-cell suppression
assay. The Ly6G+CD244+ cells significantly
inhibited antigen-specific CD8+ T-cell proliferation
compared with the Ly6G+CD244− cells at high
(1:1) and moderate (1:3) ratios (1:1: P=0.030, 1:3: P=0.017)
(Fig. 4B). T-cell suppression
meditated by the Ly6G+CD244+ cells was also
observed in the numerical evaluation of CD8+ T cells at
a high (1:1) ratio (P=0.031) (Fig.
4C). These findings suggest that the
Ly6G+CD244+ cells effectively suppressed T
cells ex vivo and thus could be functionally considered as
PMN-MDSCs.
PD nodules consist mostly of tumor
cells and scarcely of immune cells
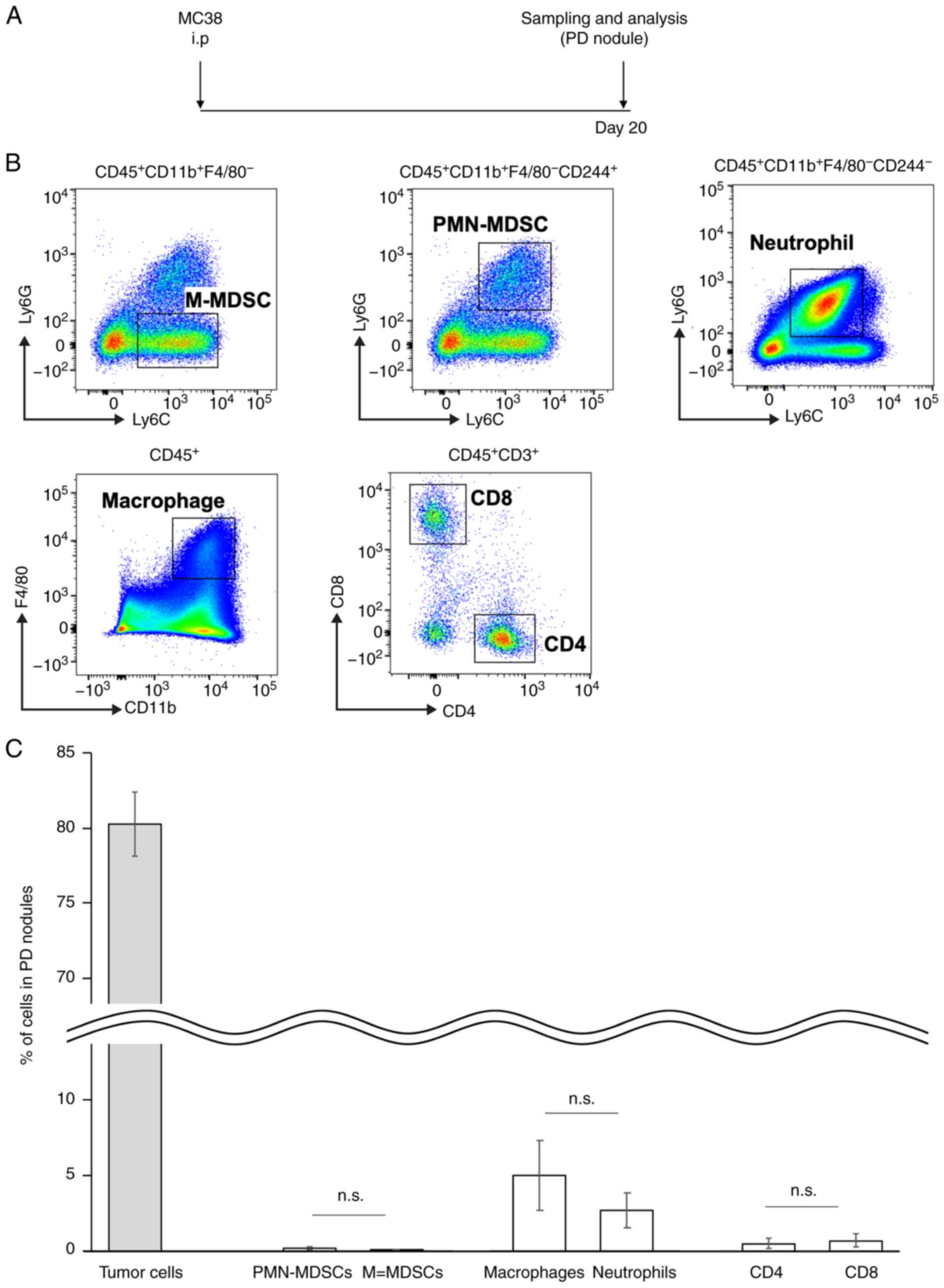
Histopathological analyses showed that immune cell
infiltration was very rare in the PD nodules (Fig. 1C). To confirm this finding from a
viewpoint of the CD244+ cells, we performed flow
cytometric analyses of the PD nodules (Fig. 5A). Representative flow cytometric
figures of the PD nodules in the MC38-based model are provided
(Fig. 5B). Consistent with the
histopathological findings (Fig. 1C),
intratumoral PMN-MDSCs were detectable but accounted for only 0.1%
of all cells (including tumor cells) in the PD nodules (Fig. 5C). Taken together, these findings
support our hypothesis that the peritoneal cavity through the
systemic circulation would be the main site of immune responses for
PD.
Tumor-derived cytokines are involved
in the PMN-MDSC induction
The finding above led us to investigate the causes
of the increase in the intraperitoneal PMN-MDSCs. To this end, we
conducted CBA assays using the ascites obtained on Day 20 to
evaluate the levels of cytokines in the peritoneal cavity
comprehensively (Fig. 6). The
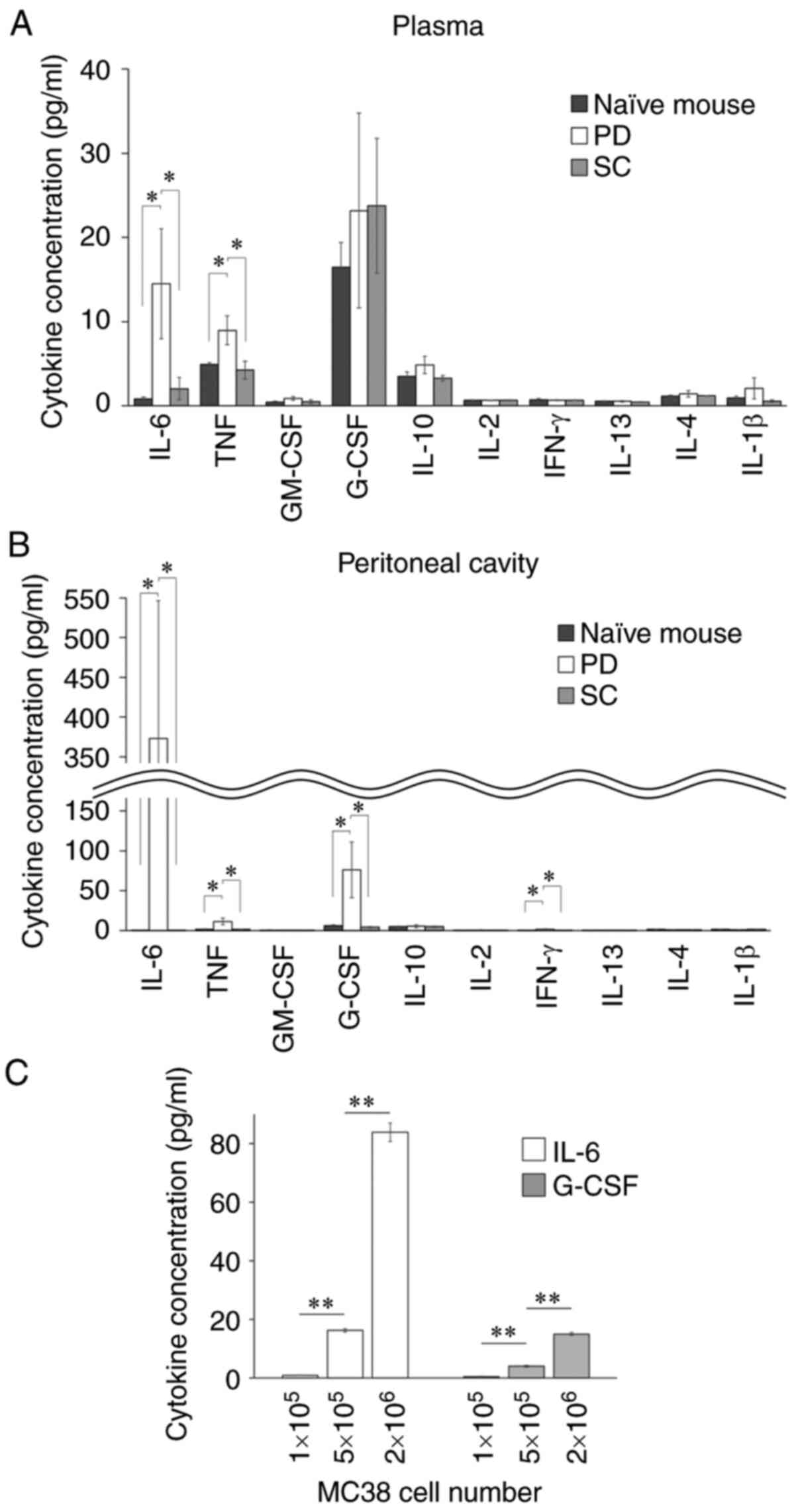
concentrations of IL-6 and TNF-α were significantly increased in
the peritoneal cavity and plasma of the PD model (Fig. 6A and B). In addition, G-CSF was
significantly increased in the peritoneal cavity (Fig. 6B). Similar to these in vivo
data, MC38 tumor cells secreted IL-6 and G-CSF in vitro, and
their production levels increased in a cell number-dependent manner
(Fig. 6C). These results suggest that
the presence of PD would induce unique immunological profiles, such
as the increases in IL-6 and G-CSF, in the peritoneal cavity.
PMN-MDSC-targeting therapy reverts the
number of T cells and suppresses the tumor progression in the PD
model
To clarify the pathological functions of PMN-MDSCs
in the PD model, we sought to deplete PMN-MDSCs in the PD model. In
this regard, since PMN-MDSCs accounted for the large majority of
Ly6G+ cells (Fig. 3) and
in vivo-compatible anti-CD244 antibody is known to exert
strong effects on natural killer (NK) and T cells inadvertently
(30), we decided to use anti-Ly6G
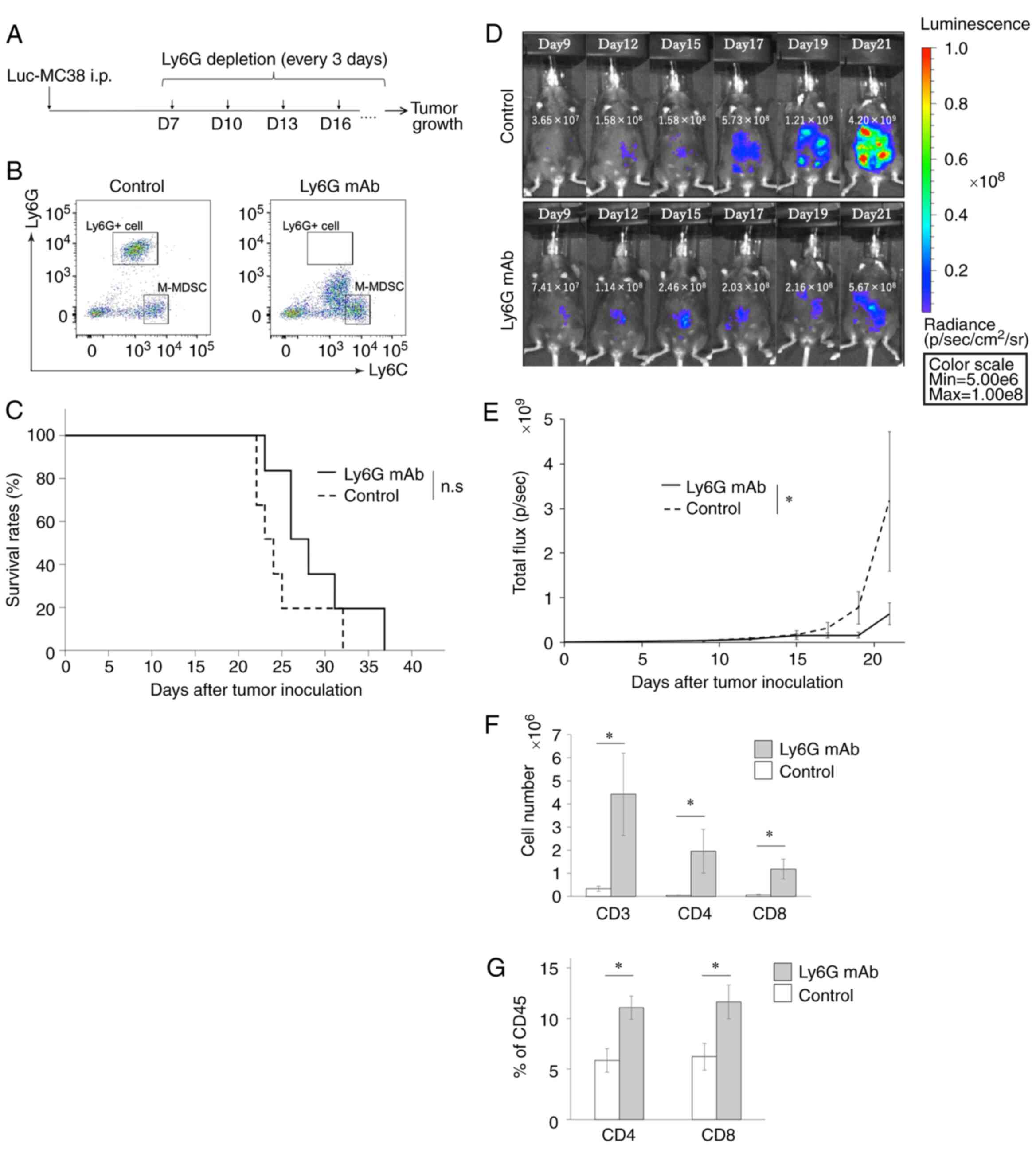
mAb to deplete PMN-MDSCs (Fig. 7A).
As expected, in vivo administration of anti-Ly6G mAb
completely depleted intraperitoneal Ly6G+ cells
(Fig. 7B). Although the treatment
with anti-Ly6G mAb did not prolong the survival of tumor-bearing
mice (Fig. 7C), tumor progression was
significantly suppressed in the mice treated with the anti-Ly6G mAb
compared with control mice until Day 21 (P=0.025) (Fig. 7D and E). We also evaluated whether
anti-Ly6G mAb would affect T-cell kinetics in the peritoneal cavity
and the peripheral blood. The treatment with anti-Ly6G mAb
significantly increased the number of both intraperitoneal
CD3+, CD4+, and CD8+ T cells of
the tumor-bearing mice (CD3: P=0.037, CD4: P=0.037, CD8: P=0.025)
(Fig. 7F). The peripheral blood
showed the similar trends (CD4: P=0.025, CD8: P=0.037) (Fig. 7G). These results suggest that
PMN-MDSCs suppress the antitumor activities of T cells to induce PD
progression and that they may be a potent therapeutic target for
PD.
Discussion
We clarified the pathological roles of peritoneal
dissemination (PD)-associated polymorphonuclear myeloid-derived
suppressor cells (PMN-MDSCs) in this study. We previously
demonstrated the associations of MDSCs with tumor progression
(13,14), which led us to hypothesize that MDSCs
may also play an important role in the PD-relevant tumor immune
microenvironment (TIME). To address this hypothesis, we first
established a PD mouse model. Based on the finding that the PD
nodules consisted scarcely of immune cells, we focused on the
peritoneal cavity, but not the PD nodule, to evaluate the
PD-relevant TIME. Among the cell population evaluated,
intraperitoneal PMN-MDSCs substantially increased in association
with PD progression. The characteristics of PMN-MDSCs were
phenotypically and functionally determined. In addition, the
concentrations of interleukin (IL)-6 and granulocyte-colony
stimulating factor (G-CSF) were significantly increased in the
peritoneal cavity, which was produced by the tumors and thought to
contribute to the increase in PMN-MDSCs. As PMN-MDSCs accounted for
the large majority of Ly6G+ cells, we conducted
Ly6G-mediated PMN-MDSC depletion in vivo. The PMN-MDSC
depletion significantly inhibited the progression of PD and
reverted both CD4+ and CD8+ T cells in the
peritoneal cavity and the peripheral blood. Taken together, these
results suggest that targeted therapy for PMN-MDSCs would provide
therapeutic values to prevent the disease progression of PD derived
from colorectal cancer (CRC).
In this study, we tested two types of murine colon
cancer cell lines (MC38 and CT26) to induce PD and observed similar
results. The findings in this study are also consistent with those
of previous studies that have analyzed the clinical samples derived
from patients with gastric cancer (31), cervical cancer (32), and breast cancer (33). In addition, human cancers have
extremely high heterogeneity, by which MDSCs can be induced through
a wide variety of tumor-relevant mechanisms (34). Based on these findings, we believe
that the findings in this study can be generalized at least to some
extent. Particularly to address the heterogeneous conditions of PD,
we are in the process of conducting an observational clinical study
using the specimens of CRC-PD patients.
Regarding the comparison of total tumor volumes in
the PD and subcutaneous inoculation (SC) models, we found it
difficult to calculate the tumor volumes from photographic data in
this study, particularly due to the multiple nature of the PD
model. PD is characterized by tumor cells spreading widely in the
abdominal cavity by the flow of ascites fluid (35). In fact, the number of PD nodules in
the abdominal cavity reached more than 10 in a substantial number
of the MC38-based PD models. In addition, the PD nodules of various
sizes were observed, some of which were fused together. In order to
compare the total tumor volumes in the SC and PD models under the
same conditions, we measured the in vivo bioluminescence
intensity using the bioluminescent MC38-luc cell line in both
models. As a result, the PD models were found to have higher tumor
volume and poorer prognosis than the SC models. Of note, it is well
known that the in vivo bioluminescence intensity reflects
the total number and quantity of luminescent materials including
living cells in host animals (36).
Such an in vivo bioluminescence detection system could be
very useful for assessing the total tumor volume in multiple tumor
models such as the PD model.
Immune cell infiltration in PD nodules was barely
detectable in our animal model. Therefore, we concluded that
detailed immunological evaluation of PD nodules was difficult to
conduct. However, other investigators have reported that immune
cells are detectable and reflect immune responses and treatment
effects (37,38). The differences are that the number of
tumor cells used in these studies was much smaller than in our
fast-growing PD model. That is, rapidly growing tumors induce areas
of hypoxia with low levels of glucose and nutrients, leading to
lactic acid accumulation and eventually inhibiting the immune cells
from infiltrating into the tumor tissue (39). In contrast, the lower the number of
cells administered, the slower the disease progression and the more
likely the immune response will occur. For this reason, the number
of infiltrating immune cells may have been high in these studies.
In this regard, further studies would be needed for an accurate
immunological evaluation of PD nodules.
It has been a long-lasting question in the research
field of MDSCs how to distinguish PMN-MDSCs from neutrophils in
mice. Since CD244 has been highlighted as a surface antigen to
identify PMN-MDSCs (11,16–18), we
tested whether Ly6G+CD244+ cells would have
immunosuppressive activity on T cells. As expected,
Ly6G+CD244+ cells showed significantly higher
gene expression levels of ARG1, NOS2, and MPO than
Ly6G+CD 244− cells. These genes are
frequently evaluated as the immunosuppressive mediators
representing PMN-MDSCs, along with positive correlations with the
corresponding protein levels (27–29).
Moreover, Ly6G+CD244+ cells suppressed T
cells ex vivo. Based on these findings, we considered
Ly6G+CD244+ cells as PMN-MDSCs in this study.
However, it has also been noted that CD244 is useful to distinguish
PMN-MDSCs from only normal neutrophils but not tumor-associated
neutrophils (11). Further studies
will be needed to determine the precise surface phenotypes of MDSC
subsets.
The murine colon cancer cell line MC38 produced
G-CSF and IL-6 at the high levels, which appeared to induce
PMN-MDSCs in the peritoneal cavity and then in the systemic
circulation. In this regard, the accumulation of MDSCs has been
shown to depend on two distinct signaling pathways; one is
associated with the expansion of immature myeloid cells and the
another is associated with their pathological activation (40). In particular, G-CSF is well known as a
cytokine associated with the differentiation and trafficking of
granulocytic lineage and the generation of PMN-MDSCs (41), whereas IL-6 would promote the systemic
accumulation of PMN-MDSCs (42). Our
cytokine profile data were consistent with these previous reports.
In the MC38-based PD model, G-CSF would induce the formation and
trafficking of immature myeloid cells into the peritoneal cavity,
and IL-6 would promote the systemic increase of MDSCs. That is, it
is assumable that PMN-MDSCs would be provided from the local
peritoneal cavity and spread systemically. Besides, both G-CSF and
IL-6 are known to activate the signaling of signal transducer and
activator of transcription 3 (STAT-3), which is strongly associated
with the induction of PMN-MDSCs (43). The precise mechanisms of PMN-MDSC
induction remain a significant issue that requires further
investigation.
In human, recombinant human G-CSF (rhG-CSF) has
become a primary therapeutic agent to prevent and recover from
chemotherapy-induced myelosuppression. Some major guidelines
recommend the use of rhG-CSF when there is approximately a 20% risk
of febrile neutropenia (44,45). In addition, a recent meta-analysis
study has shown that patients who received chemotherapy under
rhG-CSF support had better overall survival than those who did not
receive rhG-CSF support (46).
Therefore, the administration of rhG-CSF is reasonable as a
supportive therapy for those with chemotherapy-induced
myelosuppression in general. In contrast, our data of the PD model
showed tumor-derived G-CSF at high levels in the peritoneal cavity
and its association with poor prognosis of the tumor-bearing hosts
as well as increased MDSCs. Moreover, several studies based on
human gastric cancer and colon cancer have reported that high
production levels of tumor-derived G-CSF correlate with tumor
progression and poor prognosis (31,47). We
interpreted these findings with our data that the use of rhG-CSF
should be cautious when high levels of ectopic G-CSF exist
particularly with PD.
Since PMN-MDSCs accounted for the large majority of
Ly6G+ cells and in vivo-compatible anti-CD244
antibody is known to exert strong effects on natural killer (NK)
and T cells inadvertently (30), we
alternatively used anti-Ly6G mAb to deplete PMN-MDSCs in the PD
mouse model. Although the methods used in this study was not
completely specific for PMN-MDSCs, our results still suggest that
PMN-MDSCs contribute to PD progression. In this regard, it has been
shown that the continuous use of anti-Ly6G mAb induces the
reappearance of MDSCs in about a week, limiting its effectiveness
by depletion-associated extramedullary granulopoiesis (48). We consider this phenomenon to be the
cause of the suboptimal therapeutic efficacy of Ly6G depletion in
this study. It is a subject for future verification how to target
PMN-MDSCs specifically and sustainably.
PMN-MDSCs establish tumor-promoting environments
and contribute to tumor progression. The mechanism of T-cell-based
immunosuppression by PMN-MDSCs is proposed as follows. PMN-MDSCs
exert the programmed cell death-1 (PD-1)/programmed cell death
ligand-1 (PD-L1)-mediated immune checkpoint blockade (49). PMN-MDSCs also produce molecules that
exert direct immunosuppression such as arginase-1 (Arg-1), nitric
oxide synthase (NOS), and reactive oxygen species (ROS) (50) and promote tumor growth and metastatic
spread indirectly such as vascular endothelial growth factor
(VEGF), prokineticin 2 (PK2), and matrix metalloproteinase-9
(MMP-9) (51). Targeting PMN-MDSCs
would solve these issues at once and thus can be a potent
therapeutic target of cancer immunotherapy. In addition, our
finding that the Ly6G-mediated PMN-MDSC depletion reverted the
number of T cells in the peripheral blood as well as in the
peritoneal cavity suggests an important perspective; the targeted
therapy for PMN-MDSCs would enhance the therapeutic efficacy of
T-cell-based immunotherapy. In this regard, the administration of
CXCR2 inhibitor targeting PMN-MDSCs has improved the efficacy of
T-cell adoptive immunotherapy (52,53).
Although the conventional T-cell subsets of CD4, CD8, and Treg were
not associated with PD progression in this study, those with
exhaustion phenotypes, such as PD-1 or LAG3, might be associated
with PD progression (54). We are
currently in the process of addressing this aspect to seek for the
possibility to combine PMN-MDSC-targeting therapy with immune
checkpoint blockades or adoptive T-cell transfer therapies
including CAR-T cell therapy (55).
Collectively, we demonstrated that PMN-MDSCs would
suppress antitumor activities of T cells to induce PD progression
in this study. Therefore, the targeted therapy for PMN-MDSCs may
provide new therapeutic value to prevent the disease progression of
PD. Furthermore, given the recent advancement of T-cell
immunotherapy, our strategy may also provide a clue to develop new
strategies to synergize with T-cell immunotherapy for CRC-derived
PD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by JSPS KAKENHI (grant no.
20K17650).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
YS and KYamas mainly conceived and planned the
experiments. YS, MS, KYamad, KA, AW, and EF carried out the
experiments. YS, KYamas, MF, and MS contributed to the
interpretation of the results. YS took the lead in writing the
manuscript. KYamas, MF, MS, and YK critically revised the
manuscript for intellectual content. HH, SK, TO, TM, TN, and SS
provided critical feedback and helped shape the research, analysis
and manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The experimental procedures were approved by the
Ethics Committee of Kobe University (approval no. P190404).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Massalou D, Benizri E, Chevallier A,
Duranton-Tanneur V, Pedeutour F, Benchimol D and Béréder JM:
Peritoneal carcinomatosis of colorectal cancer: Novel clinical and
molecular outcomes. Am J Surg. 213:377–387. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sugarbaker PH: Colorectal cancer:
Prevention and management of metastatic disease. BioMed Res Int.
2014:7828902014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Segelman J, Granath F, Holm T, Machado M,
Mahteme H and Martling A: Incidence, prevalence and risk factors
for peritoneal carcinomatosis from colorectal cancer. Br J Surg.
99:699–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klaver YL, Simkens LH, Lemmens VE, Koopman
M, Teerenstra S, Bleichrodt RP, de Hingh IH and Punt CJ: Outcomes
of colorectal cancer patients with peritoneal carcinomatosis
treated with chemotherapy with and without targeted therapy. Eur J
Surg Oncol. 38:617–623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Franko J, Shi Q, Meyers JP, Maughan TS,
Adams RA, Seymour MT, Saltz L, Punt CJA, Koopman M, Tournigand C,
et al: Prognosis of patients with peritoneal metastatic colorectal
cancer given systemic therapy: An analysis of individual patient
data from prospective randomised trials from the analysis and
research in cancers of the digestive system (ARCAD) database.
Lancet Oncol. 17:1709–1719. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Binnewies M, Roberts EW, Kersten K, Chan
V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI,
Ostrand-Rosenberg S, Hedrick CC, et al: Understanding the tumor
immune microenvironment (TIME) for effective therapy. Nat Med.
24:541–550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fridman WH, Zitvogel L, Sautès-Fridman C
and Kroemer G: The immune contexture in cancer prognosis and
treatment. Nat Rev Clin Oncol. 14:717–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Keren L, Bosse M, Marquez D, Angoshtari R,
Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R, et al: A
Structured tumor-immune microenvironment in triple negative breast
cancer revealed by multiplexed ion beam imaging. Cell.
174:1373–1387.e19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peltanova B, Raudenska M and Masarik M:
Effect of tumor microenvironment on pathogenesis of the head and
neck squamous cell carcinoma: A systematic review. Mol Cancer.
18:632019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Veglia F, Perego M and Gabrilovich D:
Myeloid-derived suppressor cells coming of age. Nat Immunol.
19:108–119. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gabrilovich DI, Ostrand-Rosenberg S and
Bronte V: Coordinated regulation of myeloid cells by tumours. Nat
Rev Immunol. 12:253–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Otsubo D, Yamashita K, Fujita M, Nishi M,
Kimura Y, Hasegawa H, Suzuki S and Kakeji Y: Early-phase treatment
by Low-dose 5-Fluorouracil or primary tumor resection inhibits
MDSC-mediated lung metastasis formation. Anticancer Res.
35:4425–4431. 2015.PubMed/NCBI
|
|
14
|
Tanaka T, Fujita M, Hasegawa H, Arimoto A,
Nishi M, Fukuoka E, Sugita Y, Matsuda T, Sumi Y, Suzuki S, et al:
Frequency of Myeloid-derived suppressor cells in the peripheral
blood reflects the status of tumor recurrence. Anticancer Res.
37:3863–3869. 2017.PubMed/NCBI
|
|
15
|
Youn JI, Nagaraj S, Collazo M and
Gabrilovich DI: Subsets of Myeloid-Derived suppressor cells in
tumor bearing mice. J Immunol. 181:5791–5802. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fortin C, Huang X and Yang Y: NK cell
response to vaccinia virus is regulated by myeloid-derived
suppressor cells. J Immunol. 189:1843–1849. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cassetta L, Baekkevold ES, Brandau S,
Bujko A, Cassatella MA, Dorhoi A, Krieg C, Lin A, Loré K, Marini O,
et al: Deciphering myeloid-derived suppressor cells: Isolation and
markers in humans, mice and non-human primates. Cancer Immunol
Immunother. 68:687–697. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Agresta L, Hoebe KHN and Janssen EM: The
emerging role of CD244 signaling in immune cells of the tumor
microenvironment. Front Immunol. 9:28092018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM,
Wang D, Li XF and Zheng L: Monocyte/macrophage-elicited natural
killer cell dysfunction in hepatocellular carcinoma is mediated by
CD48/2B4 interactions. Hepatology. 57:1107–1116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wherry EJ and Kurachi M: Molecular and
cellular insights into T cell exhaustion. Nat Rev Immunol.
15:486–499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Youn JI, Collazo M, Shalova IN, Biswas SK
and Gabrilovich DI: Characterization of the nature of granulocytic
myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc
Biol. 91:167–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clarke P, Mann J, Simpson JF,
Rickard-Dickson K and Primus FJ: Mice transgenic for human
carcinoembryonic antigen as a model for immunotherapy. Cancer Res.
58:1469–1477. 1998.PubMed/NCBI
|
|
23
|
Ojima T, Iwahashi M, Nakamura M, Matsuda
K, Nakamori M, Ueda K, Naka T, Ishida K, Primus FJ and Yamaue H:
Successful cancer vaccine therapy for carcinoembryonic antigen
(CEA)-expressing colon cancer using genetically modified dendritic
cells that express CEA and T helper-type 1 cytokines in CEA
transgenic mice. Int J Cancer. 120:585–593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kilkenny C, Browne WJ, Cuthill IC, Emerson
M and Altman DG: Improving bioscience research reporting: The
ARRIVE guidelines for reporting animal research. PLoS Biol.
8:e10004122010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arimoto A, Yamashita K, Hasegawa H, Sugita
Y, Fukuoka E, Tanaka T, Suzuki S and Kakeji Y: Immunosuppression
induced by perioperative peritonitis promotes lung metastasis.
Anticancer Res. 38:4333–4338. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bruger AM, Dorhoi A, Esendagli G,
Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, Perecko T,
Santibanez J, Saraiva M, Van Ginderachter JA and Brandau S: How to
measure the immunosuppressive activity of MDSC: Assays, problems
and potential solutions. Cancer Immunol Immunother. 68:631–644.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shang W, Tang Z, Gao Y, Qi H, Su X, Zhang
Y and Yang R: LncRNA RNCR3 promotes Chop expression by sponging
miR-185-5p during MDSC differentiation. Oncotarget.
8:111754–111769. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schleicher U, Paduch K, Debus A, Obermeyer
S, König T, Kling JC, Ribechini E, Dudziak D, Mougiakakos D, Murray
PJ, et al: TNF-mediated restriction of arginase 1 expression in
myeloid cells triggers type 2 NO synthase activity at the site of
infection. Cell Rep. 15:1062–1075. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Agresta L, Lehn M, Lampe K, Cantrell R,
Hennies C, Szabo S, Wise-Draper T, Conforti L, Hoebe K and Janssen
EM: CD244 represents a new therapeutic target in head and neck
squamous cell carcinoma. J Immunother Cancer. 8:e0002452020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Morris KT, Khan H, Ahmad A, Weston LL,
Nofchissey RA, Pinchuk IV and Beswick EJ: G-CSF and G-CSFR are
highly expressed in human gastric and colon cancers and promote
carcinoma cell proliferation and migration. Br J Cancer.
110:1211–1220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawano M, Mabuchi S, Matsumoto Y, Sasano
T, Takahashi R, Kuroda H, Kozasa K, Hashimoto K, Isobe A, Sawada K,
et al: The significance of G-CSF expression and myeloid-derived
suppressor cells in the chemoresistance of uterine cervical cancer.
Sci Rep. 5:182172015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pilatova K, Bencsikova B, Demlova R, Valik
D and Zdrazilova-Dubska L: Myeloid-derived suppressor cells (MDSCs)
in patients with solid tumors: Considerations for granulocyte
colony-stimulating factor treatment. Cancer Immunol Immunother.
67:1919–1929. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Veglia F, Sanseviero E and Gabrilovich DI:
Myeloid-derived suppressor cells in the era of increasing myeloid
cell diversity. Nat Rev Immunol. Feb 1–2021.(Epub ahead of print).
doi: 10.1038/s41577-020-00490-y. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ceelen W, Ramsay RG, Narasimhan V, Heriot
AG and De Wever O: Targeting the tumor microenvironment in
colorectal peritoneal metastases. Trends Cancer. 6:236–246. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Toyoshima M, Tanaka Y, Matumoto M,
Yamazaki M, Nagase S, Sugamura K and Yaegashi N: Generation of a
syngeneic mouse model to study the intraperitoneal dissemination of
ovarian cancer with in vivo luciferase imaging. Luminescence.
24:324–331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Taibi A, Albouys J, Jacques J, Perrin ML,
Yardin C, Durand Fontanier S and Bardet SM: Comparison of
implantation sites for the development of peritoneal metastasis in
a colorectal cancer mouse model using non-invasive bioluminescence
imaging. PLoS One. 14:e02203602019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee YS, Lee WS, Kim CW, Lee SJ, Yang H,
Kong SJ, Ning J, Yang KM, Kang B, Kim WR, et al: Oncolytic vaccinia
virus reinvigorates peritoneal immunity and cooperates with immune
checkpoint inhibitor to suppress peritoneal carcinomatosis in colon
cancer. J Immunother Cancer. 8:e0008572020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cassim S and Pouyssegur J: Tumor
microenvironment: A metabolic player that shapes the immune
response. Int J Mol Sci. 21:1572019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Condamine T and Gabrilovich DI: Molecular
mechanisms regulating myeloid-derived suppressor cell
differentiation and function. Trends Immunol. 32:19–25. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Waight JD, Hu Q, Miller A, Liu S and
Abrams SI: Tumor-Derived G-CSF facilitates neoplastic growth
through a granulocytic myeloid-derived suppressor cell-dependent
mechanism. PLoS One. 6:e276902011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weber R, Groth C, Lasser S, Arkhypov I,
Petrova V, Altevogt P, Utikal J and Umansky V: IL-6 as a major
regulator of MDSC activity and possible target for cancer
immunotherapy. Cell Immunol. 359:1042542021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kramer ED and Abrams SI: Granulocytic
myeloid-derived suppressor cells as negative regulators of
anticancer immunity. Front Immunol. 11:19632020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Aapro MS, Cameron DA, Pettengell R,
Bohlius J, Crawford J, Ellis M, Kearney N, Lyman GH, Tjan-Heijnen
VC, Walewski J, et al: EORTC guidelines for the use of
granulocyte-colony stimulating factor to reduce the incidence of
chemotherapy-induced febrile neutropenia in adult patients with
lymphomas and solid tumours. Eur J Cancer. 42:2433–2453. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Becker PS, Griffiths EA, Alwan LM,
Bachiashvili K, Brown A, Cool R, Curtin P, Dinner S, Gojo I, Hicks
A, et al: NCCN guidelines insights: Hematopoietic growth factors,
version 1.2020. J Natl Compr Canc Netw. 18:12–22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lyman GH, Yau L, Nakov R and Krendyukov A:
Overall survival and risk of second malignancies with cancer
chemotherapy and G-CSF support. Ann Oncol. 29:1903–1910. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fan Z, Li Y, Zhao Q, Fan L, Tan B, Zuo J,
Hua K and Ji Q: Highly expressed granulocyte colony-stimulating
factor (G-CSF) and granulocyte colony-stimulating factor receptor
(G-CSFR) in human gastric cancer leads to poor survival. Med Sci
Monit. 24:1701–1711. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Moses K, Klein JC, Männ L, Klingberg A,
Gunzer M and Brandau S: Survival of residual neutrophils and
accelerated myelopoiesis limit the efficacy of antibody-mediated
depletion of Ly-6G+ cells in tumor-bearing mice. J
Leukoc Biol. 99:811–823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Law AM, Valdes-Mora F and Gallego-Ortega
D: Myeloid-derived suppressor cells as a therapeutic target for
cancer. Cells. 9:5612020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vetsika EK, Koukos A and Kotsakis A:
Myeloid-Derived suppressor cells: Major figures that shape the
immunosuppressive and angiogenic network in cancer. Cells.
8:16472019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Coffelt SB, Wellenstein MD and de Visser
KE: Neutrophils in cancer: Neutral no more. Nature Reviews Cancer.
16:431–446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Steele CW, Karim SA, Leach JDG, Bailey P,
Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z,
et al: CXCR2 inhibition profoundly suppresses metastases and
augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer
Cell. 29:832–845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun L, Clavijo PE, Robbins Y, Patel P,
Friedman J, Greene S, Das R, Silvin C, Van Waes C, Horn LA, et al:
Inhibiting myeloid-derived suppressor cell trafficking enhances T
cell immunotherapy. JCI Insight. 4:e1268532019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kurachi M: CD8+ T cell
exhaustion. Semin Immunopathol. 41:327–337. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Martinez M and Moon EK: CAR T cells for
solid tumors: New strategies for finding, infiltrating, and
surviving in the tumor microenvironment. Front Immunol. 10:1282019.
View Article : Google Scholar : PubMed/NCBI
|