Introduction
Numerous studies have indicated that cancer may be
caused both by mutations in DNA and by two specific types of
ubiquitous epigenetic modifications: DNA methylation and histone
acetylation (1,2). These two epigenetic modifications may
regulate gene expression, remodel chromatin, alter cell phenotypes
and promote cancer development (3,4). In
addition, methylation and acetylation modifications may regulate
cellular pathways that are involved in cell differentiation,
proliferation and apoptosis, which are attributed to the genesis of
leukaemia (5).
DNA methylation has been widely studied in DNA
epigenetic modification. In acute myeloid leukaemia (AML), abnormal
DNA methylation may silence the expression of tumour suppressor
genes by binding to the upstream promoter region (6). Changes in DNA methylation are
prevalent in myelodysplastic syndrome (MDS) and secondary AML and
they appear to have an important role in the transformation of MDS
to AML (7). Hence, hypomethylating
agents (HMAs) are important in the treatment of higher-risk
patients with MDS and AML, particularly in those patients who are
ineligible for haematopoietic stem cell transplantation (8,9).
5-azacitidine (5-AZA), which is a cytidine analogue prodrug, is an
HMA for the treatment of higher-risk MDS and/or AML.
Histone deacetylases (HDACs) are able to alter gene
expression and chromatin modification by inducing the deacetylation
of histones and other processes (10). In addition, the conversion of
DNA-histone complexes from an open state to a compact configuration
is closely related to gene transcription silencing (11). In addition to histone
deacetylation-induced gene silencing, DNA methylation is also
engaged in histone methylation, which is another important
epigenetic modification and may regulate proliferation, ageing,
tumourigenesis and other biological processes (12). Chidamide is an original new drug in
China and a novel selective HDAC inhibitor (HDACI). It is able to
selectively inhibit HDAC-1, −2, −3 and −10, particularly −2 and −3.
Chidamide has been researched in numerous clinical trials in the
USA and China and has been ratified to treat cutaneous T-cell
lymphoma and peripheral T-cell lymphoma in China (13). Chidamide is highly toxic to
leukaemia cells in a concentration-dependent manner, and less
toxic, more tolerated and more stable than other drugs (14). An increasing number of studies have
confirmed its antileukaemia effect. Both DNA methylation and
histone acetylation are reversible to a certain extent (2), which is worth considering for the
design of therapeutic strategies. This mechanism suggests that the
combination of HDACI and HMA may be effective against leukaemia.
Therefore, HDACI and HMA have become novel strategies for leukaemic
and epigenetic treatments in AML (15).
In the present study, the antileukaemia effects of
chidamide alone and in combination with 5-AZA on AML cells were
assessed. The results suggested that chidamide and its combination
with 5-AZA had a strong antileukaemic effect by inhibiting cell
proliferation and by inducing apoptosis of AML cells. These results
revealed that chidamide is a potential drug for leukaemia
treatment, particularly in combination with 5-AZA.
Materials and methods
Cell lines, primary AML samples and
reagents
The AML cell lines KG1a, Kasumi-1, NB4, OCI-AML3,
U937, MV4-11 and SKM1 were obtained from the Cell Biology Research
Institute (Chinese Academy of Sciences) (Table SI). U937 was validated by short
tandem repeat analysis. The cell lines were cultured in RPMI 1640
containing 10% foetal bovine serum (FBS) (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin
in an incubator at 37°C in a humidified atmosphere consisting of
95% air and 5% CO2.
Primary AML cells were isolated from peripheral
blood (PB) or bone marrow (BM) containing >50% blasts. These
specimens were cultured in IMDM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS, 50 ng/ml recombinant human (rh) stem cell
factor, 100 ng/ml fms-related receptor tyrosine kinase 3 (FLT3)
ligand, 25 ng/ml rhIL-3 and 10 ng/ml rhIL-6 (PeproTech, Inc.). The
present study was approved by the Research Ethics Committee of The
First Affiliated Hospital of Soochow University (Suzhou, China) and
written informed consent was provided by all subjects.
Chidamide (Chipscreen Biosciences Ltd.) was
dissolved in DMSO (Thermo Fisher Scientific, Inc.) to a
concentration of 20 mM to prepare a stock solution. The stock
solution was stored at −80°C. 5-AZA (MilliporeSigma) and Venetoclax
(MCE MedChemExpress) was respectively dissolved in DMSO to a 100 mM
concentration and kept at −80°C. The stock solutions were diluted
to working concentrations in subsequent experiments with growth
media.
Mononuclear cell separation
BM or PB samples were collected from 5 patients (P1:
Male, 57 years; P2: Male, 81 years; P3: Female, 55 years; P4:
Female, 75 years; P5: Male, 56 years) who had been diagnosed with
AML (non-acute promyelocytic leukaemia) at the First Affiliated
Hospital of Soochow University between January 2020 to May 2021
(Table SII). Mononuclear cells
were separated by using Ficoll solution (TBD Science) according to
the manufacturer's protocol (16).
Cell viability assay
The cytotoxic effects of chidamide and 5-AZA alone
(or the combination of the two drugs) on the AML cell lines were
determined via the use of the Cell Counting Kit-8 (CCK-8) assay
(Dojindo Laboratories, Inc.). Cell lines were seeded at a density
of 1–3×104/ml in 96-well flat-bottomed microtiter plates
at 100 µl/well and exposed to chidamide (0–20 µM) or 5-AZA (0–40
µM) at varying concentrations (either alone or in combination) for
72 h, after which they were subjected to a standard CCK-8 assay.
The plate was read at a wavelength of 490 nm using a microplate
reader (Bio-Rad Laboratories, Inc.).
Cell cycle analysis
The cells were treated with chidamide (1 µM) for 48
h. Subsequently, the cells were collected, washed with PBS and then
fixed overnight in 75% ice-cold ethanol at 4°C. The fixed cells
were then harvested, stained with propidium iodide (PI)/RNase A (BD
Pharmingen; BD Biosciences) and incubated in the dark at room
temperature for 30 min after being washed with PBS. The DNA content
was analyzed by flow cytometry (FCM) with an LSR2 instrument (BD
Biosciences). FlowJo 7.6 software (Tree Star, Inc.) was used for
data analysis.
Total RNA isolation and reverse
transcription-quantitative (RT-q)PCR
The cells were treated with chidamide (1 µM) for 72
h and total cellular RNA was extracted from the cells using
Direct-zol™ RNA MircoPrep (ZYMO Research Corp.) according to the
manufacturer's protocols. RNA was eluted with RNase-free water,
quantified at an absorbance at 260/280 nm and subjected to RT.
Total mRNA was reverse transcribed into cDNA using a SuperScript™IV
One-Step RT-PCR System (cat. no. 12594025; Thermo Fisher
Scientific, Inc.). Real-time qPCR was performed using TaqMan master
mix (Thermo Fisher Scientific, Inc.) with 0.2 mM TaqMan probe
(Thermo Fisher Scientific, Inc.). The real-time qPCR conditions
were as follows: 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 20
sec and 40 cycles at 95°C for 1 sec and 60°C for 20 sec. Signals
were detected with a QuantStudio 7 Flex Real-Time PCR system
(Thermo Fisher Scientific, Inc.). Relative expression levels were
determined by normalizing to GAPDH levels. GAPDH-qPCR Taqman probe
(cat. no. Hs02786624_g14453320; Thermo Fisher Scientific, Inc.) and
early growth response 1 (EGR1)-qPCR Taqman probe (cat. no.
Hs00152928_m14331182; Thermo Fisher Scientific, Inc.) were used.
The results were analyzed using the 2−ΔΔCq method, in
which ∆Cq=Cq (target gene)-Cq (internal reference) and ∆∆Cq=∆Cq
(sample)-∆Cq (control) (17). Each
sample was measured in triplicate.
Cell apoptosis assay
The effects of chidamide alone or combined with
5-AZA on apoptosis of AML cell lines were analysed by using FCM
analysis. In brief, cells were harvested, washed with cold PBS and
resuspended in 400 µl Annexin Binding Buffer. Subsequently, 1 µl
allophycocyanin (APC)-conjugated Annexin V and 5 µl PI (BD
Pharmingen; BD Biosciences) were added to each sample. Stained
samples were analysed by using FCM with an LSR2 instrument (BD
Biosciences).
AML cell differentiation analysis
AML cell lines and AML patient samples were treated
with chidamide for three days prior to analysis.
Phycoerythrin/cyanin 7-conjugated mouse anti-human CD11b monoclonal
antibody (clone ICRF44; cat. no. 301322; BioLegend, Inc.) and
APC-conjugated mouse anti-human CD86 monoclonal antibody (clone
IT2.2; cat. no. 305412; BioLegend, Inc.) were used for staining at
a 1:200 dilution (30 min at 4°C). Stained samples were analysed by
using FCM with an LSR2 instrument (BD Biosciences). The results
were then analysed with the use of FlowJo 7.6 software (TreeStar,
Inc.).
Western blot analysis
Cultured cells were harvested, washed with PBS and
then lysed in RIPA buffer. The protein lysates were clarified via
centrifugation at 12,000 × g for 30 min at 4°C and the supernatant
was collected. The protein concentration was measured by using a
BCA Protein Assay Kit (Beyotime Institute of Biotechnology). Equal
amounts (30 µg per lane) of protein were separated by using 12%
SDS/PAGE, after which they were electrotransferred onto a PVDF
membrane (MilliporeSigma). The membranes were blocked with 5%
skimmed milk (BD Biosciences) and incubated with primary antibodies
at 4°C overnight. The primary antibodies were as follows: BCL-2
(cat. no. sc-7382; dilution, 1:500; Santa Cruz Biotechnology,
Inc.), BAX (cat. no. sc-7480; dilution, 1:500; Santa Cruz
Biotechnology, Inc.), BCL-XL (cat. no. sc-8392; dilution, 1:500;
Santa Cruz Biotechnology, Inc.) and MCL-1 (cat. no. sc-12756;
dilution, 1:500; Santa Cruz Biotechnology, Inc.). GAPDH (cat. no.
97166; dilution, 1:1,000; Cell Signaling Technology, Inc.) was used
as a loading control. The membrane was then incubated with
secondary antibodies (cat. no. A0216; dilution, 1:1,000; Beyotime
Institute of Biotechnology) for 1 h at room temperature, and
visualized with the use of an enhanced chemiluminescent western
blotting detection reagent (cat. no. RPN2209; Cytiva) via an Gel
Doc™ EZ imaging system (Bio-Rad Laboratories, Inc.).
Statistical analysis
All of the experiments were performed in triplicate
wells as three independent replicates. All of the data are
presented as the mean ± standard deviation. One-way ANOVA was used
to compare multiple independent groups by GraphPad Prism 5
(GraphPad Software, Inc.). Further statistical comparisons were
performed by using Bonferroni's and Dunnett's test and the
corresponding bar charts or linear graphs were drawn by using
GraphPad Prism 5 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference. The
half-maximal inhibitory concentration (IC50) values were
calculated with GraphPad. The combination index (CI) value was
calculated from the fraction-affected value of each combination
(according to the Chou-Talalay method) by using CompuSyn software
1.0.1 (ComboSyn, Inc.).
Results
Chidamide and 5-AZA synergistically
inhibit the proliferation of AML cell lines and primary cultured
AML cells
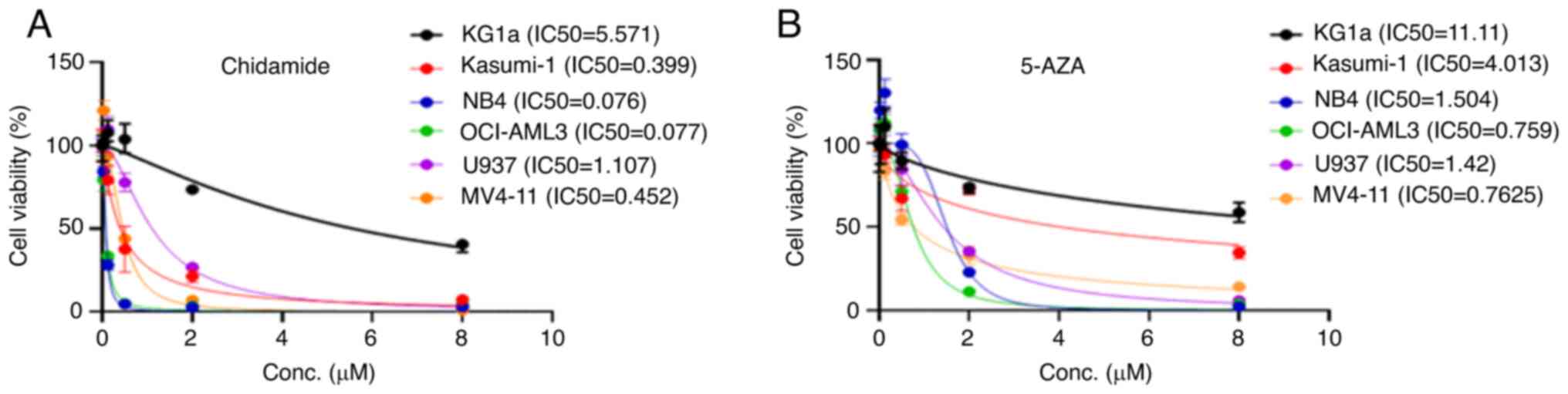
To determine the effect of chidamide and 5-AZA on
proliferation, the IC50 values for chidamide and 5-AZA
alone were first determined at different concentrations when
applied to the AML cell lines M1-M5 for 72 h. The M1-M5 cell lines
represent different AML subtypes in accordance with the
French-American-British (FAB) classification (Table SI). Chidamide and 5-AZA caused
growth inhibition of the M1-M5 cell lines in a dose-dependent
manner (Fig. 1A and B). Chidamide
markedly inhibited AML cell proliferation at low concentrations
(except for KG1a). By contrast, KG1a and Kasumi-1 cells were
insensitive to 5-AZA compared with the other cell lines.
Next, the effect of the chidamide and 5-AZA
combination on cell viability was evaluated. Cell lines were
treated with different concentrations, in accordance with the
IC50 of each drug. Chidamide plus 5-AZA inhibited AML
cell proliferation in a dose-dependent manner and the combination
had a stronger inhibitory effect than either drug alone (Fig. 2A-F). In addition, chidamide and
5-AZA (alone or in combination) had marked cytotoxic effects on the
proliferation of primary cultured AML cells from patients and the
inhibitory effect was dose-dependent (Fig. 2G and H). The calculation of the CI
value suggested that chidamide combined with 5-AZA also had a clear
synergistic effect at their suitable concentrations (Fig. 2I). Furthermore, the results of the
cell cycle analysis indicated that chidamide was able to cause G1
phase arrest in the MV4-11 cell line (Fig. S1).
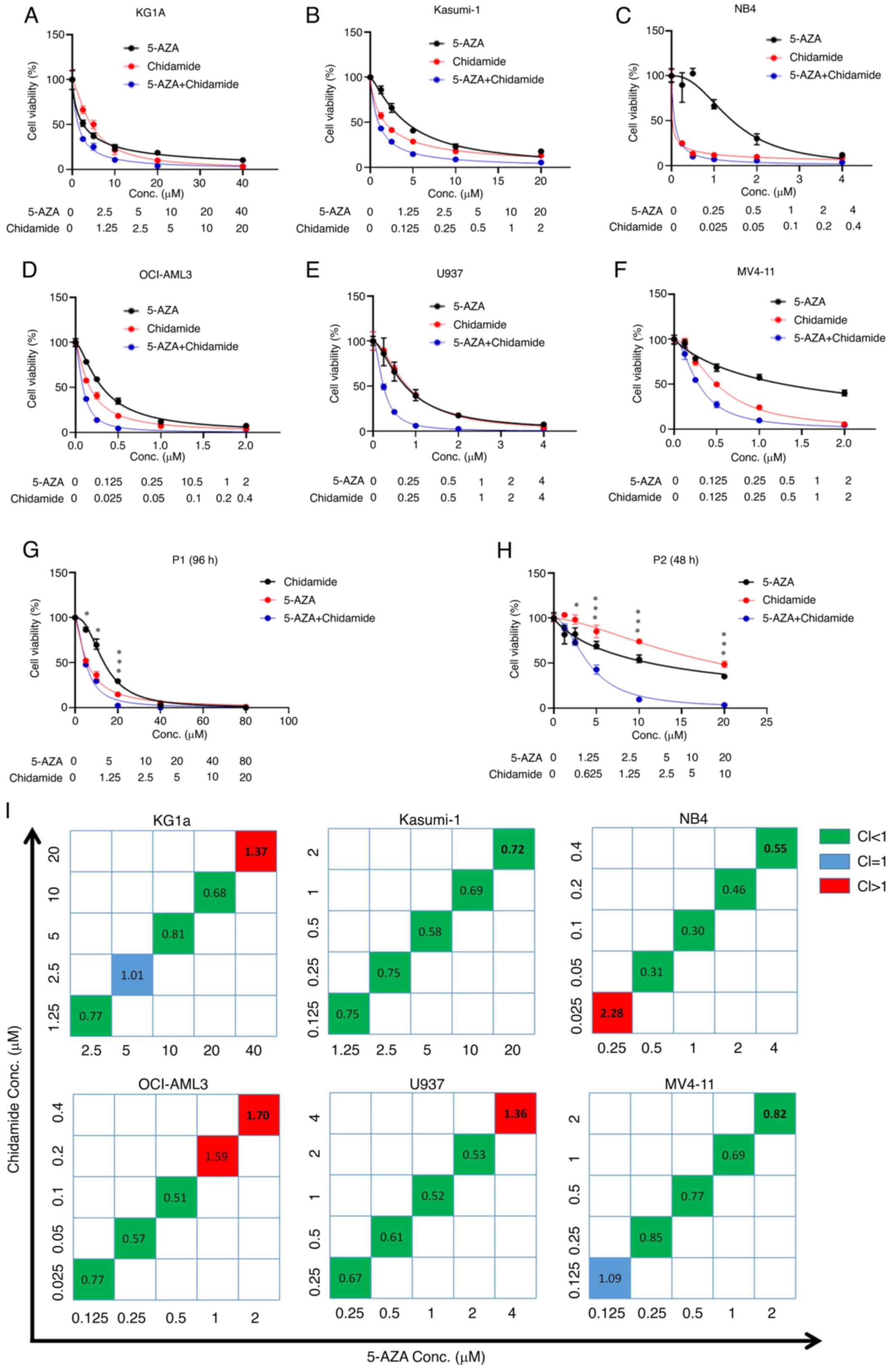 | Figure 2.Chidamide, 5-AZA, and their
combination inhibit cell proliferation in AML cells. (A) KG1a, (B)
Kasumi-1, (C) NB4, (D) OCI-AML3, (E) U937 and (F) MV4-11 cell lines
were exposed to chidamide, 5-AZA and their combination for 72 h and
the cell viability was subsequently determined by using a Cell
Counting Kit-8 assay. The curves represent the dose-dependent
effects of chidamide and 5-AZA on cell proliferation. (G and H) The
two drugs in combination (5-AZA+Chidamide) exhibited a significant
proliferation inhibition effect compared with each single drug
(5-AZA and Chidamide) in AML primary samples of (G) P1 and (H) P2.
*P<0.05, ***P<0.001. (I) CI values for the two drugs in the
KG1a, Kasumi-1, NB4, OCI-AML3, U937 and MV4-11 cell lines. The
horizontal axis represents different concentrations of 5-AZA and
the vertical axis represents different concentrations of chidamide.
The CI value reflects the degree of drug interaction: CI<1, CI=1
or CI>1 indicated synergistic, additive or antagonistic effects,
respectively. Different colours represent different CI values. P1,
patient 1; 5-AZA, 5-azacitidine; AML, acute myeloid leukaemia;
Conc., concentration; CI, combination index. |
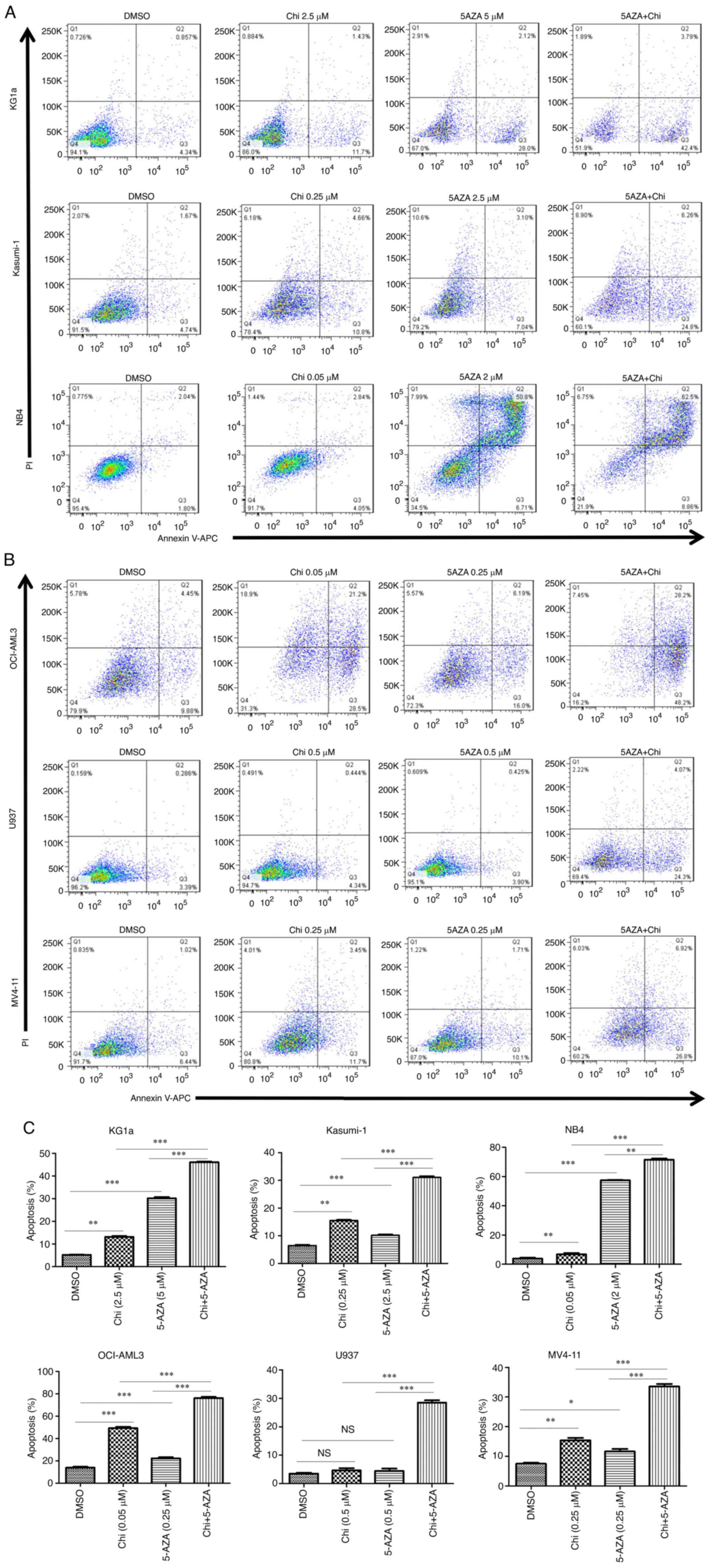
Apoptosis is significantly induced by
chidamide plus 5-AZA in AML cell lines
An FCM analysis was performed to determine the
ability of the drugs to induce apoptosis in AML cells. It was
observed that the percentage of Annexin V-positive cells
significantly increased in the M1-M5 cell line at 72 h after the
use of chidamide or 5-AZA treatment as well as with the combination
treatment (Fig. 3A and B). Of
note, treatment with chidamide plus 5-AZA induced significantly
higher rates of apoptosis than single treatments with either drug
(Fig. 3C). The results also
suggested that chidamide plus 5-AZA not only increased the early
apoptotic population (Annexin V+/PI−) in
KG1a, Kasumi-1, U937 and MV4-11 cells [as well as the late
apoptotic population (Annexin V+/PI+) in NB4
cells], but also caused increases in both early and late apoptotic
populations in OCI-AML3 cells.
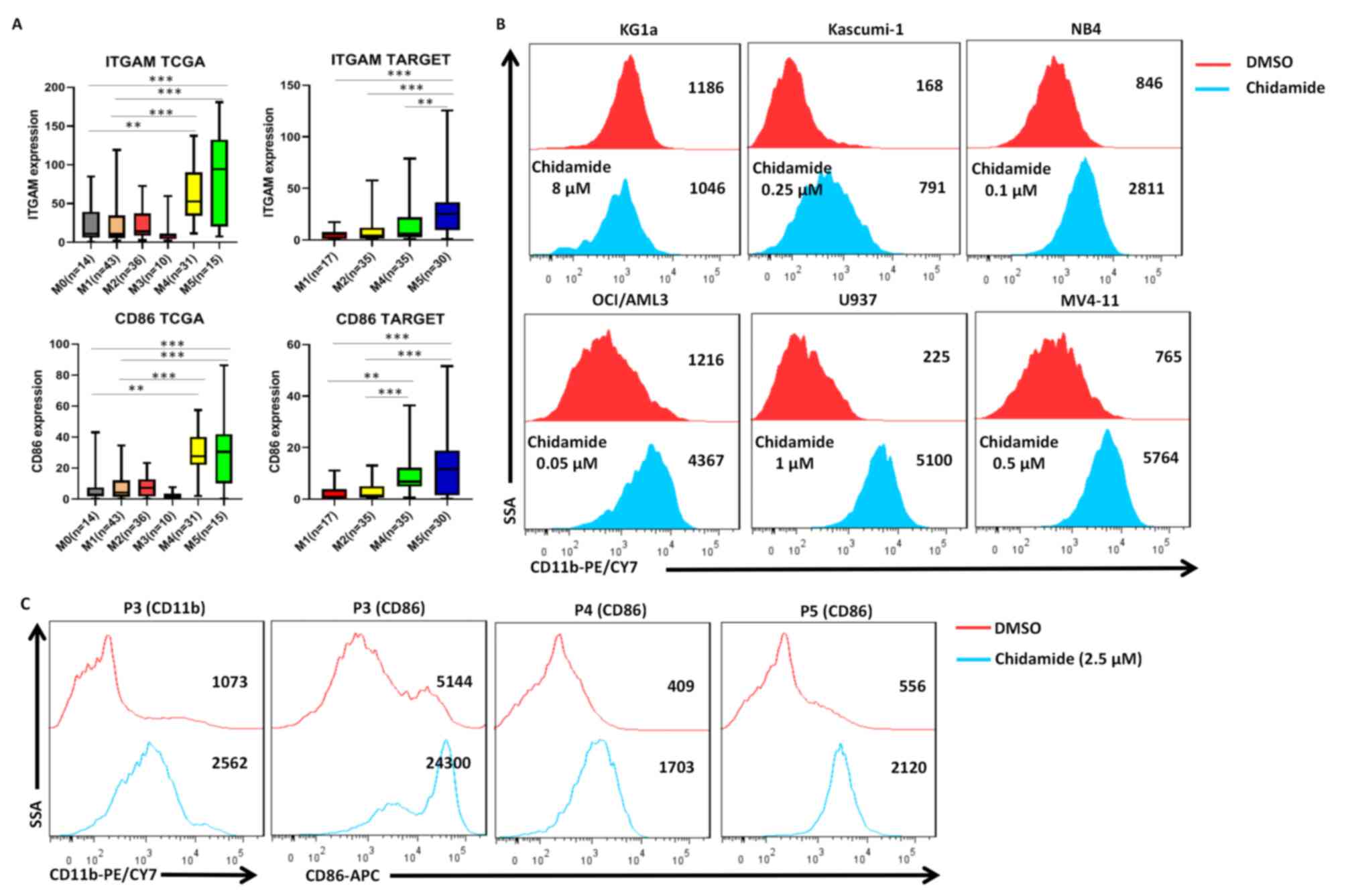
Chidamide induces the differentiation
of AML cell lines and primary AML samples
To investigate the effect of chidamide on AML cell
differentiation, the ability of the drug to upregulate the
differentiation markers CD11b (integrin subunit αM) and CD86 was
assessed in AML cells. CD11b is a myeloid differentiation marker
and CD86 is a monocytic/dendritic differentiation marker. Analysis
of The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) and TAGERT (https://ocg.cancer.gov/programs/target)
databases indicated that from the M1 to M5 subtypes of AML, the
expression levels of CD11b and CD86 gradually increased at
different stages of cell differentiation and maturation (Fig. 4A). Chidamide induced the expression
of CD11b in Kasumi-1, NB4, OCI-AML3, U937 and MV4-11 cells
(Fig. 4B). Chidamide also
upregulated the expression of CD86 (4-fold increase) in primary AML
cells but the effect on CD11b (2-fold increase) expression was only
slight (Fig. 4C). To investigate
the mechanism of differentiation, additional experiments were
performed, which indicated that chidamide upregulates the
expression of EGR-1 in U937 cells (Fig. S2).
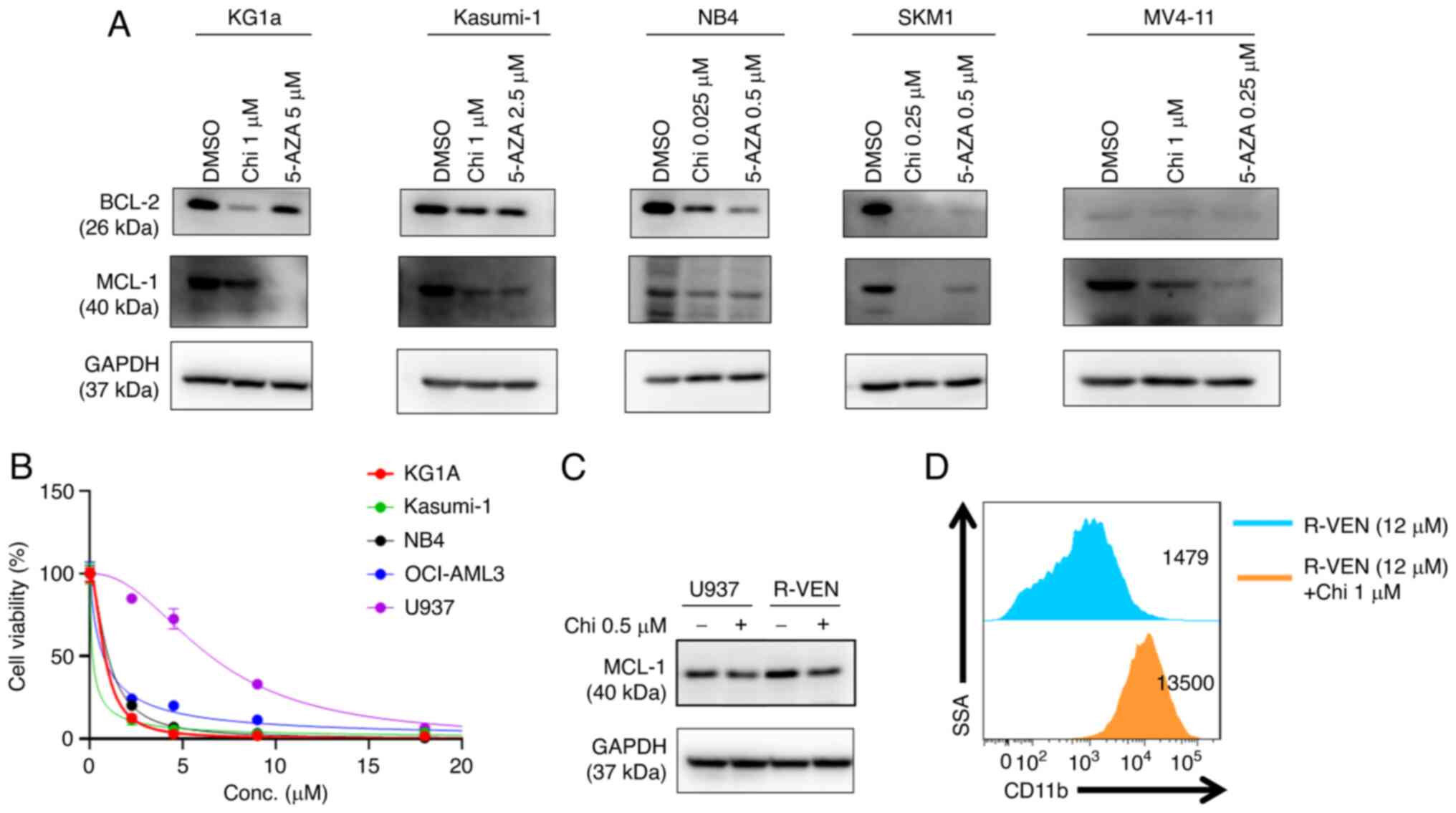
Chidamide and 5-AZA downregulate the
antiapoptotic proteins BCL-2 and MCL-1 in AML cell lines
BCL-2 family proteins are critical regulators of
apoptotic pathways. To understand the molecular mechanism
underlying chidamide- and 5-AZA-induced apoptosis, western blot
assays were performed, which indicated that AML cell lines
exhibited decreased levels of antiapoptotic proteins (BCL-2 and
MCL-1) in response to the single drug treatments after 72 h
(Fig. 5A). Chidamide also
downregulated the expression of BCL-XL and upregulated BAX in
MV4-11 and Kasumi-1 (Fig.
S3).
Chidamide downregulates BCL-2 and
induces the differentiation of the venetoclax-resistant U937 cell
line
Venetoclax markedly inhibited the proliferation of
different AML cell lines (M1-M5) at low concentrations (except for
U937; IC50=6 µM) (Fig.
5B); thus, the U937 cell line was selected for further
analysis. Next, a venetoclax-resistant U937 cell line (R-VEN) was
established by gradually increasing the concentration of venetoclax
to 12 µM in vitro. Western blot analysis suggested that the
expression of MCL-1 protein increased in R-VEN; however, exposure
to chidamide led to a modest reduction in MCL-1 expression in R-VEN
cells (Fig. 5C). Furthermore,
chidamide upregulated the expression of CD11b in the R-VEN cell
line (Fig. 5D).
Discussion
From the 1970s until 2017, the progress in the
frontline treatment of AML was limited. In other words, since the
establishment of ‘7+3′ chemotherapy (cytarabine and anthracycline)
in the mid-1970s, the development of agents for patients with AML
has proven to be a daunting challenge (18). With improvements in supportive
care, long-term survival has improved in younger patients with AML,
with a 5-year overall survival (OS) in the range of 40–50%.
However, among patients above the age of 60 years who make up the
bulk of AML cases, the long-term outlook is dismal, with a 5-year
OS of 10–20% (19). Therefore,
there remains to be a clear requirement for newer therapies and a
more individualized approach for the treatment of AML.
Genes that may be silenced by abnormal methylation
or acetylation modifications are promising targets for cancer
therapies, particularly in AML (20). In clinical practice, using HDACIs
and HMAs in combination or with other antitumour drugs may be an
effective antileukaemia strategy. To explore this, clinical trials
of HMA combined with HDACI or other drugs for AML have been
performed (21–23). HDACI plus HMA has already
demonstrated promising effects in the clinical treatment of AML
(11,24,25).
In the present study, different types of AML cell
lines were used as models, and chidamide and 5-AZA alone or in
combination inhibited the proliferation of the M1-M5 AML cell lines
in a dose-dependent manner. Different cell lines represent
different subtypes of the AML FAB classification. MV4-11 represent
patients harbouring the FLT3-internal tandem duplication mutation,
which has the highest mutation frequency in AML. When compared with
other AML cell lines, both drugs had a weak effect on the growth
inhibition of KG1A cells and the IC50 values of
chidamide and 5-AZA were 5.57 and 11.11 µM, respectively. The
reason may be the slow growth rate of the cell itself; therefore,
it is not sensitive to chemotherapy drugs. However, it remains
elusive why Kasumi-1 was insensitive to 5-AZA compared with other
cell lines; this may possibly be due to special cytogenetics
(chromosome and gene mutations). When the two drugs were combined,
the inhibitory effect on proliferation was more potent than that of
a single drug and the significant synergistic effect was confirmed
by the CI value at certain suitable concentrations. It was also
observed that chidamide and 5-AZA alone or in combination markedly
inhibited the proliferation of AML cells from patient samples;
however, the time-point of the test was important, as it was
observed that 72 h was too toxic to cells and the synergistic
effect was not obvious, and the incubation time was thus decreased
to 24 h, at which the synergistic effect was more obvious. In the
subsequent experiment, the minimum value of the CI was not selected
as the combination concentration, but the intermediate
concentration combination was chosen for apoptosis in order to
obtain a significant difference in the results. When measuring
differentiation, a concentration close to the IC50 was
selected, as changes in the expression rate of CD11b were most
obvious with this concentration. Meanwhile, the chidamide-induced
cell death did not affect the results of the differentiation
assay.
Numerous studies have confirmed that HDACI induces
cell cycle arrest in G0/G1 phase in vitro (13,26,27).
The experimental results of the present study suggested that
chidamide is able to cause G1 phase arrest. HDACI and HMA are able
to overcome drug resistance by reactivating silenced tumour
suppressor genes and inducing cell cycle arrest (28). When chidamide was combined with
5-AZA, chidamide potently and synergistically enhanced the
cytotoxicity in AML cell lines. This synergistic cytotoxicity was
obvious, as the two agents were able to cooperatively induce cell
apoptosis. The present study indicated that the single drug-induced
inhibition was incomplete; however, when chidamide plus 5-AZA was
added, the inhibition was significantly improved. Different cell
lines had different apoptosis types; specifically, certain cell
lines exhibited early apoptosis, while others exhibited late
apoptosis, and certain cells exhibited both types of apoptosis.
The FAB classification system is based on morphology
for defining specific immunotypes (29). From M1 to M5, the maturity of cells
increased and the expression levels of CD11b and CD86 also tended
to increase, which was also confirmed by the results from the TCGA
and TARGET databases. CD11b is considered a myeloid differentiation
marker of leukaemia cells (30).
For instance, the expression of CD11b may be significantly
upregulated when AML-M3 cells are treated with ATRA and a similar
change may also be observed in U937 cells treated with Brequinar
(31). CD86 is considered a
monocytic/dendritic differentiation marker (32). The present results indicated that
the expression of CD11b on Kasumi-1, NB4, OCI-AML3, U937 and MV4-11
cells was upregulated (except for KG1a cells) after treatment with
chidamide. Perhaps KG1a has different biological characteristics
from other AML cell lines, thus making them more primitive and more
difficult to differentiate, based on drug induction. Further
research on the combined effect of chidamide and 5-AZA on the
differentiation of AML cells requires to be performed. EGR-1 is a
differentiation-related transcription factor. The results of the
present study indicated that chidamide was able to upregulate the
expression of EGR-1 in U937. However, when primary cultured cells
from patients with AML were treated with chidamide in vitro,
it was observed that both CD11b and CD86 were upregulated, thus
indicating that the cells further differentiated into
monocytic/dendritic cells. As most patients are diagnosed with M4
or M5, they are more likely to exhibit monocytic/dendritic
differentiation. It was speculated that this may be related to the
subtypes of AML patients, which requires further exploration.
BCL-2 family proteins are important for cell fate,
as they may regulate cells in two ways: Anti- and pro-apoptosis
(33,34). Our previous research also found
that chidamide is able to induce apoptosis in AML cell lines via
antiapoptotic BCL-2 family proteins (BCL-2 and MCL-1) (35). However, in the present study, no
significant synergistic effect of chidamide combined with 5-AZA to
downregulate the BCL-2 and MCL-1 was observed (data not shown). In
addition, chidamide was also able to downregulate the expression of
BCL-XL and upregulate BAX in MV4-11 and Kasumi-1. Various HDACIs
(in combination with decitabine) shortened tumour cell survival at
the mRNA and protein levels (36).
The results of the present study demonstrated that chidamide and
5-AZA alone were able to degrade the antiapoptotic proteins BCL-2
and MCL-1 in several AML cell lines. Of note, it was observed that
the expression of MCL-1 was compensatorily increased in the
venetoclax-resistant U937 cell line (35). Chidamide degraded MCL-1 in
venetoclax-resistant U937 cells and upregulated the expression of
CD11b, and venetoclax slightly promoted CD11b expression in
venetoclax-resistant U937 cells as compared with U937 (data not
shown). There is reason to believe that the downregulation of MCL-1
may be a promising strategy for the treatment of
venetoclax-resistant leukaemia. Further investigations will be
performed in in vivo models (e.g. PDX animal models or an
increased number of patient samples).
Other studies have reported that chidamide is able
to degrade the AML1-ETO fusion gene and also inhibit leukaemia
cells with mixed lineage leukemia (MLL) rearrangements (37,38).
Chidamide may become a novel option in the treatment of certain
subtypes of AML, such as t(8;21) and MLL rearrangement, in the
future.
It has been demonstrated that antitumour drugs have
multiple functions and targets. HDACI combined with 5-AZA may have
multiple pathways for inducing cell apoptosis, differentiation and
proliferation inhibition. In conclusion, chidamide in combination
with 5-AZA has a synergistic antileukaemia effect in vitro
and may be considered a therapeutic strategy for AML.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Social
Development-Clinical Frontier Project of Jiangsu Province (grant
no. BE2018652), the National Natural Science Foundation of China
(grant no. 81970138), Translational Research Grant of the National
Clinical Research Center for Hematology (grant no. 2020ZKMB05) and
the Project of State Key Laboratory of Radiation Medicine and
Protection (grant no. GZK1202002).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HYQ contributed conceptualization, funding
acquisition, supervision, resources, study design and confirmed the
authenticity of all the raw data. SLX contributed to
conceptualization, funding acquisition, supervision, resources,
study design and confirmed the authenticity of all the raw data. ZL
designed and performed the experiments. JZ, MZ and JLL performed
the experiments. QCQ and JHF analyzed data. ZL wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The First
Affiliated Hospital of Soochow University (Suzhou, China) and all
patients provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bhatla T, Wang J, Morrison DJ, Raetz EA,
Burke MJ, Brown P and Carroll WL: Epigenetic reprogramming reverses
the relapse-specific gene expression signature and restores
chemosensitivity in childhood B-lymphoblastic leukemia. Blood.
119:5201–5210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roy DM, Walsh LA and Chan TA: Driver
mutations of cancer epigenomes. Protein Cell. 5:265–296. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun Y, Chen BR and Deshpande A: Epigenetic
regulators in the development, maintenance, and therapeutic
targeting of acute myeloid leukemia. Front Oncol. 8:412018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Figueroa ME, Lugthart S, Li Y,
Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J,
van Putten W, Skrabanek L, et al: DNA methylation signatures
identify biologically distinct subtypes in acute myeloid leukemia.
Cancer Cell. 17:13–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang Y, Dunbar A, Gondek LP, Mohan S,
Rataul M, O'Keefe C, Sekeres M, Saunthararajah Y and Maciejewski
JP: Aberrant DNA methylation is a dominant mechanism in MDS
progression to AML. Blood. 113:1315–1325. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fey MF and Buske C; ESMO Guidelines
Working Group, : Acute myeloblastic leukaemias in adult patients:
ESMO clinical practice guidelines for diagnosis, treatment and
follow-up. Ann Oncol. 24 (Suppl 6):vi138–vi143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fenaux P, Haase D, Santini V, Sanz GF,
Platzbecker U and Mey U; ESMO Guidelines Committee. Electronic
address: Clinicalguidelines@esmo.org: Myelodysplastic syndromes, :
ESMO clinical practice guidelines for diagnosis, treatment and
follow-up†☆. Ann Oncol. 32:142–156. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seto E and Yoshida M: Erasers of histone
acetylation: The histone deacetylase enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan P, Wei A, Mithraprabhu S, Cummings N,
Liu HB, Perugini M, Reed K, Avery S, Patil S, Walker P, et al: Dual
epigenetic targeting with panobinostat and azacitidine in acute
myeloid leukemia and high-risk myelodysplastic syndrome. Blood
Cancer J. 4:e1702014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Viré E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden
JM, et al: The polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and differentiation in
human leukaemia cells. Biochem J. 443:735–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Zhang P and Liu Y: Chidamide
tablets: HDAC inhibition to treat lymphoma. Drugs Today (Barc).
53:167–176. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu F, Guo H, Shi M, Liu S, Wei M, Sun K
and Chen Y: A combination of low-dose decitabine and chidamide
resulted in synergistic effects on the proliferation and apoptosis
of human myeloid leukemia cell lines. Am J Transl Res.
11:7644–7655. 2019.PubMed/NCBI
|
|
16
|
Jaatinen T and Laine J: Isolation of
mononuclear cells from human cord blood by ficoll-paque density
gradient. Curr Protoc Stem Cell Biol Chapte. Jun 1–2007.(Epub ahead
of print). doi: 10.1002/9780470151808.sc02a01s1.
|
|
17
|
Ambavaram MM and Pereira A: Setting up
reverse transcription quantitative-PCR experiments. Methods Mol
Biol. 678:45–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pollyea DA: Therapeutic advances in
first-line management of acute myeloid leukemia. J Natl Compr Canc
Netw. 17:1441–1443. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European LeukemiaNet. Blood. 115:453–474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ganesan A, Nolan L, Crabb SJ and Packham
G: Epigenetic therapy: Histone acetylation, DNA methylation and
anti-cancer drug discovery. Curr Cancer Drug Targets. 9:963–981.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anzai H, Frost P and Abbruzzese JL:
Synergistic cytotoxicity with 2′-deoxy-5-azacytidine and topotecan
in vitro and in vivo. Cancer Res. 52:2180–2185. 1992.PubMed/NCBI
|
|
22
|
Garcia-Manero G, Kantarjian HM,
Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M,
Wierda WG, Ravandi F, Koller C, et al: Phase 1/2 study of the
combination of 5-aza-2′-deoxycytidine with valproic acid in
patients with leukemia. Blood. 108:3271–3279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Najem SA, Khawaja G, Hodroj MH, Babikian P
and Rizk S: Adjuvant epigenetic therapy of decitabine and
suberoylanilide hydroxamic acid exerts anti-neoplastic effects in
acute myeloid leukemia cells. Cells. 8:14802019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bewersdorf JP, Shallis R, Stahl M and
Zeidan AM: Epigenetic therapy combinations in acute myeloid
leukemia: What are the options? Ther Adv Hematol.
10:20406207188166982019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lübbert M and Kuendgen A: Combining DNA
methyltransferase and histone deacetylase inhibition to treat acute
myeloid leukemia/myelodysplastic syndrome: Achievements and
challenges. Cancer. 121:498–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Z, Ding K, Li L, Liu H, Wang Y, Liu C
and Fu R: A novel histone deacetylase inhibitor Chidamide induces
G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed
Pharmacother. 83:1032–1037. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sandor V, Senderowicz A, Mertins S,
Sackett D, Sausville E, Blagosklonny MV and Bates SE: P21-dependent
g(1)arrest with downregulation of cyclin D1 and upregulation of
cyclin E by the histone deacetylase inhibitor FR901228. Br J
Cancer. 83:817–825. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vijayaraghavalu S, Dermawan JK, Cheriyath
V and Labhasetwar V: Highly synergistic effect of sequential
treatment with epigenetic and anticancer drugs to overcome drug
resistance in breast cancer cells is mediated via activation of p21
gene expression leading to G2/M cycle arrest. Mol Pharm.
10:337–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hassan K, Bukhari KP, Zafar A, Malik MZ
and Akhtar MJ: Acute leukaemia in children-French-American-British
(FAB) classification and its relation to clinical features. J Pak
Med Assoc. 42:29–31. 1992.PubMed/NCBI
|
|
30
|
Heo SK, Noh EK, Yoon DJ, Jo JC, Koh S,
Baek JH, Park JH, Min YJ and Kim H: Rosmarinic acid potentiates
ATRA-induced macrophage differentiation in acute promyelocytic
leukemia NB4 cells. Eur J Pharmacol. 747:36–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sykes DB, Kfoury YS, Mercier FE, Wawer MJ,
Law JM, Haynes MK, Lewis TA, Schajnovitz A, Jain E, Lee D, et al:
Inhibition of dihydroorotate dehydrogenase overcomes
differentiation blockade in acute myeloid leukemia. Cell.
167:171–186 e15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Re F, Arpinati M, Testoni N, Ricci P,
Terragna C, Preda P, Ruggeri D, Senese B, Chirumbolo G, Martelli V,
et al: Expression of CD86 in acute myelogenous leukemia is a marker
of dendritic/monocytic lineage. Exp Hematol. 30:126–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krishna S, Low IC and Pervaiz S:
Regulation of mitochondrial metabolism: Yet another facet in the
biology of the oncoprotein Bcl-2. Biochem J. 435:545–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ebrahim AS, Sabbagh H, Liddane A, Raufi A,
Kandouz M and Al-Katib A: Hematologic malignancies: Newer
strategies to counter the BCL-2 protein. J Cancer Res Clin Oncol.
142:2013–2022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang BR, Wan CL, Liu SB, Qiu QC, Wu TM,
Wang J, Li YY, Ge SS, Qiu Y, Shen XD, et al: A combined histone
deacetylases targeting strategy to overcome venetoclax plus
azacitidine regimen resistance in acute myeloid leukaemia: Three
case reports. Front Oncol. 11:7979412021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaminskyy VO, Surova OV, Vaculova A and
Zhivotovsky B: Combined inhibition of DNA methyltransferase and
histone deacetylase restores caspase-8 expression and sensitizes
SCLC cells to TRAIL. Carcinogenesis. 32:1450–1458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Al- Harbi S, Aljurf M, Mohty M, Almohareb
F and Ahmed SOA: An update on the molecular pathogenesis and
potential therapeutic targeting of AML with
t(8;21)(q22;q22.1);RUNX1-RUNX1T1. Blood Adv. 4:229–238. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ahmad K, Katryniok C, Scholz B, Merkens J,
Löscher D, Marschalek R and Steinhilber D: Inhibition of class I
HDACs abrogates the dominant effect of MLL-AF4 by activation of
wild-type MLL. Oncogenesis. 3:e1272014. View Article : Google Scholar : PubMed/NCBI
|