Introduction
Rhabdomyosarcoma (RMS) is an extremely aggressive
paediatric tumour that can occur in any part of the body. The
embryonal (ERMS) and alveolar (ARMS) histological subtypes of RMS
are differentiated on the basis of distinct genetic alterations
that may play a role in the pathogenesis of these tumours (1,2). The
most common alterations are represented by p53 and K-Ras or N-Ras
mutations (3–5). Ras is a small GTP-binding protein
that lies upstream of several signalling pathways, including
Raf/MEKs/ERKs and PI3K/AKT, both of which play the role of
mediators in cell survival and proliferation (5–8). The
abnormal expression of Ras leads to the upregulation of these
pathways and, consequently, to tumorigenesis (9). The constitutive activation of ERKs,
which results from Ras mutations in RMS cells, leads to the reduced
capacity of myodifferentiation due to the inhibition of p38
signalling (10), which is known
to regulate the expression of specific skeletal muscle genes
(11,12). Indeed, MEK/ERK inhibition in RD
cells has been shown to induce the p38-dependent rescue of the
myogenic program that is also related to a marked c-Myc
downregulation (13,14). The deregulated expression of Myc
family proteins (c-, L-, N-) has been found to play a pivotal role
in the transformation from the normal phenotype to the malignant
phenotype. Indeed, Myc family members are aberrantly activated in a
wide range of human haematological malignancies and solid tumours
(15). In particular, the c-Myc
transcription factor controls numerous cellular functions,
including cell cycle progression, cell growth, genomic instability,
angiogenesis and apoptosis (16),
and exerts a positive and negative effect on differentiation
(17,18), thereby proving to be at a
crossroads of several signalling pathways.
The functional interaction between Ras and Myc has
long been known to enhance the accumulation of transcriptionally
active Myc (19). Since MEK/ERK
inhibition in ERMS plays an anti-oncogenic and pro-myogenic role
(14,20) and MEK/ERK inhibitors affect p38
MAPK (13), the present study
aimed to investigate whether p38 reverses the transformed phenotype
by inducing Myc downregulation in the ERMS cell system. The
upstream activators of p38 are MKK6 and the closely related MKK3
(21). It is noteworthy that all
p38 MAPKs are common substrates of MKK6 and MKK3 kinases. However,
p38 activation by one or more MKK kinases is dependent on the type
of stimulus, specific cell type and strength of the stimulus
(22). Notably, both MKK3 and MKK6
play a dual role by either promoting or suppressing cancer
(23).
Given the potential involvement of MKK3 and MKK6 in
myogenic fate, the ambiguity of their specific roles, the balance
between MKK3 and MKK6, as well as the interplay with mutant p53 in
directing the final biological outcome, the present study aimed to
investigate the effects of MKK6 and MKK3 kinases in the
anti-oncogenic function and myogenic differentiation induced by
MEK/ERK inhibition in ERMS cellular models. Herein, it is
demonstrated that MEK/ERK inhibitors concomitantly decrease Myc
expression and induce myogenic differentiation through MKK6/p38/AKT
pathways. The specific AKT1 isoform is essential for initiation of
differentiation and myoblast mobility, while AKT2 has been proven
to be essential for myotube maturation (24). Of note, it has been demonstrated
that during myogenesis, the p38 pathway activation involves the
concurrent activation of AKT. For all these reasons, the present
study investigated whether AKT activation is induced during the
pathological myogenesis of ERMS-derived cells and examined its
association with p38 activation (25). More importantly, the present study
demonstrates that MEK/ERK inhibitors restore MKK6/p38 and AKT
pathway activation, which is a novel finding in RMS tumours and, as
it occurs in normal myogenesis and in later stages of myotubes
development, AKT is activated (24). Lastly, these results are in
accordance with previous findings on the mechanisms leading to
myogenic differentiation in embryonic development and in adult
muscle (26).
Materials and methods
Cell culture and treatments
The ERMS RD (cat. no. CCL-136™; ATCC)
cell line was tested and authenticated by ATCC for the expression
of myoglobin and myosin ATPase cellular products. The ERMS TE671
(cat. no. HTL97021) cell line was obtained from the Interlab Cell
Line Collection in 2006. The TE671 cells, hereafter indicated as TE
cells, were tested and authenticated by the Interlab Cell Line
Collection for the expression of nicotinic acetylcholine receptor,
acetylcholine receptor and peripheral type benzodiazepine receptor.
The cells were cultured in Dulbeccos' modified Eagles' medium
(DMEM), supplemented with glutamine, gentamicin (Gibco; Thermo
Fisher Scientific, Inc.) and 10% heat-inactivated foetal bovine
serum (HyClone; Cytiva). All cell lines were maintained at 37°C in
5% CO2.
At 1 day after plating, the cells were treated with
5 µM SB203580 (cat. no. S8307; MilliporeSigma) for 1 day, 10 µM
U0126 MEK inhibitor for 3 h, overnight (O/N), 1 day or 3 days (cat.
no. V1121; Promega Corporation) or 10 nM trametinib for 3 h,
overnight (O/N) or 3 days (cat. no. SC-364639; Santa Cruz
Biotechnology, Inc.).
Plasmid transfection
Cells were seeded at 1,5×106 cells/well
in 6-well plates. At 1 day after plating, the RD or TE cells were
transfected with 4 µg/well of specific plasmid using Lipofectamine
2000® (cat. no. 11668019; Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
following plasmids were used: CMV (cat. no. 16440; Addgene, Inc.),
MKK3-Ala (dnMKK3, cat. no. 14669; Addgene, Inc.), MKK3-Glu (caMKK3,
cat. no. 14670; Addgene, Inc.), expressing dominant negative and
constitutively active MKK3 isoforms, respectively; the MKK6-EE
(caMKK6-EE) constitutively active form of MKK6 was a gift from
Professor Puri Pier Lorenzo (Development, Aging and Regeneration
Program, Sanford Burnham Prebys Medical Discovery Institute, La
Jolla, CA, USA) (10). Plasmids
expressing shRNA specific for p38α knockdown and puromycin
resistance for transfected cell selection were obtained from
OriGene Technologies, Inc. shRNAs were cloned in the pRS plasmid
under the U6 promoter, with puromycin as selectable marker and
ampicillin as bacterial resistance. A combination of two different
shRNAs for p38 (shp38) was used (cat. no. TR320309), specifically
‘C’ with the sequence, 5′-CAGTGACTTTACAGGAGGTTGTGGATGCT-3′, and ‘D’
with the sequence, 5′-CCAGTAGTCAGAAGCAGGTTCTTGATGTC-3′. As a
negative control, non-effective shRNA was used [scramble (SCR),
cat. no. TR30012] with the sequence,
5′-GCACTACCAGAGCTAACTCAGATAGTACT-3′. To enrich the cell populations
in transfected cells, p-BABE-puro (cat. no. 1764-DNA.cg; Addgene,
Inc.) was co-transfected at a 1:3 ratio with plasmids lacking
puromycin resistance in order to perform 2–3 days of selection to
remove untransfected cells. Following 6–8 h of transfection, the
cells were cultured in complete medium for one night before medium
containing 2.5 µg/ml of puromycin (cat. no. P7225-25MG;
MilliporeSigma) was added for selection purposes. Semi-stable cell
lines were obtained at the end of the puromycin treatment.
Total lysate preparation and western
blot analysis
Total lysates were obtained after scraping the RD or
TE cells in RIPA buffer, modified as follows: 20 mM Tris HCl pH
7.6, 140 mM NaCl, 0.5% IGEPAL (NP40), 2 mM EDTA, 0.5% DOC and 0.5%
SDS supplemented with protease and phosphatase inhibitor (cat. no.
11836153001 and cat. no. 04906845001, respectively; Roche
Diagnostics), sonicated for 30 sec. Following Lowry or Bradford
quantification, 50–100 µg of lysates were processed for western
blot analysis. Total proteins were separated on 8, 10 or 12% SDS
PAGE and blotted onto nitrocellulose (cat. no. 16533; Schleicher
& Schuell GmbH) or PVDF membranes (cat. no. 10600029; Amersham;
Cytiva). Filters were blocked with 5% non-fat dry milk or 3% BSA
for 1 h at room temperature and incubated overnight at 4°C with the
following primary antibodies: Anti-Myc (cat. no. sc-40; 1:300),
anti-ERK-PO4 E-4 (cat. no. sc-7383; 1:500), anti-ERK1/2 C-9 (cat.
no. sc-514302; 1:500), anti-p38-PO4 (cat. no. sc-166182; 1:1,000),
anti-p38 (cat. no. sc-535; 1:500), anti-MKK3 (cat. no. sc-961;
1:500), anti-cyclin D1 (cat. no. sc-20044; 1:1,000), anti-p21 (cat.
no. sc-6246; 1:200), anti-GAPDH (cat. no. sc-47724; 1:500) and
anti-tubulin (cat. no. sc-5286; 1:500) (all from Santa Cruz
Biotechnology, Inc.); anti MKK6-PO4 D8E9 (MEK3/6) (cat. no. 12280;
1:1,000), anti-MKK6 D31D1 (cat. no. 8550; 1:1,000), anti-AKT-PO4
Thr308 (cat. no. 4056; 1:1,000), anti-AKT-PO4 Ser473 (cat. no.
9271; 1:1,000) and AKT (cat. no. 9272; 1:1,000) (all from Cell
Signalling Technology, Inc.); anti-myosin heavy chain (MHC; cat.
no. MF20; 1:300) and anti-myogenin (cat. no. F5D; 1:300) were
monoclonal from hybridoma supernatant (all from Developmental
Studies Hybridoma Bank). Filters were then incubated for 1 h at
room temperature in 2% non-fat dry milk or 3% BSA with the
following secondary antibodies: peroxidase-conjugate sheep
anti-mouse (cat. no. A90-146B; 1:2,000) or donkey anti-rabbit IgG
(cat. no. A120-108B; 1:2,000) (both from Bethyl Laboratories,
Inc.). Immunocomplexes were detected by means of ECL Chemidoc XRS+
acquisition (Bio-Rad Laboratories, Inc.). All experiments were
performed three times unless otherwise indicated. Representative
western blot images are shown and densitometric analysis was
performed using ImageJ software version 1.53k (National Institutes
of Health) and the results are reported in each respective figure
as the mean ± standard deviation (SD).
Cell proliferation assay
The changes in the proliferative potential of RD
cells (overexpressing CMV, caMKK3 or caMKK6, or transfected with
shp38 or SCR shRNA) were analysed. Specifically, RD cells
overexpressing caMKK6 were treated with or without SB203580, whilst
p38-silenced (shp38) RD cells were treated with or without U0126.
At the end of puromycin selection of transfected cells or the
specific treatments, cells were counted using the trypan blue (cat.
no. T10282; Invitrogen; Thermo Fisher Scientific, Inc.) exclusion
method according to the manufacturer's instructions. Briefly, the
cell suspension was added to trypan blue stain in a 1:1 mixture and
incubated for 2 min at room temperature. The results are plotted as
the mean ± SD of three independent transfections.
Cell morphology and
immunofluorescence
To observe the morphological changes in RD cells
under the different experimental conditions [overexpressing caMKK3
or caMKK6 genes, overexpressing caMKK6, and treated with or without
SB203580, p38-silenced (shp38) treated with or without U0126], the
specific samples were photographed under a phase contrast
microscope (Nikon Eclipse TS100; Nikon Corporation) at ×20
magnification.
For immunofluorescence assays, the cells were fixed
in 4% paraformaldehyde for 10 min at room temperature and washed;
non-specific binding sites were blocked with 3% BSA in PBS for 20
min at room temperature. The cells were then incubated for 1 h at
room temperature with a 1:100 dilution of the anti-MHC (cat. no.
MF20), or anti-Myc monoclonal antibody (cat. no. sc-40; 1:100).
After rinsing with PBS, the cells were incubated with anti-mouse
IgG-Cy3 (cat. no. A90-516C3) or anti-mouse IgG-Cy2 (cat. no.
A90-516C2) antibodies (all from Bethyl Laboratories, Inc.) and DAPI
(MilliporeSigma). Staining was visualised on a Zeiss Axioskop 2
Plus microscope (Carl Zeiss AG). The experiments were performed
twice.
Bioinformatics analysis
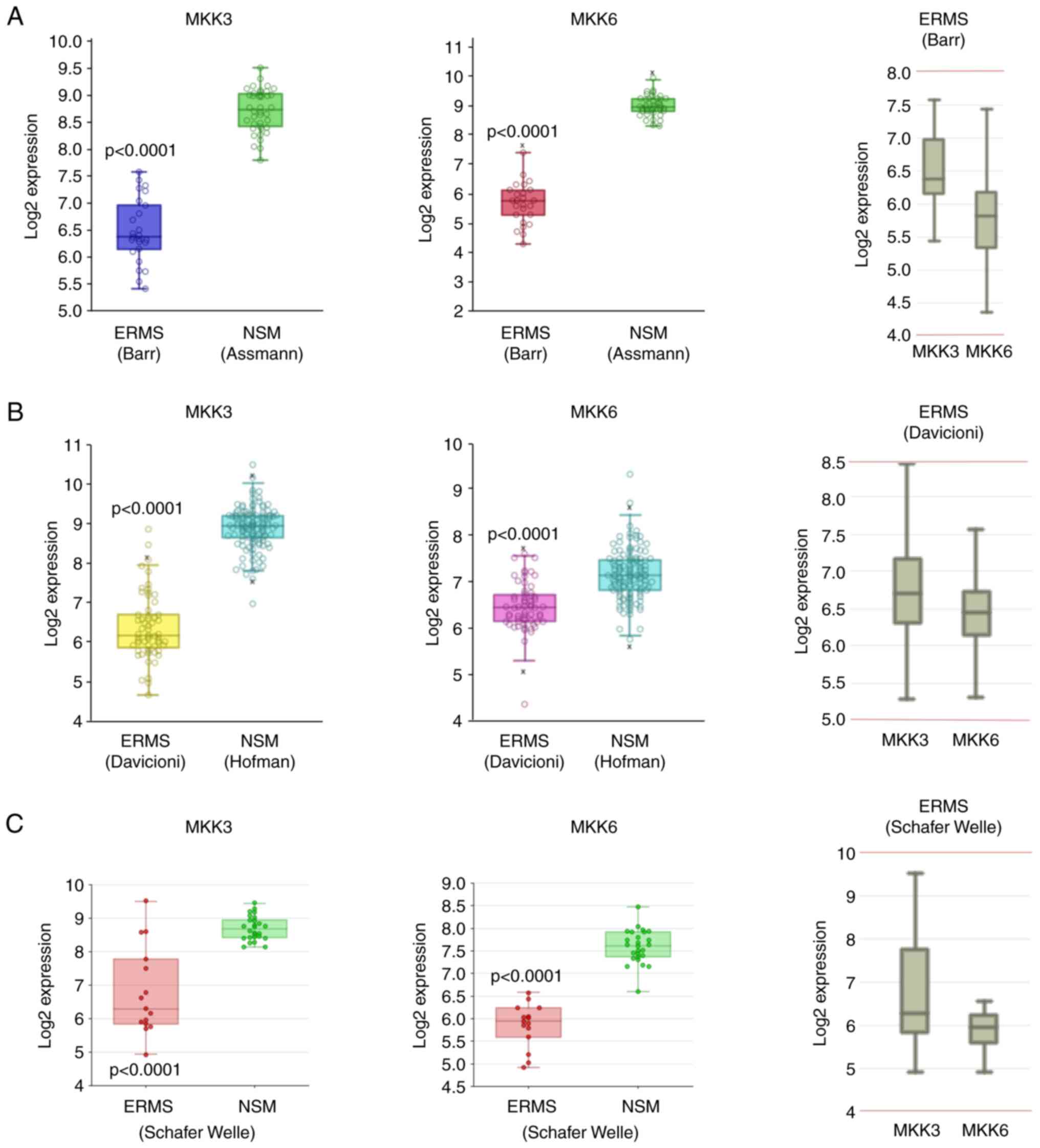
MKK3 and MKK6 gene expression analysis across
different public databases of RMS primary biopsies was performed by
interrogating the R2-Genomics Analysis and Visualization Platform
(http://r2.amc.nl). Specifically, to compare the
expression levels of MKK3 and MKK6 in a large cohort of RMS tumours
and normal skeletal muscle (NSM), five different datasets were
analysed: Barr (RMS, GSE66533), Davicioni (RMS) (27), Schafer Welle (RMS and NSM), Assmann
(NSM, GSE9103) and Hofman (NSM, GSE3307), for a total of 100 RMS
tumour biopsies and 187 NSM tissues. Based on the specific
characteristics of each dataset, a different approach of analysis
was used as specified below.
The MegaSampler algorithm (http://r2.amc.nl) was used to compare the expression
levels of MKK3 and MKK6 genes from multiple datasets, which are on
the same chip type and are normalised by the same algorithm. In
particular, the Barr-MAS5.0-u133p2 dataset (n=25), filtered for the
exclusion of PAX-FOXO fusion positive tumour samples, was compared
with the Assmann-MAS5.0-u133p2 dataset (n=40), whilst the
Daviconi-MAS5.0-u133a dataset (n=60), filtered for the exclusion of
RMS subtypes other than ERMS, was compared with the
Hofman-MAS5.0-u133a dataset (n=121).
As regards the Schafer Welle-MAS5.0-u133a dataset,
MKK3 and MKK6 gene expression levels were extracted and correlated
only in the ERMS subtype and NSM samples, by excluding ARMS
biopsies.
The box dot plot of MKK3 or MKK6 expression in ERMS
biopsies and NSM generated from R2-Genomics Analysis and
Visualization Platform was downloaded and formatted for
publication. To calculate whether the means of the expression
levels between the selected datasets differed significantly,
one-way ANOVA was performed using the R2 platform.
To evaluate the relative expression of MKK3 and MKK6
in each RMS dataset, the ‘multiple gene’ option was used by
interrogating the Barr, Davicioni or Schafer Welle datasets.
Statistical analyses
Statistical analyses were performed using the
Student's t-test, one-way ANOVA or two-way ANOVA. Dunnett's post
hoc test or Tukey's post hoc test were applied for multiple
comparisons. A probability value of P<0.05 was considered to
indicate a statistically significant difference.
Results
MKK6, but not MKK3 induces p38
activation and the myogenic differentiation of RD cells
Taking into account the data available on the
promyogenic role of MKK6 (10),
the present study aimed to investigate the possible differential
roles played by MKK3 and MKK6 in the ERMS reversal of the
transformed phenotype. MKK3 and MKK6 expression levels in ERMS
primary biopsies compared to NSM were obtained by interrogating
three different public transcriptomic datasets deposited on the
R2-Genomics Analysis and Visualization Platform (http://r2.amc.nl). The bioinformatics analysis
highlighted that MKK3 and MKK6 expression was lower in patients
with ERMS compared to NSM (Fig.
1). Moreover, the relative expression of MKK3 and MKK6
differed, with the MKK3 levels being higher than the MKK6 levels in
ERMS primary biopsies (Fig. 1,
right panels). With the purpose of studying a possible distinct
role of MKK3 and MKK6 kinases, the RD cells were co-transfected
with CMV, dnMKK3 (MKK3-Ala), caMKK3 (MKK3-Glu) or caMKK6 (MKK6-EE)
expression plasmids together with a puromycin-positive vector (as
described in the ‘Materials and methods’ section) so as to allow
transfected cells to be specifically selected and enriched by
puromycin treatment. The transfection efficiency was assessed by
analysing the MKK3 and MKK6 expression levels using western blot
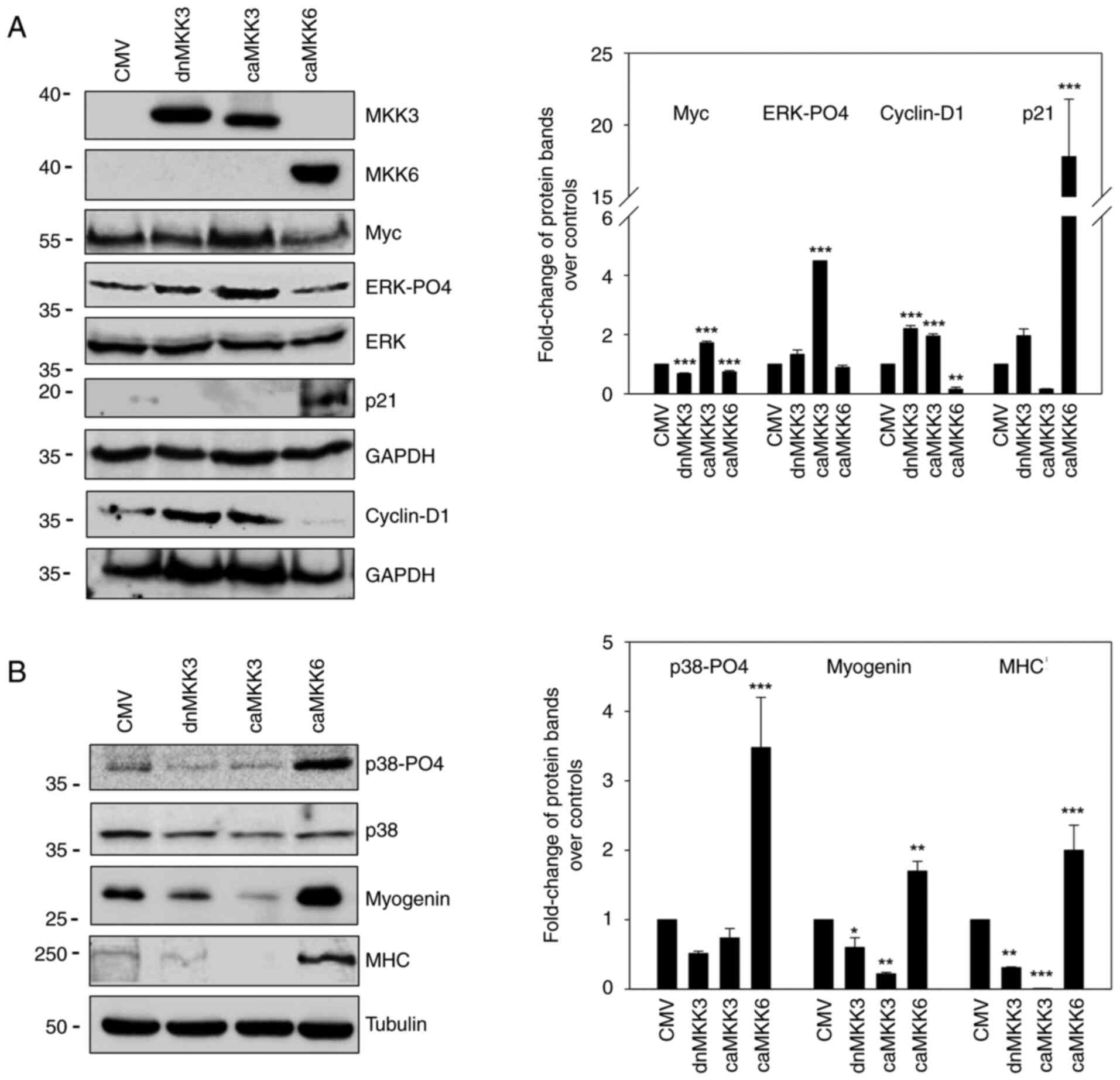
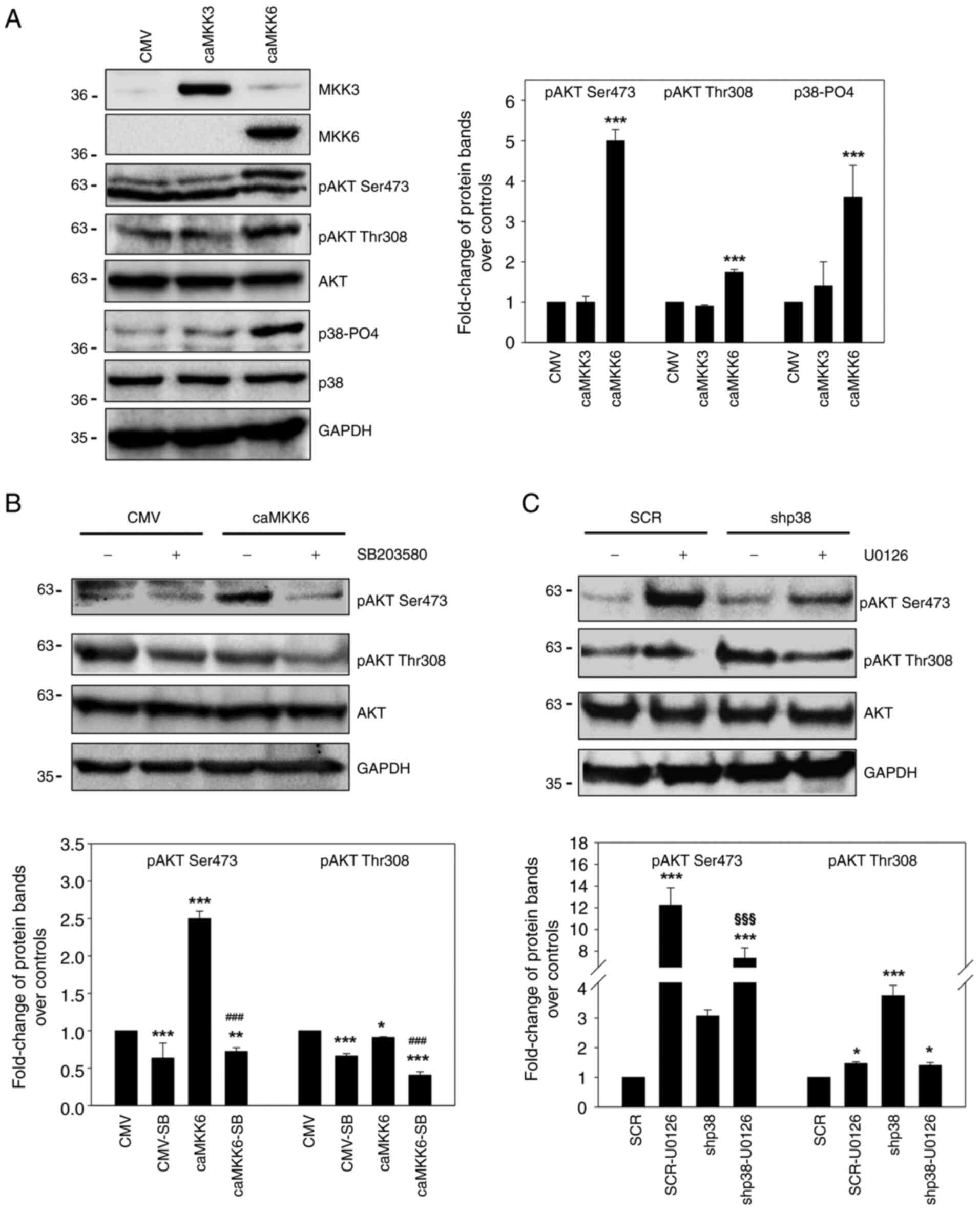
assays, as shown in Fig. 2A. While
caMKK6 induced Myc (P<0.001) and ERK-PO4 downregulation, caMKK3
led to an increase in Myc (P<0.001) and ERK-PO4 (P<0.001)
expression levels, whereas dnMKK3 did not (Fig. 2A). Notably, the reduced
proliferative potential of RD cells induced by caMKK6 was
demonstrated by the decrease in cyclin D1 expression (P=0.001),
which was markedly upregulated in caMKK3-transfected cells
(P<0.001), and the concomitant marked increase in p21 protein
levels (P<0.001) only in caMKK6-transfected cells (Fig. 2A).
To assess the distinct role of MKK3 and MKK6 in the
induction of myogenic differentiation, specific markers were
examined using western blot analysis. As shown in Fig. 2B, p38-PO4 (P<0.001), myogenin
(P=0.005) and MHC (P<0.001) expression levels were markedly
induced by caMKK6; however, this was not observed with dnMKK3 or
caMKK3 transfection.
The evident contrasting role played by MKK3 and MKK6
in RD cells was also confirmed by measuring the growth potential of
CMV-MKK3- and MKK6-transfected cells at 4 days following
transfection. In accordance with the observed alteration in the
cyclin D1 and p21 expression levels, trypan blue dye exclusion
assay revealed a consistent decrease in cell proliferation only in
RD cells transfected with caMKK6 (P=0.002), whilst
caMKK3-transfected cells maintained a high proliferative state
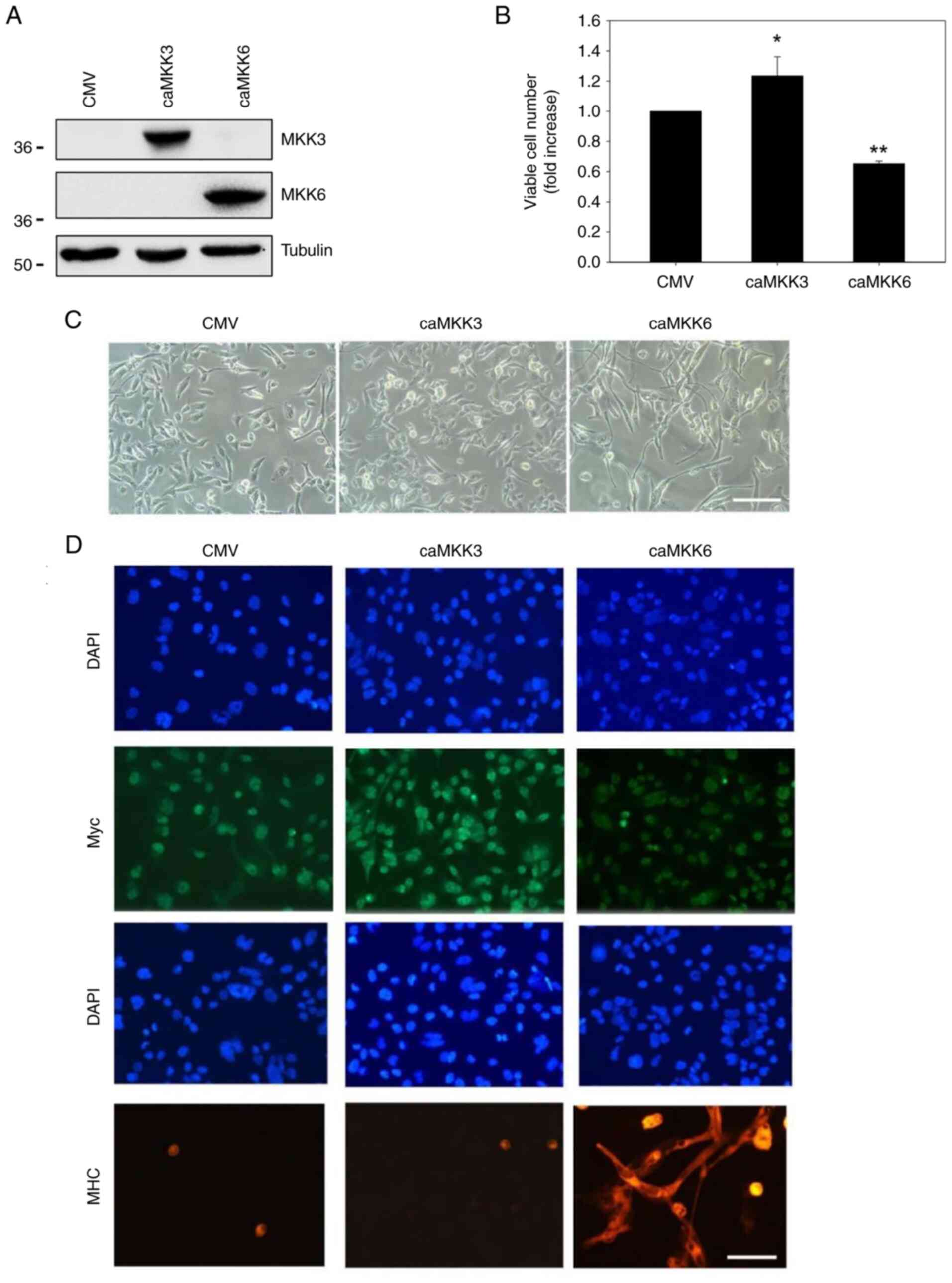
compared to the mock-transfected control cells (P=0.01) (Fig. 3B). The transfection efficiency was
confirmed by analysing the MKK3 and MKK6 protein levels using
western blot analysis (Fig. 3A).
Moreover, morphological investigations in caMKK6- or
caMKK3-transfected RD cells confirmed that caMKK6 reduced the
number of cells and induced an elongated myogenic-like morphology,
whereas caMKK3 increased the number of cells without inducing any
morphological changes when compared with the empty
vector-transfected cells (Fig.
3C). Finally, immunofluorescence analyses detected decreased
staining for Myc in caMKK6-transfected cells whereas staining in
caMKK3-transfected cells was increased (Fig. 3C). By contrast, the expression of
the myogenic marker MHC, was markedly induced in caMKK6-, but not
in caMKK3-transfected cells. Taken together these results indicate
that the MKK6/p38 cascade, but not MKK3, triggers growth arrest and
induces myogenic differentiation at the morphological and
biochemical level by reducing both ERK-PO4 and Myc expression.
Effects of p38 on the pro-myogenic and
anti-oncogenic action of MKK6 and MEK inhibitor
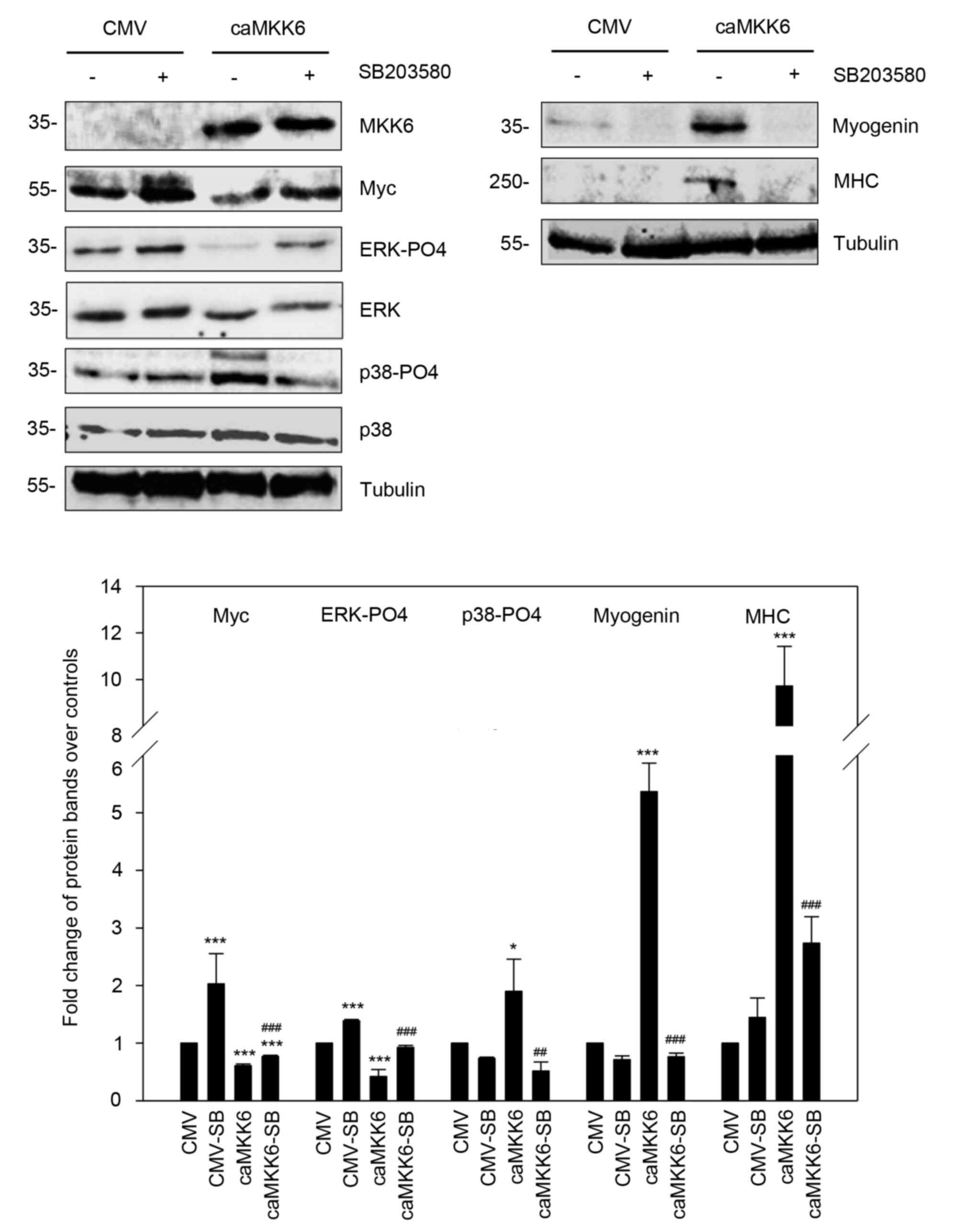
To demonstrate the function of p38 in mediating the
anti-oncogenic and pro-myogenic action of MKK6, caMKK6-transcted RD
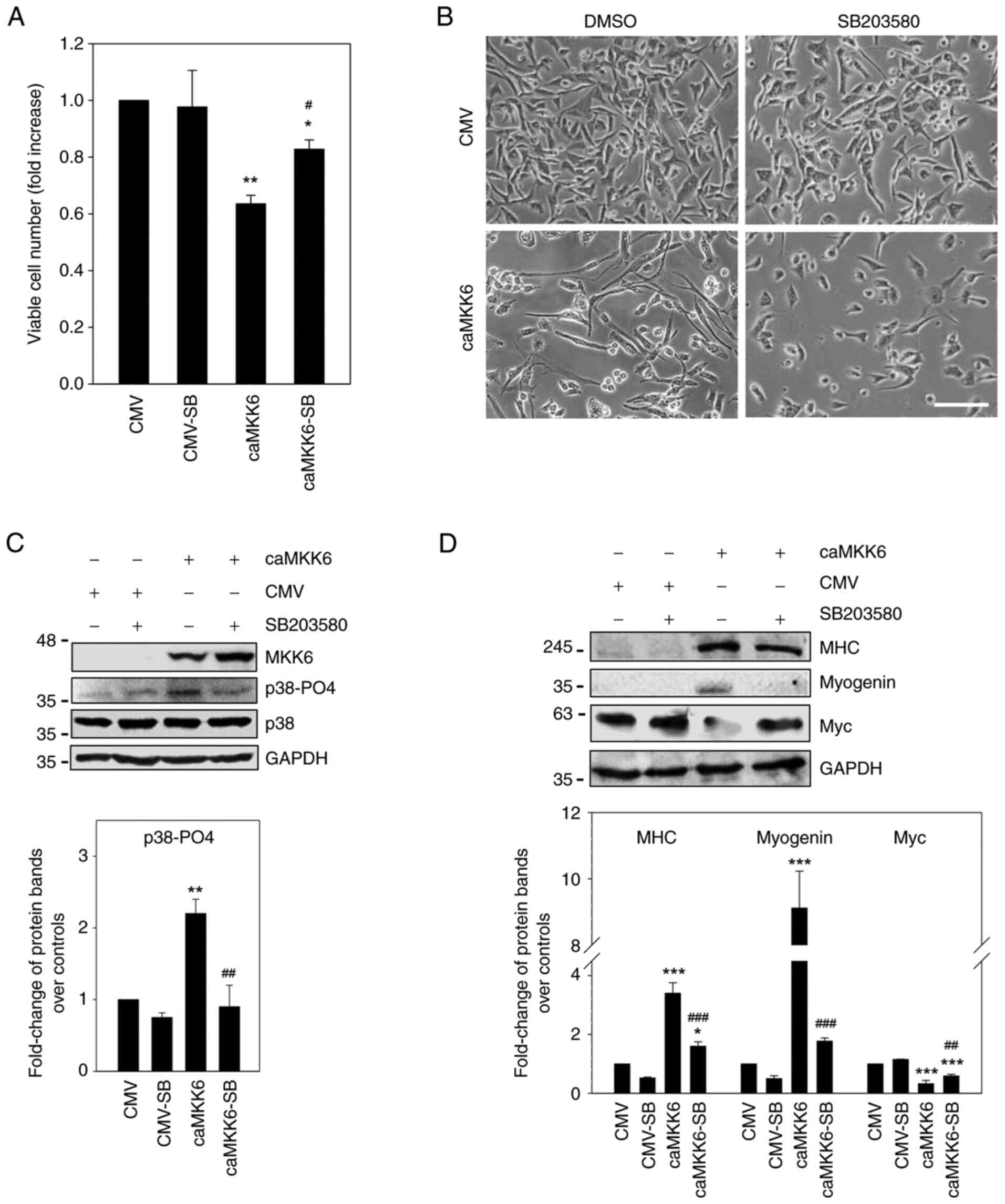
cells were treated with or without SB203580 (Fig. 4). The investigation of the effects
of p38 inhibition on the proliferative potential and morphology of
RD cells revealed that 5 µM SB203580 exposure alone did not induce
significant changes in cell proliferation and morphology (Fig. 4A and B). By contrast, the p38
inhibitor was able to counteract the decrease in cell proliferation
observed in RD cells transfected with caMKK6 by ~20% (P=0.04), as
indicated by the trypan blue dye exclusion assay, and to revert the
MKK6-induced myogenic differentiation. Indeed, MKK6-overexpressing
RD cells treated with SB203580 exhibited less elongated cellular
bodies compared to MKK6-overexpressing cells not treated with the
inhibitor (Fig. 4A and B). In
agreement with these results, it was found that the p38-PO4 levels
were markedly increased in caMKK6-transfected cells (P=0.002) and
were markedly downregulated by SB203580 treatment (P=0.002)
(Fig. 4C). On the other hand, CMV
mock-transfected RD cells exhibited a barely detectable p38-PO4
basal level, thus making it difficult to observe alterations in
this kinase. When caMKK6-transfected cells were treated with 5 µM
of the p38 inhibitor, SB203580, the expression levels of MHC
(P<0.001) and myogenin (P<0.001) significantly decreased and
were not detected in the CMV-transfected cells (Fig. 4D).
Moreover, the Myc expression levels were
significantly decreased in the caMKK6-transfected cells in
comparison to the CMV mock-transfected control cells (P<0.001)
(Fig. 4D); however, the decrease
in Myc expression was counteracted by treatment with SB203580
(P<0.001 vs. CMV; P=0.004 vs. caMKK6) (Fig. 4D), whilst in the CMV
mock-transfected control cells, the effect of p38 inhibitor on Myc
expression was minimal (Fig. 4D).
The effects of the p38 inhibitor on the ERK-PO4 and Myc levels were
also examined in untransfected RD cells and in cells treated with
the MEK inhibitor (Fig. S1A).
To verify whether the absence of p38 affects the
anti-oncogenic action of MEK inhibitor, p38-silenced (shp38) RD
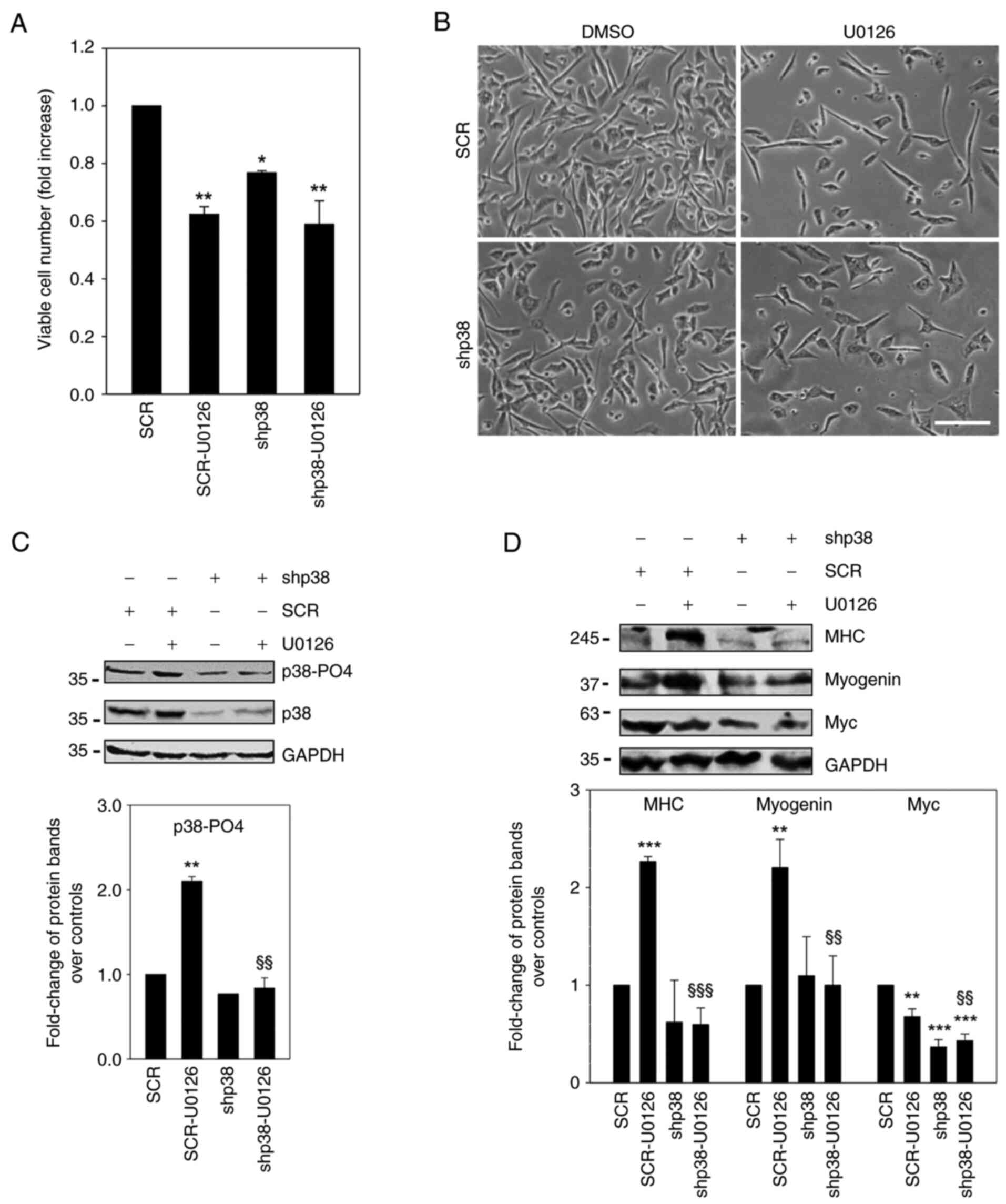
cells were treated with or without U0126 (Fig. 5). By investigating the
proliferative potential, it was found that p38 silencing reduced
the proliferation rate of RD cells by ~20% (Fig. 5A, shp38 vs. SCR, P=0.02), whilst
U0126 treatment reduced this by ~40% (Fig. 5A, SCR-U0126 vs. SCR, P=0.003). The
effect of U0126 on the proliferation of p38-silenced cells was
visibly diminished (shp38-U0126 vs. shp38). By analysing the
morphology, it was observed that U0126 induced a more elongated
shape in the SCR-treated cells, whilst these same effects were
attenuated in p38-silenced cells (Fig.
5B). As shown in Fig. 5C, the
U0126-mediated upregulation of phospho-active p38 was reduced in
p38-silenced cells (shp38-U0126 vs. SCR-U0126, P=0.004) but not in
SCR-U0126 cells (Fig. 5C), since
the total p38 expression was markedly downregulated by its specific
silencing. In p38-silenced cells, U0126 treatment led to barely
visible MHC (P<0.001 vs. SCR-U0126) and myogenin (P=0.004 vs.
SCR-U0126) expression levels (Fig.
5D), whereas it induced the high expression of both proteins in
SCR-transfected cells (Fig. 5D),
as well as in untransfected cells (Fig. S1B). Moreover, the
U0126-mediated-Myc downregulation was visibly attenuated in
p38-silenced cells compared to untreated cells (Fig. 5D).
Taken together, these different approaches on RD
cells (MKK6 enforced expression with or without SB203580 treatment
and p38 silencing with or without U0126 exposure), indicate the
contribution of MKK6 kinase in the activation of p38, which in turn
is crucial in inducing the expression of myogenic markers and in
counteracting the ERMS oncogenic phenotype. Notably, the
p38-silenced RD cells were less responsive in processing
anti-oncogenic signals induced by U0126, thereby revealing a
contribution of p38 in orchestrating myogenic phenotype expression,
including growth arrest.
MKK6 induces pro-myogenic and
anti-oncogenic effects mediated by the p38 pathway in the TE
ERMS-derived cell line
The present study then examined the effects of
caMKK6 in another embryonal RMS cell line that the authors had
previously used (28) due to its
sensitivity to the differentiating effects of MEK inhibitor in both
in vitro and in vivo assays. The enforced expression of MKK6 in TE
cells induced MAPK-p38 activation (P=0.04), whilst both ERK-PO4
(P<0.001) and Myc (P<0.001) levels decreased (Fig. 6). The reduction in ERK-PO4 and Myc
levels was reversed by SB203580 (P<0.001 and P=0.02 vs. caMKK6).
MHC (P<0.001) and myogenin (P<0.001) expression levels were
also induced by MKK6 overexpression, and abolished by p38 silencing
(P<0.001 and P<0.001 vs. caMKK6). These data and those
aforementioned suggest that the MKK6/p38 axis transmits
anti-oncogenic and pro-myogenic specific signals in ERMS-derived
cell lines.
MEK/ERK inhibitors in ERMS cell lines
mimic MKK6-induced differentiation pathways
A growing body of evidence suggests that the MEK/ERK
pathways play a positive role in the differentiation of certain
cell types (29,30). However, in pathological myogenic
differentiation, the MEK/ERK pathways have been found to be
involved in myogenic differentiation induced by Myc inactivation
(14). To date, and to the best of
our knowledge, no data are yet available on the mechanisms through
which MEK/ERK inhibitors induce Myc downregulation, growth arrest
and myogenic differentiation in ERMS cells. The present study thus
investigated whether the treatment of RD cells with the MEK
inhibitors induces MKK6 kinase activation and, as a consequence,
p38 activation. Trametinib, a second generation MEK inhibitor with
nanomolar activity, was also used, which specifically inhibits ERK
and affects the p38 pathways in the same manner as RD cells were
affected by U0126 treatment (Fig.
S2). The results of this experiment confirmed that the U0126
data were not due to off-target effects. However, since most of the
data collected were based on experiments that included U0126
treatment, the U0126 inhibitor was used in the majority of the
experiments presented herein.
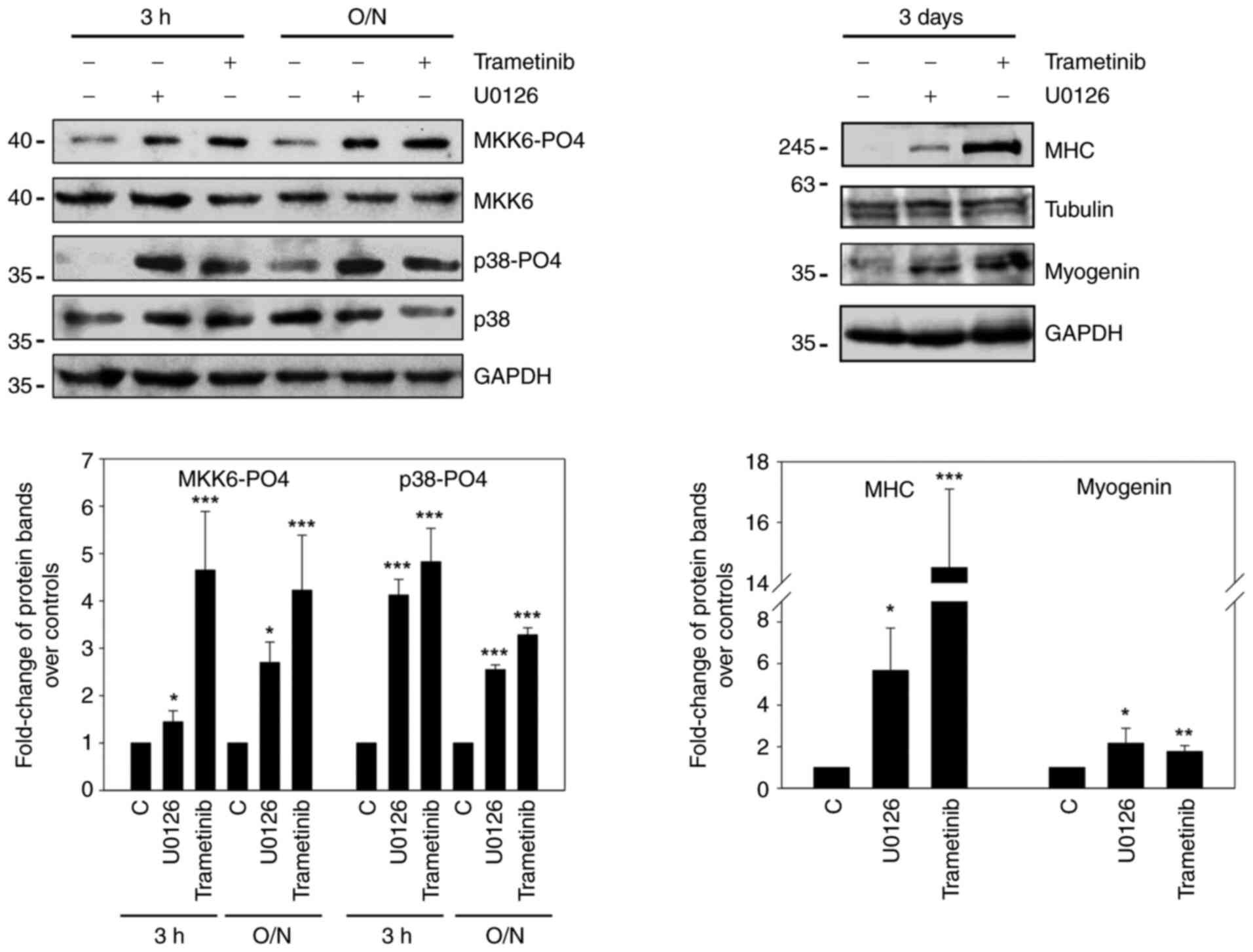
It was hypothesised that MEK inhibitors mimic the
myodifferentiation pattern mediated by caMKK6 by transmitting
anti-oncogenic signals. The treatment of RD and TE cells with U0126
or trametinib induced the MKK6/p38 pathways (Figs. 7 and S3). The results demonstrated that
MKK6-PO4 was detectable after short (3 h), as well as prolonged
(O/N) treatments with both MEK inhibitors in RD cells (P=0.04 and
P<0.001; P=0.02 and P<0.001) (Fig. 7) and particularly after U0126
exposure in TE cells (P<0.001; P=0.009) (Fig. S3). Moreover, p38-PO4 expression
was markedly increased in U0126- and trametinib-treated cells
following the same time course as MKK6, particularly in RD cells
(P<0.001 and P<0.001; P<0.001 and P<0.001) (Fig. 7).
Myogenin and MHC proteins were detected following
the use of both inhibitors only after prolonged treatments (3 days)
in both RD (P=0.02 and P=0.007; P=0.04 and P<0.001) (Fig. 7) and TE (P=0.004 and P=0.002;
P=0.04 and P=0.001) cells (Fig.
S3).
These results indicate, for the first time, to the
best of our knowledge, that MEK/ERK inhibition causes the
concomitant activation of the MKK6/p38 cascade and myogenic
program, a finding that has not previously been reported in ERMS
cells.
AKT is part of the myogenic pathway
induced by the MKK6/p38 axis
The deregulated expression of Myc has been reported
to affect PI3K/AKT activation by enhancing p38 in cells treated
with toxic agents (31). In
addition, the activation of the p38 pathway affects AKT at the
transcriptional and protein levels (25). Thus, the present study investigated
whether the MEK inhibitor-mediated downregulation of Myc in the
ERMS system was linked to the concomitant variation of AKT-PO4
expression and whether p38 activation may contribute to the
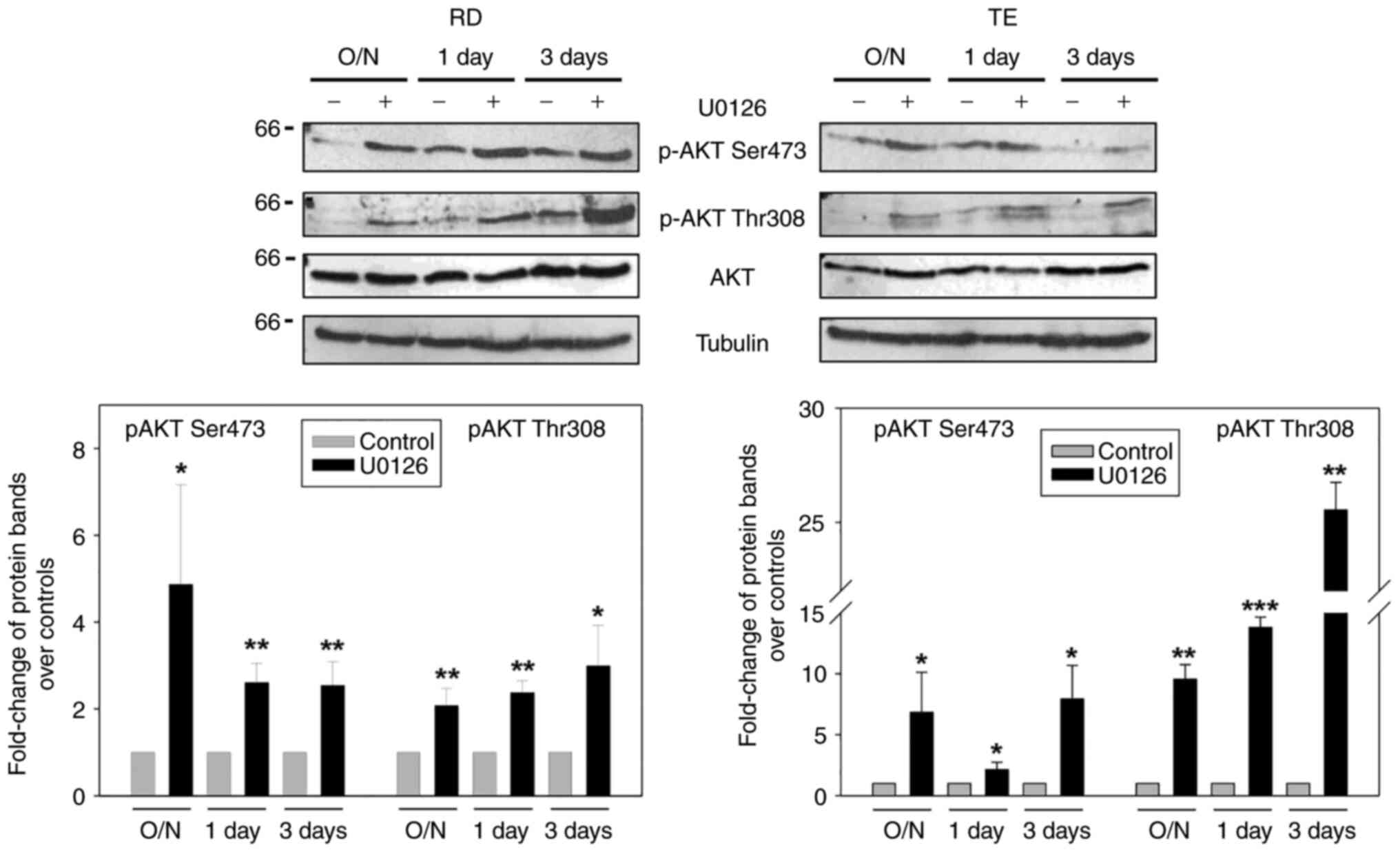
phospho-AKT modulation. For this purpose, RD and TE cells were
treated for different periods of time with 10 µM U0126, and AKT-PO4
Ser473 and Thr308 levels were analysed (Fig. 8).
While the expression levels of the two isoforms of
phospho-AKT were very low in the control cells, particularly
AKT-PO4 Thr308, these levels significantly increased when the MEK
inhibitor was added to both ERMS lines at the early and late
treatment times [AKT-PO4 Ser473: P=0.04, P=0.004 and P=0.008 (RD
cells); P=0.04, P=0.03 and P=0.01 (TE cells); AKT-PO4 Thr308:
P=0.009, P=0.001 and P=0.02 (RD cells); P=0.009, P<0.001 and
P=0.001 (TE cells)]. AKT activation was also confirmed by treating
the RD cells with 10 nM trametinib (data not shown). Notably, the
AKT-PO4 levels increased only in cells overexpressing MKK6
(P<0.001 and P<0.001), but not with the caMKK3 vector,
similarly to p38-PO4 levels (P<0.001) (Fig. 9A); this suggested that MKK6-induced
AKT activation may be mediated by p38.
Indeed, caMKK6 induced the phosphorylated form of
AKT in both Ser473 and Thr308 (Fig. 9A
and B) and the reduction in AKT activation following treatment
with the p38 inhibitor, SB203580 (P<0.001 and P<0.001 vs.
caMKK6), demonstrated the positive contribution of MAPK p38 to the
activation of both AKT isoforms in MKK6 overexpressing cells
(Fig. 9B).
Likewise, the silencing of p38 prevented the
activation of AKT mediated by U0126-induced p38 activation (AKT-PO4
Ser473 P<0.001 vs. SCR-U0126), which was present in the SCR
control-transfected cells following U0126 treatment (Fig. 9C). These results indicate that AKT
is activated when the myogenic program is induced either by
pharmacological treatment or by the enforced expression of the
MKK6/p38 pathway.
Discussion
To the best of our knowledge, the present study is
the first to demonstrate the differential role of the MKK class of
MAP kinases, namely MKK6 and MKK3, in the ERMS cell system,
specifically in the RD and TE cell lines. Both MKK3 and MKK6 are
upstream kinases of p38, the difference between these kinases being
that MKK3 markedly stimulates JNK, which is known to counteract
myogenesis (32). Though both
kinases are upstream pathways of p38 (33). The present study demonstrated, by
using constitutively active isoforms (ca) of both, that caMKK6
markedly enhanced phospho-active p38, growth arrest and myogenic
differentiation. By contrast, caMKK3 failed to induce active p38
and maintained the RD cells in a proliferative state. This finding
was demonstrated by the induction of proliferative markers in
caMKK3-transfected cells and the downregulation of these markers in
MKK6-overexpressing cells. When examining the effect of dnMKK3,
which cannot be activated due to the replacement of Ser-189 and
Thr-193 with Ala, it was found that it was unable to control Myc
upregulation and ERK-PO4 kinase activation, whilst it still
functioned in the control of cyclin D1 specific expression. This is
intriguing data and future research is required to provide further
insight into the related molecular mechanisms. Since the present
study did not extend the investigation to other aspects aimed at
deeply analysing the proliferative role of MKK3 in ERMS-derived
cells, this point remains an open question, thus representing a
limitation of the present study. The analysis of the growth
potential of RD cells confirmed that caMKK3 sustains the
proliferative phenotype, whereas caMKK6 abrogates it, as is shown
by the biochemical data. The overexpression of Myc, phospho-active
ERK and cyclin D1, under the enforced expression of caMKK3 which
failed to activate p38, is indicative of a permanent proliferative
state. This result supports the reported role of p38 in the cell
cycle exit of myoblasts (34).
Indeed, Perdiguero et al (34) reported that the genetic deficiency
of p38α in myoblasts confers enhanced growth and results in defects
in the withdrawal from the cell cycle, as well as in the formation
of multinucleated myotubes. In agreement with the key role played
by p38 in controlling the proliferative potential of RMS cells, the
authors of the present study recently demonstrated that the
inhibition of p38 activity, by using SB203580, increases the
clonogenic ability of RD cells (35). In line with the role of p38 in
controlling the growth and differentiation of RMS tumours, it was
found that the anti-growth and promyogenic responses to U0126 were
attenuated in the p38-silenced cells compared to SCR-transfected
cells, thus corroborating p38 to actively participate in the
antioncogenic responses induced by MEK/ERK inhibition.
Myc and myosin in RD cells transfected with caMKK3,
caMKK6 or an empty vector (CMV) displayed a morphological pattern
of cells that undergo myogenic differentiation with MKK6
overexpression and an increased transformed phenotype expression
was observed in MKK3-overexpressing cells.
Finally, the meaning of the data of the two kinases
in ERMS cell lines is coherent with those obtained by interrogating
public transcriptomics datasets on RMS tumours. Indeed,
bioinformatics analysis demonstrated that the expression levels of
MKK3 and MKK6 were lower in patients with ERMS compared to normal
muscle. In addition, in ERMS, MKK3 expression was higher than that
of MKK6, this suggesting that for the reversal of the ERMS
oncogenic phenotype, the induction of MKK6 is strictly
required.
In both RD and TE cell lines, MKK6/p38 pathway
activation induced the downregulation of Myc and ERK-PO4
accompanied by increased MHC and myogenin expression. SB203580
treatment specifically abolished myogenic marker expression in both
ERMS cell lines and counteracted the decrease in cell proliferation
induced by caMKK6 ectopic expression.
Exploring the hypothesis that MEK inhibitors can
mimic the effect of the enforced expression of caMKK6, it was
demonstrated that one of the early responses to the MEK inhibitors
U0126 or trametinib in both RD and TE cells was the activation of
MKK6.
It has previously been reported that the enforced
induction of MKK6/p38 pathways restores the myogenic
differentiation of ERMS cell lines (10). The present study demonstrates for
the first time, to the best of our knowledge, that extracellular
signals, such as those induced by the MEK/ERK inhibitor within the
pathological myogenesis of ERMS, restore the activation of
endogenous MKK6/p38.
N-Ras and mutant p53, which both sustain the ERMS
phenotype, are known to be related to p38 MAPK (36). The present study found a functional
connection between MKK6, ERK, p38 and AKT that is orchestrated by
the MEK/ERK inhibition or by the overexpression of caMKK6 (MKK6-EE)
though not by caMKK3 (MKK3-Glu) expression. The functional
connection between MKK6, ERK, p38 and AKT reflects that reported in
a study on normal myoblasts, in which myogenesis was induced in
differentiating medium (37).
Serra et al (37)
demonstrated the role played by responses at the chromatin level
and the specific role of kinases in mediating the formation of
transcription complexes that are active in muscle during the
specific regeneration program. When they investigated myogenic
precursors by focusing on p38 and ERK in a normal myogenic program,
they found that p38 was required for the expression of
MyoD-responsive genes and that ERK plays a biphasic role, peaking
in undifferentiated and post-mitotic myoblasts.
Importantly, the effects of the MEK/ERK inhibitors
on RMS is further supported by the similarity between the data of
the present study and those of the aforementioned authors (37), as is shown by the ability of these
inhibitors to induce a pathway in tumour cells, just as occurs in
the normal myogenic program.
The fact that p38 activation is responsible for the
concomitant decreases in ERK-PO4 and Myc levels demonstrates that
the oncogenic functional partnering of active ERK and Myc (19) can be disrupted by activated p38
kinase. The concomitant ERK-PO4 and Myc downregulation also
demonstrates that the increase in p38 activity and the attenuation
of ERK activity promote the transition from the proliferation to
the differentiation of ERMS cells.
While the crosstalk between ERK and p38 has
previously been reported by the authors of the present study
(13), as well as by other authors
(38,39), the dependence of Myc levels on
active p38 has not. The present study demonstrated that the
activation of MKK6 by either MEK inhibitors or by caMKK6
overexpression altered the p38/ERK ratio towards a differentiation
state, thereby suggesting that ERK and p38 kinases are regulated by
a critical ratio during pathological myogenic differentiation.
In agreement with this, the balance of p38 and
ERK-PO4 regulate cell differentiation in osteosarcoma and the use
of MEK inhibitor has been suggested as a candidate for reverting
malignant phenotype in this system (40). In keeping with this hypothesis, it
is not surprising that SB203580 p38 inhibitor not only inhibits
p38, but enhances ERK-PO4 and Myc expression, thereby corroborating
the inverse association between ERK and p38 proteins in ERMS. Since
Myc inactivation alone leads to growth arrest and myogenic
differentiation of cultured ERMS cells (14), the MEK/ERK inhibitor can recruit
the kinases capable of mediating Myc degradation, which in turn
release myodifferentiation signals. Indeed, Myc accumulation is one
of the oncogenic and anti-myogenic responses to Ras/MEK/ERK
overactivation. Likewise, following MEK/ERK inhibition by either
MEK/ERK inhibitor or caMKK6 enforced expression, the induction of
AKT signalling is rapid and p38-dependent. The activation of AKT
may be consequent to a variation in Myc expression (31). Indeed, Myc is known to impair
PI3K/AKT activation levels (31),
which suggests that PI3K/AKT may be released when Myc levels are
reduced. AKT activation may play an important role in the restored
myogenic program by MEK/ERK inhibition, which leads to the
re-establishment of the MKK6/p38/AKT cascades. It is noteworthy
that the activation of p38 by the inhibition of ERK has been
reported to be linked to the apoptotic action of p38, which is
modulated by the concomitant PI3K/AKT module (41). In targeted knockdown and in genetic
knockout experiments, AKT proteins have been implicated in myogenic
differentiation and myofiber maturation (24). The controlled activation of AKT in
proliferating ERMS cells has the potential to be an additional
promising option in differentiation therapy for the myogenic rescue
of RMS tumours.
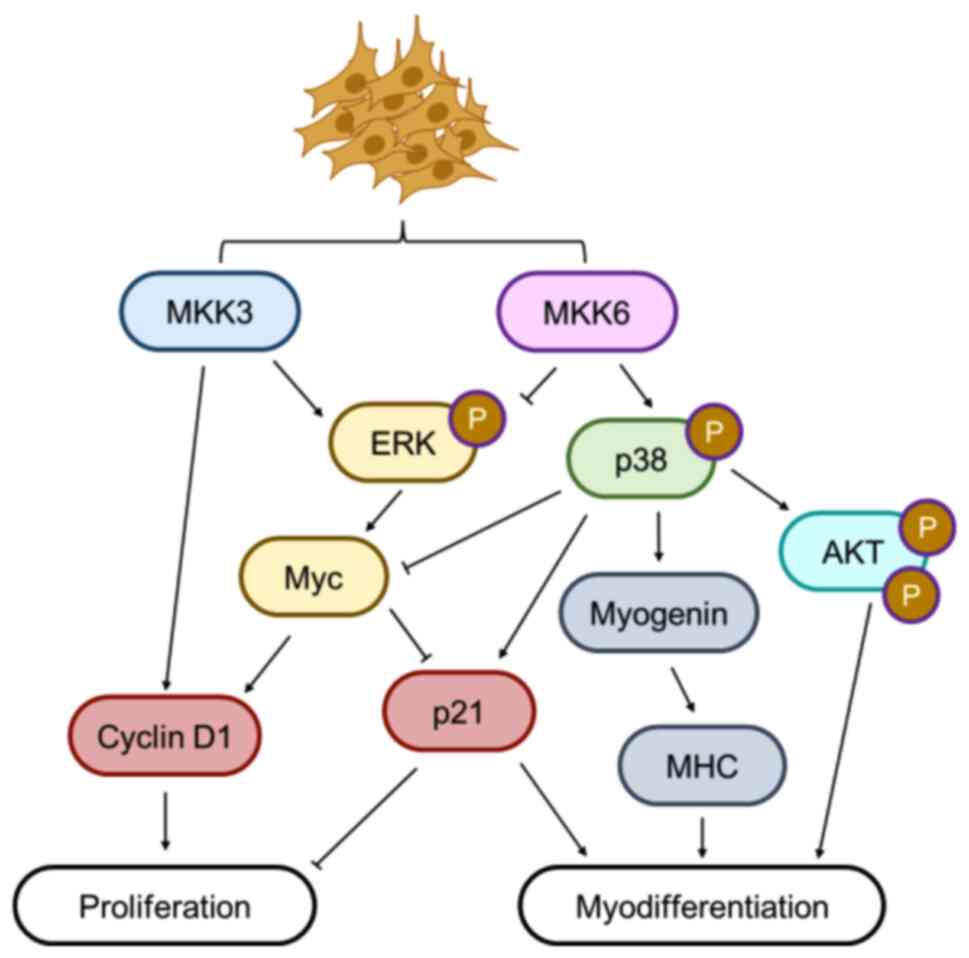
The present study defined the differential role of
MKK kinases in controlling growth arrest and myogenic
differentiation and in maintaining the proliferative phenotype in
ERMS cells. In detail, MKK6 activation (by MEK inhibitor treatment
or ectopic expression) triggers both p38 and AKT activation, which
mediate the expression of myogenic markers, such as MHC and
myogenin. Conversely, Myc, which is known to counteract myogenic
programs is downregulated together with ERK-PO4 contributing to the
growth arrest (Fig. 10). The
finding that MEK/ERK inhibitors recover the MKK6/p38 axis inducing
myogenic differentiation in RMS may lead to the use of them in
combined and more advanced therapies for this aggressive solid
tumour and prevent its dissemination. From a therapeutic point of
view, the MEK/ERK inhibitor-based therapies had limited success
(42); nevertheless, since ERK and
Myc functional partnering in ERMS cells are disrupted by MKK6/p38
activation from MEK inhibitors, their concomitant inhibition lends
itself to exploitation at the therapeutic level.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Lewis
Baker (Sapienza Language Centre, Sapienza University of Rome) for
reviewing the English language in the manuscript. The authors are
grateful to Professor Pier Lorenzo Puri (Development, Aging and
Regeneration Program, Sanford Burnham Prebys Medical Discovery
Institute, La Jolla, CA, USA) who provided the MKK6EE vector. The
authors would also like to thank Dr Giulia Vitali (Department of
Experimental Medicine, Sapienza University of Rome) for providing
supporting with the western blot analyses.
Funding
The present study was partially funded by 'Progetti di Ricerca
di Ateneo 2018′ n. RM1181642707D875, Sapienza University of Rome
Italy. The research leading to these results received funding from
AIRC under IG 2020-ID.24696 project. The present study was also
supported by ‘Io, domani…’ Associazione per la lotta contro i
tumori infantili Onlus (Rome, Italy) with a postdoctoral
fellowship.
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request or from R2-Genomics Analysis and Visualization Platform
website (http://r2.amc.nl).
Authors' contributions
BMZ, ADR, FMa, CC, SC, GB and FMe conceptualised and
designed the study. ADR, BMZ, SC, AB, LS, CC, BLO and FMe performed
the experiments and/or analysed and interpreted the data. SC
carried out the bioinformatics and statistical analyses. BMZ, MB
and CM were responsible for data curation. BMZ wrote the original
draft of the manuscript. BMZ, SC and FMe critically reviewed and
edited the manuscript. BMZ and SC prepared the figures. FMa, FMe
and CM provided funds. authors have read and approved the final
manuscript. ADR and BMZ confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shern JF, Chen L, Chmielecki J, Wei JS,
Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et
al: Comprehensive genomic analysis of rhabdomyosarcoma reveals a
landscape of alterations affecting a common genetic axis in
fusion-positive and fusion-negative tumors. Cancer Discov.
4:216–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun X, Guo W, Shen JK, Mankin HJ, Hornicek
FJ and Duan Z: Rhabdomyosarcoma: Advances in molecular and cellular
biology. Sarcoma. 2015:2320102015. View Article : Google Scholar
|
|
3
|
Chardin P, Yeramian P, Madaule P and
Tavitian A: N-ras gene activation in the RD human rhabdomyosarcoma
cell line. Int J Cancer. 35:647–652. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Felix CA, Kappel CC, Mitsudomi T, Nau MM,
Tsokos M, Crouch GD, Nisen PD, Winick NJ and Helman LJ: Frequency
and diversity of p53 mutations in childhood rhabdomyosarcoma.
Cancer Res. 52:2243–2247. 1992.PubMed/NCBI
|
|
5
|
McCubrey JA, Steelman LS, Abrams SL, Lee
JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA,
D'Assoro AB, et al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT
pathways in malignant transformation and drug resistance. Adv
Enzyme Regul. 46:249–279. 2006. View Article : Google Scholar
|
|
6
|
Rodriguez-Viciana P, Warne PH, Dhand R,
Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD and Downward J:
Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature.
370:527–532. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Castellano E and Downward J: RAS
Interaction with PI3K: More than just another effector pathway.
Genes Cancer. 2:261–274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Puri PL, Wu Z, Zhang P, Wood LD, Bhakta
KS, Han J, Feramisco JR, Karin M and Wang JY: Induction of terminal
differentiation by constitutive activation of p38 MAP kinase in
human rhabdomyosarcoma cells. Genes Dev. 14:574–584. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lluís F, Perdiguero E, Nebreda AR and
Muñoz-Cánoves P: Regulation of skeletal muscle gene expression by
p38 MAP kinases. Trends Cell Biol. 16:36–44. 2006. View Article : Google Scholar
|
|
12
|
Martínez-Limón A, Joaquin M, Caballero M,
Posas F and de Nadal E: The p38 pathway: From biology to cancer
therapy. Int J Mol Sci. 21:19132020. View Article : Google Scholar
|
|
13
|
Mauro A, Ciccarelli C, De Cesaris P,
Scoglio A, Bouché M, Molinaro M, Aquino A and Zani BM:
PKCalpha-mediated ERK, JNK and p38 activation regulates the
myogenic program in human rhabdomyosarcoma cells. J Cell Sci.
115:3587–3599. 2002. View Article : Google Scholar
|
|
14
|
Marampon F, Ciccarelli C and Zani BM:
Down-regulation of c-Myc following MEK/ERK inhibition halts the
expression of malignant phenotype in rhabdomyosarcoma and in non
muscle-derived human tumors. Mol Cancer. 5:312006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malempati S, Tibbitts D, Cunningham M,
Akkari Y, Olson S, Fan G and Sears RC: Aberrant stabilization of
c-Myc protein in some lymphoblastic leukemias. Leukemia.
20:1572–1581. 2006. View Article : Google Scholar
|
|
16
|
Gartel AL and Shchors K: Mechanisms of
c-myc-mediated transcriptional repression of growth arrest genes.
Exp Cell Res. 283:17–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iavarone A and Lasorella A: Myc and
differentiation: Going against the current. EMBO Rep. 15:324–325.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zinin N, Adameyko I, Wilhelm M, Fritz N,
Uhlen P, Ernfors P and Henriksson MA: MYC proteins promote neuronal
differentiation by controlling the mode of progenitor cell
division. EMBO Rep. 15:383–391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sears R, Leone G, DeGregori J and Nevins
JR: Ras enhances Myc protein stability. Mol Cell. 3:169–179. 1999.
View Article : Google Scholar
|
|
20
|
Megiorni F, Camero S, Ceccarelli S,
McDowell HP, Mannarino O, Marampon F, Pizer B, Shukla R, Pizzuti A,
Marchese C, et al: DNMT3B in vitro knocking-down is able to reverse
embryonal rhabdomyosarcoma cell phenotype through inhibition of
proliferation and induction of myogenic differentiation.
Oncotarget. 7:79342–79356. 2016. View Article : Google Scholar
|
|
21
|
Han J, Lee JD, Jiang Y, Li Z, Feng L and
Ulevitch RJ: Characterization of the structure and function of a
novel MAP kinase kinase (MKK6). J Biol Chem. 271:2886–2891. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Remy G, Risco AM, Iñesta-Vaquera FA,
González-Terán B, Sabio G, Davis RJ and Cuenda A: Differential
activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal.
22:660–667. 2010. View Article : Google Scholar
|
|
23
|
Stramucci L, Pranteda A and Bossi G:
Insights of crosstalk between p53 protein and the MKK3/MKK6/p38
MAPK signaling pathway in cancer. Cancers (Basel). 10:1312018.
View Article : Google Scholar
|
|
24
|
Gardner S, Anguiano M and Rotwein P:
Defining Akt actions in muscle differentiation. Am J Physiol Cell
Physiol. 303:C1292–C1300. 2012. View Article : Google Scholar
|
|
25
|
Cabane C, Coldefy AS, Yeow K and Dérijard
B: The p38 pathway regulates Akt both at the protein and
transcriptional activation levels during myogenesis. Cell Signal.
16:1405–1415. 2004. View Article : Google Scholar
|
|
26
|
Bhatnagar S and Kumar A, Makonchuk DY, Li
H and Kumar A: Transforming growth factor-beta-activated kinase 1
is an essential regulator of myogenic differentiation. J Biol Chem.
285:6401–6411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Davicioni E, Finckenstein FG, Shahbazian
V, Buckley JD, Triche TJ and Anderson MJ: Identification of a
PAX-FKHR gene expression signature that defines molecular classes
and determines the prognosis of alveolar rhabdomyosarcomas. Cancer
Res. 66:6936–6946. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marampon F, Gravina GL, Di Rocco A,
Bonfili P, Di Staso M, Fardella C, Polidoro L, Ciccarelli C,
Festuccia C, Popov VM, et al: MEK/ERK inhibitor U0126 increases the
radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by
downregulating growth and DNA repair signals. Mol Cancer Ther.
10:159–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miranda MB, McGuire TF and Johnson DE:
Importance of MEK-1/-2 signaling in monocytic and granulocytic
differentiation of myeloid cell lines. Leukemia. 16:683–692. 2002.
View Article : Google Scholar
|
|
30
|
Kurtzeborn K, Kwon HN and Kuure S:
MAPK/ERK signaling in regulation of renal differentiation. Int J
Mol Sci. 20:17792019. View Article : Google Scholar
|
|
31
|
Bellmann K, Martel J, Poirier DJ, Labrie
MM and Landry J: Downregulation of the PI3K/Akt survival pathway in
cells with deregulated expression of c-Myc. Apoptosis.
11:1311–1319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie SJ, Li JH, Chen HF, Tan YY, Liu SR,
Zhang Y, Xu H, Yang JH, Liu S, Zheng LL, et al: Inhibition of the
JNK/MAPK signaling pathway by myogenesis-associated miRNAs is
required for skeletal muscle development. Cell Death Differ.
25:1581–1597. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Raingeaud J, Whitmarsh AJ, Barrett T,
Dérijard B and Davis RJ: MKK3- and MKK6-regulated gene expression
is mediated by the p38 mitogen-activated protein kinase signal
transduction pathway. Mol Cell Biol. 16:1247–1255. 1996. View Article : Google Scholar
|
|
34
|
Perdiguero E, Ruiz-Bonilla V, Serrano AL
and Munoz-Canoves P: Genetic deficiency of p38alpha reveals its
critical role in myoblast cell cycle exit: The p38alpha-JNK
connection. Cell Cycle. 6:1298–1303. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Camero S, Vitali G, Pontecorvi P,
Ceccarelli S, Anastasiadou E, Cicchetti F, Flex E, Pomella S,
Cassandri M, Rota R, et al: DNMT3A and DNMT3B targeting as an
effective radiosensitizing strategy in embryonal rhabdomyosarcoma.
Cells. 10:29562021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gaestel M: MAPK-activated protein kinases
(MKs): Novel insights and challenges. Front Cell Dev Biol.
3:882015.
|
|
37
|
Serra C, Palacios D, Mozzetta C, Forcales
SV, Morantte I, Ripani M, Jones DR, Du K, Jhala US, Simone C and
Puri PL: Functional interdependence at the chromatin level between
the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle
differentiation. Mol Cell. 28:200–213. 2007. View Article : Google Scholar
|
|
38
|
Wu Z, Woodring PJ, Bhakta KS, Tamura K,
Wen F, Feramisco JR, Karin M, Wang JY and Puri PL: p38 and
extracellular signal-regulated kinases regulate the myogenic
program at multiple steps. Mol Cell Biol. 20:3951–3964. 2000.
View Article : Google Scholar
|
|
39
|
Xiao YQ, Malcolm K, Worthen GS, Gardai S,
Schiemann WP, Fadok VA, Bratton DL and Henson PM: Cross-talk
between ERK and p38 MAPK mediates selective suppression of
pro-inflammatory cytokines by transforming growth factor-beta. J
Biol Chem. 277:14884–14893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shimo T, Matsumura S, Ibaragi S, Isowa S,
Kishimoto K, Mese H, Nishiyama A and Sasaki A: Specific inhibitor
of MEK-mediated cross-talk between ERK and p38 MAPK during
differentiation of human osteosarcoma cells. J Cell Commun Signal.
1:103–111. 2007. View Article : Google Scholar
|
|
41
|
Berra E, Diaz-Meco MT and Moscat J: The
activation of p38 and apoptosis by the inhibition of Erk is
antagonized by the phosphoinositide 3-kinase/Akt pathway. J Biol
Chem. 273:10792–10797. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marampon F, Ciccarelli C and Zani BM:
Biological rationale for targeting MEK/ERK pathways in anti-cancer
therapy and to potentiate tumour responses to radiation. Int J Mol
Sci. 20:25302019. View Article : Google Scholar
|