Introduction
Lung cancer is a malignant tumor with the second
highest morbidity rate and the leading mortality rate worldwide
(1). EGFR tyrosine kinase
inhibitor (TKI) therapy has made a great progress in non-small cell
lung cancer treatment with a longer patient survival time and fewer
adverse effects. However, acquired resistance is inevitable after a
period of time treatment (2). The
third generation EGFR-TKIs have met the same fate, though they were
designed to surmount some resistant mutations (3). Tumor cells are spared from drug
attack after obtaining resistant phenotype, either by on-target
resistance, such as ATP affinity alteration by EGFR tyrosine kinase
domain mutation (mainly T790M), or by off-target resistance, such
as bypassing signaling transduction pathway activation (4). Either way, tumor resistance
acquirement is inseparable with tumor microenvironment (TME).
Macrophages and other cells of the TME may provide various growth
factors and extracellular signals to orchestrate between tumor
cells and host body, and thus mold the malignancy of tumor cells
(5).
Macrophages coordinate immunity and tumor
development with their constant migration and renewal. They are
associated with carcinogenesis, tumor cell proliferation,
metastasis and even influence the response to anti-tumor therapy
(6–10). Though some research has indicated
that EGFR-TKIs treatment interferes with macrophage immunoreaction,
it is still unclear how macrophages are involved in acquirement of
resistance to EGFR-TKIs (11,12).
A number of studies have focused on macrophage polarization. It is
considered that M2 macrophages assist in obtaining secondary
resistance to gefitinib, and high recruitment of M1 macrophages is
associated with good response to gefitinib and longer survival
(13–16). However, macrophage phenotypes
change with TME changes, and the M1 and M2 specific marker may be
co-expressed in some macrophages (6,13,17).
Moreover, short-lived macrophages need to constantly renew
themselves, and in either renewal way, new macrophages can polarize
into M1 and M2 macrophages (6,18).
It would be time consuming if new macrophages didn't perform
protection until polarization.
Macrophage renewal ways are mainly determined by
their origins. Resident macrophages renew by local proliferation,
which are from embryonic precursors of the yolk sac (6,17).
Monocyte-derived macrophages are recruited at the tumor site after
emigrating through the blood stream, originating from hematopoietic
stem cells (6,19). Macrophage refreshing potentially
leads to various temporal and spatial distributions, and the
distribution environment instead will affect their functions
(20,21). Resident macrophages are more
involved in tumor initiation, whereas monocyte-derived macrophages
are more associated with tumor metastasis (6,13,22).
Although the origin of the macrophage endows some inherence
characters, macrophages with the same origin may possess functional
heterogeneity (20). The
functional status of macrophages is closely associated with their
different renewal processes. Previous studies have demonstrated
that monocyte-derived macrophages commonly recruited in
intratumoral area and intratumoral macrophages contribute to the
epithelial-mesenchymal transition phenotype of tumor cells, which
is a mechanism of secondary resistance (7,20,22–25).
Notably, macrophage refreshing affects secondary resistance to
EGFR-TKIs.
The present study induced resistance to gefitinib in
PC-9 cells with an EGFR deletion mutation by alternative treatment
with gefitinib and recovery in drug-free medium for ~10 months in
macrophage co-culture systems. Renewal modes were stimulated by
controlling macrophage co-culture ways as follows: i) Co-culture
started before PC-9 cells treatment; meaning that co-culture
continued all along from treatment to recovery, and was used as
self-renewal resident macrophages (Mr); and ii) co-culture started
after PC-9 cells treatment; meaning that co-culture presented only
in recovery, was used as migrated macrophages (Mm). The aim of the
present study was to investigate the dynamic role of macrophages in
the acquired resistance to gefitinib in PC-9 cells, which are
sensitive to gefitinib. To the best of our knowledge, this is the
first report to build an in vitro model to simulate
macrophage renewal ways.
Materials and methods
Reagents and cell lines
Gefitinib powder was purchased from Selleck
Chemicals (cat. no. S1025). The human lung cancer cell line PC-9
(cat. no. 90071810; European Collection of Authenticated Cell
Cultures; STR analysis was used to confirm the cell line STR loci
including D5S818, D13S317, D7S820, D16SS539, VWA, TH01, AM, TPOX
and CSE1PO were all matched) with EGFR-mutation (exon 19 deletion,
E746-A750 deletion) and the human acute monocyte leukemia cell line
THP-1 (TIB-202™; American Type Culture Collection) were used. Both
of them were provided by Cobioer Biosciences Co., Ltd. PC-9 cells
were cultured in RPMI 1640 medium (Hyclone; Cytiva) with 10% fetal
calf serum (PAN Biotech UK, Ltd.). The THP-1 cells were cultured in
RPMI 1640 medium with 10% fetal calf serum supplemented with 0.05
mM 2-mercaptoethanol (Sigma-Aldrich; Merck KGaA) and differentiated
into macrophages using 320 nM phorbol-12-myristate-13-acetate (PMA;
Abcam) for 48 h at 37°C. All cells were maintained in a 5%
CO2 humidified incubator at 37°C.
Inducing gefitinib-resistant sublines
of PC-9 cells with and without macrophages co-culture
PC-9 cells and macrophages differentiated from THP-1
cells were co-cultured in Transwell system with 3-µm Biopore™
membrane (MilliporeSigma) inserted in six-well plates. PC-9 cells
were seeded into lower chamber (5×105 cells/well) and
macrophages in the upper chamber (6.25×104 cells/well)
in RPMI-1640 medium supplemented with 10% fetal calf serum, 100
U/ml penicillin and 100 µg/ml streptomycin. Gefitinib-resistance
was induced in PC-9 cells by alternating gefitinib treatment for 24
h and recovery in drug-free medium at 37°C. When PC-9 cells were at
50–70% confluency, they were sub-cultured 1–2 times, then the
processes of treatment and recovery were repeated. Gefitinib was
added with increasing concentration from 5 to 35 µM by 5 µM each
step. Macrophages were renewed when co-cultured PC-9 cells were
passaged. Local self-renewal macrophages and migrated macrophages
were stimulated using two co-culture ways. In one group, co-culture
of macrophages and PC-9 cells began before gefitinib treatment,
therefore, macrophages accompanied by PC-9 cells all along, during
both gefitinib treatment or recovery in drug-free medium.
Macrophages in this group were called resident macrophages (Mr). In
the other group, co-culture began after gefitinib treatment during
the first recovery in drug-free medium. Macrophages only presented
in the drug free medium recovery process. Macrophages in this group
were called migrant macrophages (Mm). As a control group, resistant
was induced in PC-9 cells without co-culture with macrophages
(Fig S1). After 10 months, PC-9
cells induced in Mr co-culture, in Mm co-culture and without
co-culture were marked as PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR,
respectively.
Cell proliferation curves
The cell viability was measured by trypan blue dye
(Spectrum Laboratory Products, Inc.) exclusion method (26). Cell counting was performed manually
using a hematocytometer. PC-9 cell growth co-cultured with
macrophage in different ratios were recorded and charted with cell
proliferation curves. Growth of PC-9 cells and the three
gefitinib-resistance sublines (PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR)
under the circumstances of different gefitinib concentration were
also measured using cell proliferation curves. Cells were counted
in triplicate.
MTT assay
The growth inhibition of PC-9 cells and three
gefitinib-resistance sublines (PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR)
to gefitinib was detected using MTT (Sigma-Aldrich; Merck KGaA)
assay. After treated with gefitinib in two-time serial dilution
(20–1.25 µM for resistance sublines, 0.04–0.0025 µM for PC-9 cells)
at 37°C for 48 h, growth inhibition was detected using MTT assay,
as described previously (27).
Optical density measurements were repeated in triplicate. Growth
inhibition ratios of the four types of cells at different
concentration of gefitinib were charted.
Flow cytometry analysis
The cell cycle (G2, S and
G0/G1 phase) distributions were analyzed
using flow cytometry. The parental PC-9 cells and three resistant
sublines (PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR) were treated with or
without gefitinib (5 µM) for 24 h at 37°C, then harvested and fixed
in 70% ethanol at 4°C overnight. After staining with PI/RNase
solution (Nanjing KeyGen Biotech Co., Ltd.) for 30 min in a 37°C
water bath, according to the manufacturer's instructions, cell
cycle distribution was detected using a FACSCalibur™ flow cytometer
(BD Biosciences).
Macrophage phenotypes were identified using
fluorescence antibodies anti-human leukocyte antigen (HLA)-DR-FITC
(1:50; cat. no. 130-095-295; Miltenyi Biotec GmbH) for M1 and
CD206-PE for M2 (1:50; cat. no. 130-095-220; Miltenyi Biotec GmbH).
Phenotype alteration of macrophages before and after co-culture
with PC-9 cells as well as after treatment with low concentration
gefitinib (5 µM) were analyzed using a FACSCalibur™ flow cytometer
(BD Biosciences). After the debris was excluded with forward and
side scatter, the data were presented using x- and y-axis
biexponential display. Cells close to the x-axis were corrected.
The cell distribution was compensated to provide an even visual.
The gate of negative and positive of markers was set manually,
since the quadrant gate was shifted after compensation. Because the
shift mainly occurred in x-axis, the gate was set according to FITC
channel histogram. The same gate was applied in all groups. All
analysis was performed using BD FACSDiva software (version 8.0; BD
Biosciences). All processes were performed using the manufacturer's
protocol and checked by an expert in flow cytometry technology.
High-resolution melting analysis and
sequencing
Genomic DNA of parental PC-9 cells and all resistant
sublines (1×106 cells) were extracted using a
E.Z.N.A.® tissue DNA kit (Omega Bio-Tek, Inc.) according
to manufacturer's protocol for cultured cells. PCR was performed in
a total volume of 10 µl reaction mixture with a 15 µl mineral oil
overlay in each tube on a 72-well rotor. The reaction mixture
contained 1× PCR buffer (Takara Bio, Inc.) with MgCl2
(2.0 mM for EGFR exon 19 and 20; 2.5 mM for EGFR exon 18 and 21),
200 µM dNTPs, 0.25 U Hot Start Ex Taq polymerase (Takara Bio,
Inc.), 10 ng genomic DNA, 0.5 µM primers and 1× LCGreen
PLUS® (BioFire Diagnostics). Primer sequences are
presented in Table SI. PCR was
performed on a Rotor-Gene Q (Qiagen, Inc.) instrument with 35
cycles of 98°C for 20 sec, 58–67°C for 20 sec (58°C for exon 19;
62°C for exon 18; 67°C for exons 20, 21) and 72°C for 20 sec. Then
high-resolution melting (HRM) was run from 60–95°C and other
parameters were defaulted. Instrumental commercial software
(Rotor-Gene Q Series software; version 2.3.4; Qiagen, Inc.) was
used for HRM analysis (28,29).
EGFR exon 18–21 fragments for sequencing were
amplified on a Mastercycler (Eppendorf) with 35 cycles of 98°C for
30 sec, 61°C for 30 sec and 72°C for 30 sec. The reaction mixture
contained 1× PCR buffer (Takara Bio, Inc.) with 2.0 mM
MgCl2, 200 µM dNTPs, 0.25 U Hot Start Ex Taq polymerase
(Takara Bio, Inc.), 10 ng genomic DNA and 0.5 µM primers. Primer
sequences are presented in Table
SI. In order to double check the results from HRM analysis and
confirm deletion mutation sequence of the 4 fragments, Sanger
sequencing of PCR products was performed blindly, which was
performed by General Biol., Inc. (www.generalbiol.com).
Western blot analysis
PC-9 cells, PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR were
collected, and treated with cell lysis buffer (Beyotime Institute
of Biotechnology). Whole proteins were quantified using a
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology). Aliquots of cell lysate containing 60 µg protein
were separated by 10% SDS-PAGE (Beyotime Institute of
Biotechnology) and transferred to polyvinylidene difluoride
membranes (MilliporeSigma). After blocking with 5% skim milk for 2
h at room temperature, the membranes were incubated with anti-EGFR
(rabbit monoclonal antibody; 1:1,000; cat. no. #4267; Cell
Signaling Technology, Inc.), anti-phosphorylated (p)-EGFR (rabbit
monoclonal antibody, Try1068; 1:1,000; cat. no. #3777; Cell
Signaling Technology, Inc.), anti-HER2 (rabbit polyclonal antibody;
1:2,000; cat. no. 18299-1-AP; ProteinTech Group, Inc.),
anti-insulin-like growth factor 1 receptor (IGF-1R; mouse
monoclonal antibody; 1:2,000; cat. no. 66283-1-Ig; ProteinTech
Group, Inc.), anti-TGF-βII receptor (TGF-βIIR; rabbit monoclonal
antibody; 1:1,000; cat. no. ab184948; Abcam) and anti-β actin
(mouse monoclonal antibody; 1:500; cat. no. TA328071; OriGene
Technologies, Inc.) at 4°C overnight. Afterwards, they were treated
with IRDye 800CW goat anti-rabbit (cat. no. #926-32211) and
anti-mouse (cat. no. #926-32210) secondary IgG antibodies (1:5,000;
LI-COR Biosciences) for 1 h at room temperature, and the blots were
visualized using an Odyssey CLX (LI-COR Biosciences). The
gray-level of bands were analyzed using Image Studio (version 4.0;
LI-COR Biosciences).
Immunofluorescence staining
PC-9 cells, PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR were
cultured on coverslips. After fixing with 4% formaldehyde in PBS
for 15 min and blocking with 50 µl goat serum (OriGene
Technologies, Inc.) for 30 min at room temperature, cells were
incubated with anti-E-cadherin (mouse monoclonal antibody; 1:50;
60335-1-lg; ProteinTech Group, Inc.) and anti-Vimentin (rabbit
polyclonal antibody; 1:50; 10366-1-AP; ProteinTech Group, Inc.) at
4°C overnight, respectively. Then they were incubated with
Rhodamine (TRITC)-conjugated goat anti-rabbit IgG (1:50; cat. no.
SA00007-2; ProteinTech Group, Inc.) and fluorescein
(FITC)-conjugated goat anti-mouse IgG (1:100; cat. no. SA00013-1;
ProteinTech Group, Inc.) for 1 h at room temperature, and DAPI
staining solution 5 min at room temperature. Samples were imaged
using an immunofluorescence microscope (Leica DMI 3000B; Leica
Microsystems, Inc.).
Enzyme-linked immunosorbent assay
(ELISA)
TGF-β1 levels in cell culture supernatants of
transient treatment and long term induction were detected. TGF-β1
levels were measured two consecutive days after discarding
gefitinib using a Human TGFβ1 ELISA kit (cat. no. ab100647; Abcam)
according to the manufacture instruction. In the transient
treatment group, PC-9 cells in Mr co-culture (PC-9/Mr), in Mm
co-culture (PC-9/Mm) as well as without co-culture (PC-9) were
treated with 5 µM gefitinib at 37°C for 24 h. In the long term
induction group, three gefitinib-resistant sublines were treated
with 35 µM gefitinib at 37°C for 24 h.
Statistical analysis
All data are presented as mean ± standard deviation
(SD). One way analysis of variance (ANOVA) and Tukey's HSD test
were used to compare the difference of cell numbers in various
environments and cell cycle distribution among four cell types. All
calculations were performed using Excel (Microsoft Office 2013;
Microsoft Corporation) or R software version 3.5.2 (30). P<0.05 was considered to indicate
a statistically significant difference. All of the assays were
repeated at least two times.
Results
Macrophages affect acquirement of PC-9
cell gefitinib-resistance
Macrophages and PC-9 cells were co-cultured in a
Transwell system using two approaches: Co-culture starting before
(Mr) or after gefitinib treatment (Mm) to induce PC-9 cell
resistance to gefitinib. The macrophages differentiated from THP1
cells and were not polarized specifically. PC-9 cells proliferation
was inhibited in the co-culture system, compared with those without
co-culture. The inhibition was stronger with increasing macrophage
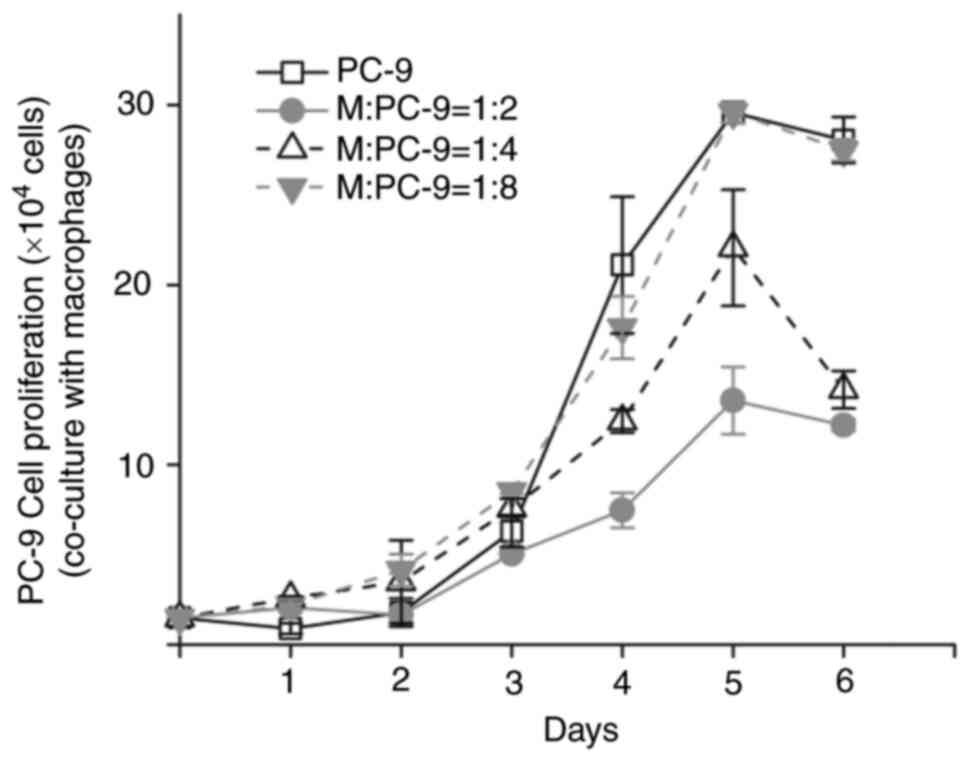
percentage (Fig. 1 and Table SII). When the ratio of macrophages
to PC-9 cells was 1:8, PC-9 cell proliferation in the co-culture
system was not markedly different compared with the control PC-9
cells. Therefore, the co-culture system was built at a ratio of 1:8
(M:PC-9).
Following gefitinib treatment in each step, PC-9
cells in Mr and Mm co-culture as well as without co-culture
recovered at different speeds. It took about 9–13 days for PC-9
cells to recover and reach confluency in Mm co-culture, about 13–17
days in Mr co-culture, and about 10–14 days without co-culture
after each step treatment from 5 to 30 µM gefitinib (data not
shown). When treated with 35 µM gefitinib for 24 h, PC-9/Mm/GR
cells took 8.9±2.0 days to reach confluency again in drug-free
medium, which was significantly shorter compared with the time
needed by PC-9/Mr/GR cells (14.7±2.7 days) and PC-9/GR cells
(11.0±1.7 days). By contrast, parental PC-9 cells could not recover
even after 60 days (Data was not shown). After 10 months, the
PC-9/Mr/GR, PC-9/Mm/GR and PC-9/GR sublines all acquired resistance
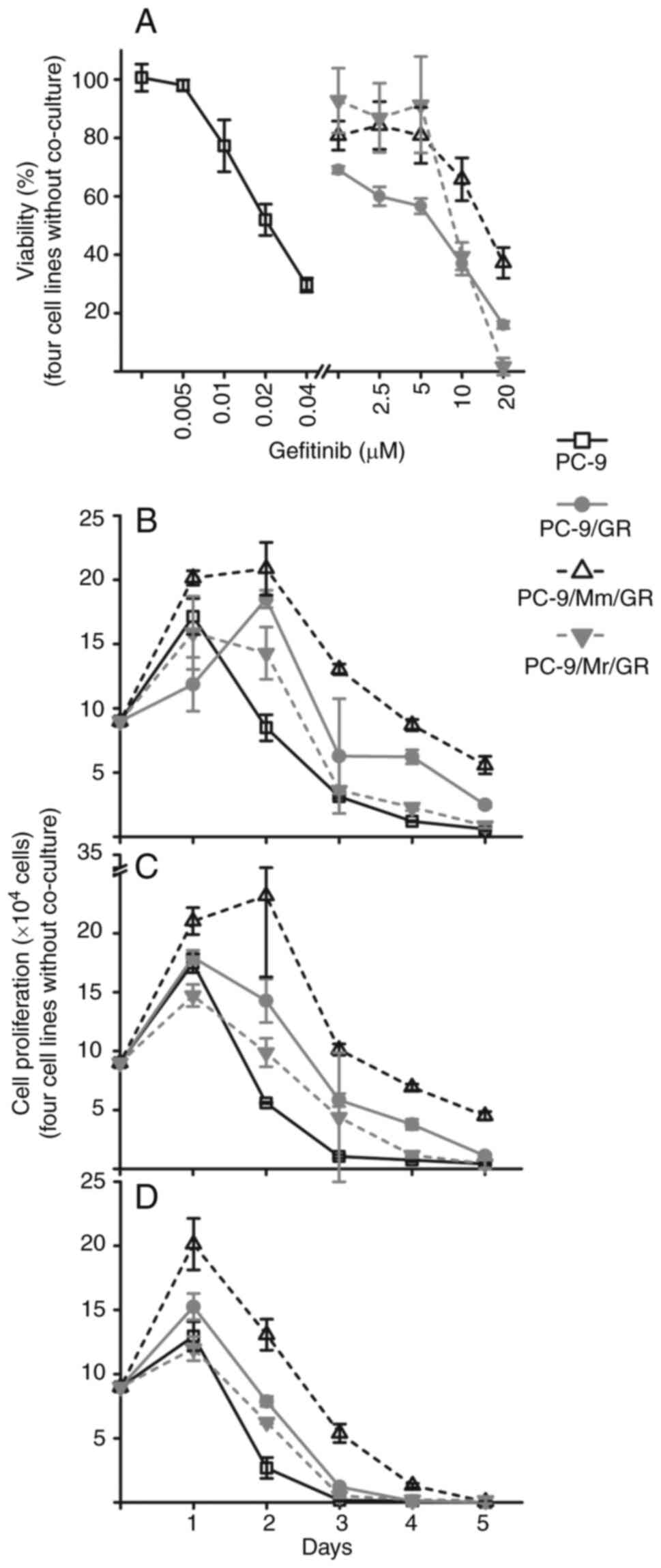
to gefitinib as shown in Fig. 2A
and Table SIII. Half maximal
inhibition concentration (IC50) of PC-9 was ~0.02 µM.
PC-9/Mm/GR had the largest IC50 at ~20 µM.
IC50 of PC-9/GR was slightly deceased compared with that
of PC-9/Mr/GR, and both of them located between 5 and 10 µM. At low
concentration of gefitinib (<10 µM), PC-9/Mr/GR had a higher
viability compared with PC-9/GR (Fig.
2A).
PC-9/Mm/GR had the strongest resistance ability.
PC-9/Mr/GR displayed stronger resistant ability compared with
PC-9/GR at low concentration of gefitinib (<10 µM). The
proliferation curves of the three resistant sublines and the
parental PC-9 cells in the presence of 5, 10 and 20 µM gefitinib
are presented in Fig. 2B-D and
Table SIV. PC-9 cells had
proliferative ability only on day 1 in three gefitinib
concentration environments, which then began to be inhibited. By
contrast, the three resistant sublines could keep proliferating on
Day 2 in 5 µM gefitinib environment, while PC-9/Mm/GR proliferated
on Day 2 even in 10 µM gefitinib environment. PC-9/Mm/GR maintained
the largest cell numbers from Day 1 in three gefitinib
environments. The three gefitinib-resistant sublines had a similar
proliferation trend to that of parental PC-9 cells in a gefitinib
environment. PC-9/Mm/GR showed the strongest proliferation ability
from the beginning, and PC-9/GR demonstrated increased
proliferation compared with PC-9/Mr/GR.
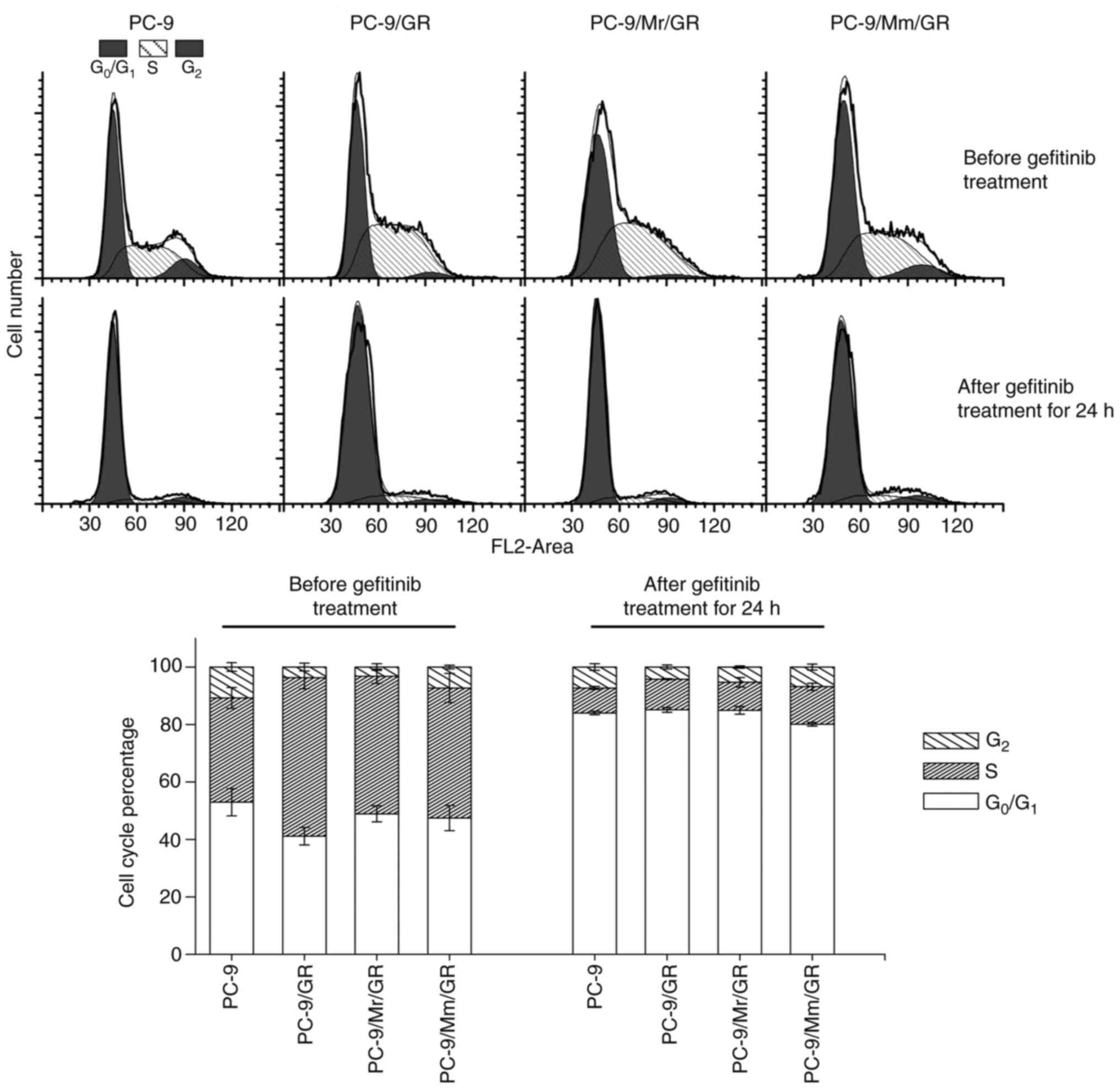
Features of gefitinib-resistant
sublines are induced in three ways
Gefitinib-resistant sublines induced in three ways
displayed different features. PC-9/Mm/GR had similar a cell cycle
distribution to that of parental PC-9 cells (the G2, S
and G0/G1 phase percentage was 10.7±1.5% vs.
7.3±0.6%, P=0.075; 36.3±3.6% vs. 45.3±5.0%, P=0.112 and 52.9±4.7%
vs. 47.5±4.4%, P=0.433, respectively in PC-9 vs. PC-9/Mm/GR cells),
while PC-9/Mr/GR and PC-9/GR showed a significantly higher
percentage of S phase and lower percentage of G2 phase
(specifically, the G2, S and G0/G1
phase percentage was 3.2±1.1%, P=0.001; 47.9±2.5%, P=0.024 and
48.9±2.8%, P=0.577, respectively, in PC-9/Mr/GR cells, and
3.8±1.4%, P=0.001; 55.1±3.8%, P=0.002 and 41.1±3.0%, P=0.024,
respectively, in PC-9/GR cells) compared with those of parental
PC-9 cells (Fig. 3 and Tables SV). After treatment with 5 µM
gefitinib for 24 h, the majority of cells, PC-9 cells and three
resistant sublines, tended to stay in G0/G1
phase and decreased markedly in S phase. The cell cycle
distribution was comparable among parental PC-9 cells and the three
resistant sublines. PC-9/Mm/GR displayed the strongest
proliferation activity with the largest S and G2 phase
percentages (19.9±0.6% in PC-9/Mm/GR vs. 16.0±0.6% in PC-9, P=0.003
and 14.9±0.8% in PC-9/GR vs. PC-9, P=0.487 and 15.1±1.3% in
PC-9/Mr/GR vs. PC-9, P=0.610; Tables
SVI).
After 10 months of induction, no new mutation on the
EGFR exon 18–21 was revealed in any of the three resistant
sublines, except for harboring a deletion mutation on exon 19 from
parental PC-9 cells (Figs. S2 and
S3). However, resistant sublines
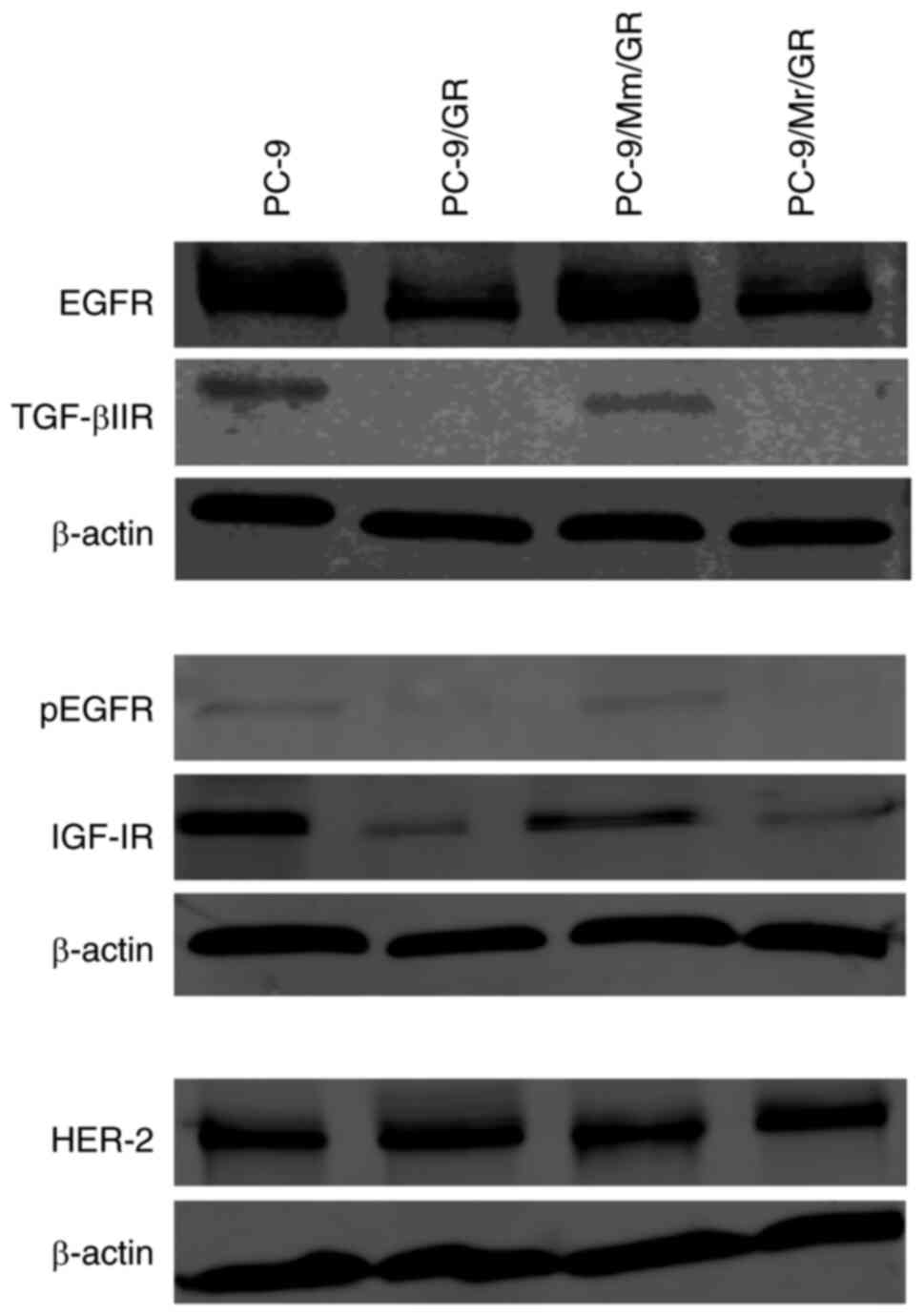
exhibited varied phenotypes. Compared with the findings in parental
PC-9 cells, PC-9/GR and PC-9/Mr/GR lost expression of certain
membrane receptors, including EGFR, p-EGFR, IFG-1R and TGF-βIIR
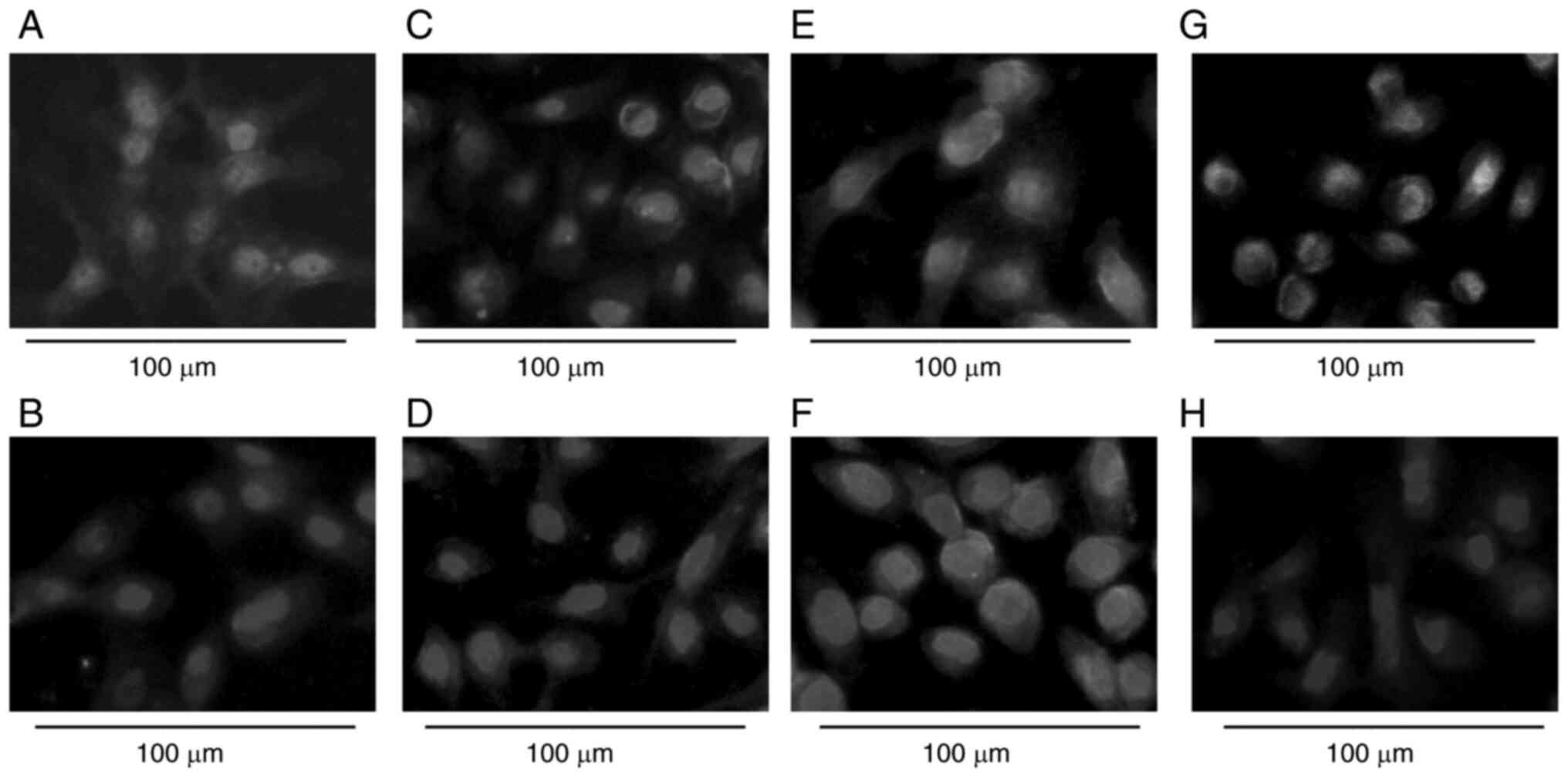
(Fig. 4). E-cadherin expression
was also decreased, while vimentin expression did not markedly
change (Figs. 5A-D, G, H and
S4A-D, G and H). PC-9/Mm/GR
maintained EGFR, p-EGFR, IFG-1R and TGF-βIIR expression similarly
to parental PC-9 cells (Fig. 4).
In addition, PC-9/Mm/GR sustained both E-cadherin and vimentin
expression (Figs. 5E and F and
S4E and F).
The TGF-β content in the supernatant of PC-9 cells
in Mr and Mm transient co-culture as well as without co-culture was
measured for 2 days continuously after treatment with gefitinib (5
µM) for 24 h. In the supernatant of PC-9 cells in Mr co-culture
(PC-9/Mr), the TGF-β content remained close to the basic level
(1,114.9 pg/ml). However, in the supernatant of PC-9 cells without
co-culture (PC-9) and in Mm co-culture (PC-9/Mm), the TGF-β content
increased and was higher compared with that of PC-9/Mr in days 1
(P=0.0131) and 2 (P=0.007) after discarding the drugs (Fig. 6A and Table SVII). The TGF-β level in the
supernatant of the gefitinib-resistant sublines PC-9/Mr/GR,
PC-9/Mm/GR and PC-9/GR was also detected. After treatment with
gefitinib (35 µM) for 24 h, the TGF-β level in the supernatant was
similar among the three sublines at day 1 after discarding the
drugs (P=0.381). However, on day 2, the TGF-β level decreased
significantly in PC-9/Mr/GR compared with the other two sublines
(P=0.027; Fig. 6B and Table SVIII).
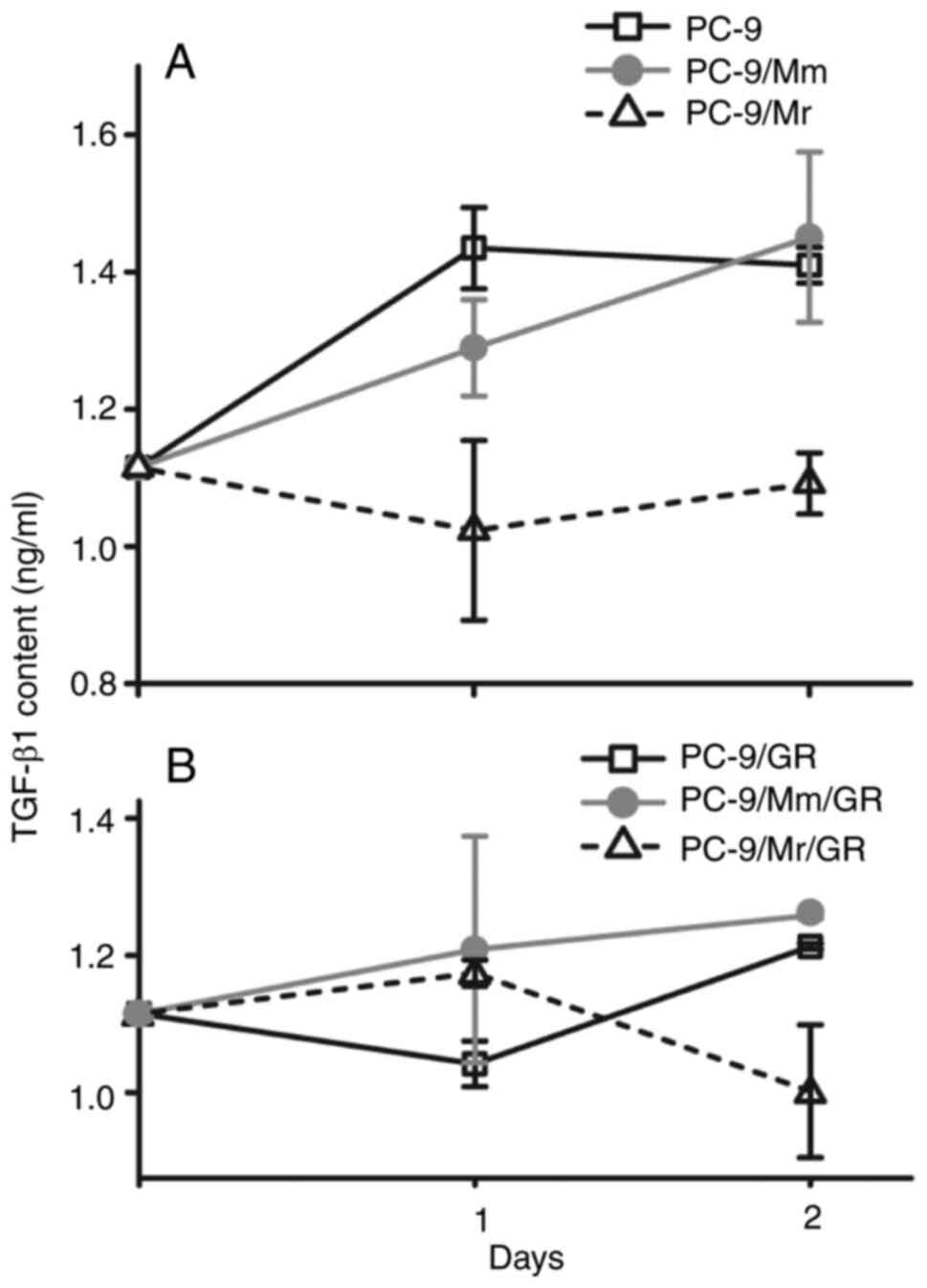 | Figure 6.TGF-β levels in the supernatant of
PC-9 and three resistant sublines. (A) After PC-9 cells alone and
transient co-culture in Mr way (PC-9/Mr) or Mm way (PC-9/Mm) were
treated with gefitinib (5 µM) for 24 h, TGF-β levels in the
supernatant of PC-9/Mr were significantly lower compared that in
others at the first and second day after discarding drugs (P=0.0131
and P=0.007, respectively). (B) After resistant sublines of
PC-9/GR, PC-9/Mr/GR and PC-9/Mm/GR were treated with gefitinib (35
µM), TGF-β level was not significantly different among three groups
(P=0.381) at the first day, while it was significantly lower in
PC-9/Mr/GR compared with that of others at the second day
(P=0.027). Mr, resident macrophage; Mm, migrated macrophage;
PC-9/Mr, PC-9 cells co-cultured with Mr; PC-9/Mm, PC-9 cells
co-cultured with Mm; PC-9/Mr/GR, gefitinib-resistant sublines
induced with PC-9 cells in Mr co-culture; PC-9/Mm/GR,
gefitinib-resistant sublines induced with PC-9 cells in Mm
co-culture. |
Macrophage polarization in co-culture
systems
Following differentiation from THP-1 with PMA, ~25%
of macrophages expressed HLA-DR while nearly none expressed CD206.
As shown in Fig. 7, the percentage
of macrophages with HLA-DR expression increased slightly when
treated with gefitinib for 24 h. When co-cultured with PC-9 cells,
the percentage of macrophages with HLA-DR expression decreased to
4.9 and 1.6% at a ratio of macrophages to PC-9 cells of 1:8 and
1:1, respectively, whereas no CD206-expressing macrophages
appeared. In either co-culture way and in either co-culture cell
ratio, there were not macrophages polarized to M1 (HLA-DR positive)
or M2 (CD206-positive).
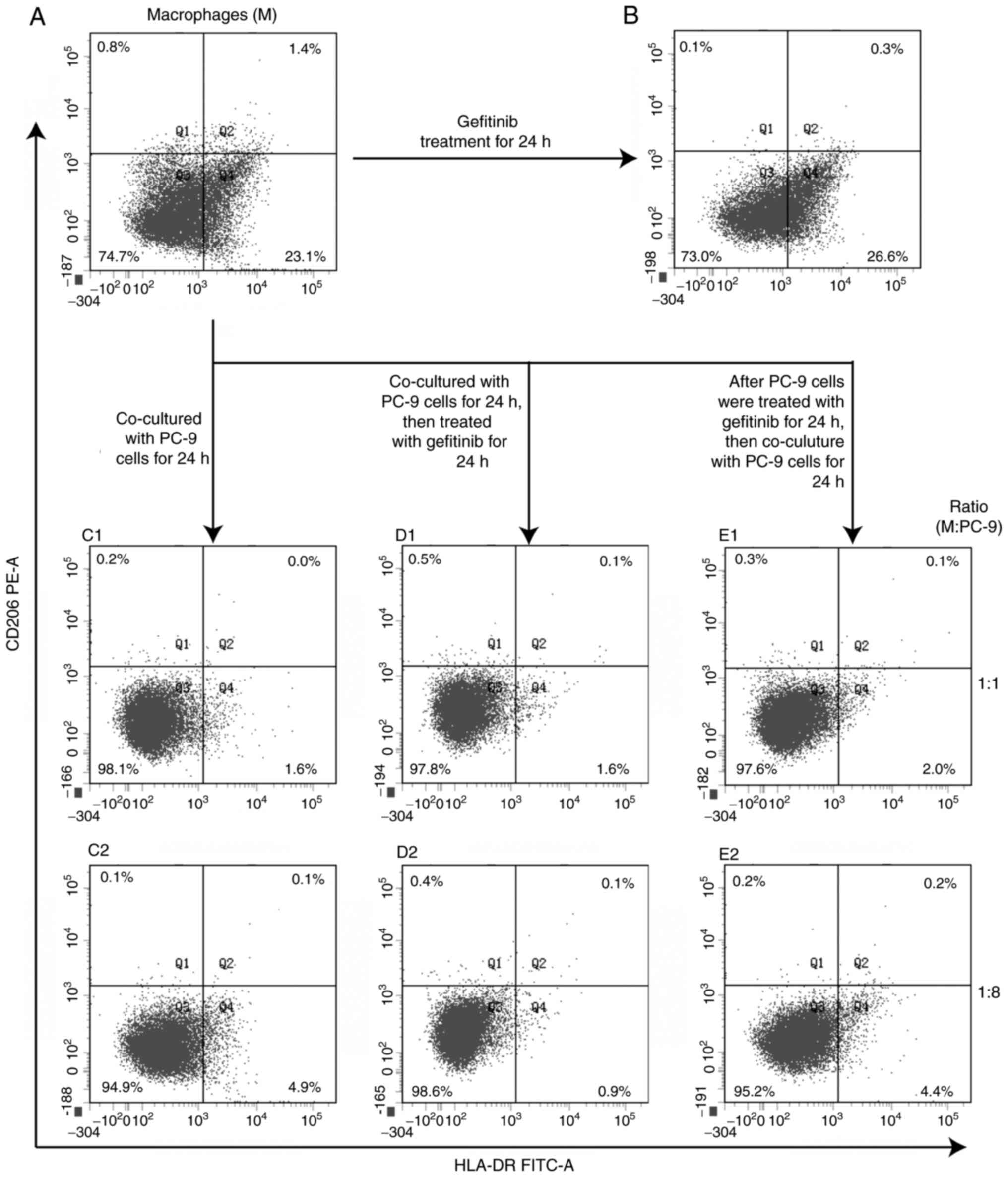 | Figure 7.Macrophage polarization after
gefitinib treatment and co-culture with PC-9 cells. HLA-DR (M1-like
macrophage marker) and CD206 (M2-like macrophage marker) positive
macrophages were detected with flow cytometry. (A) After THP-1 was
differentiated with PMA (320 nM) for 48 h, there were ~1/4
HLA-DR-positive macrophages, while very few CD206 positive cells.
(B) Macrophages differentiated from THP-1 were treated with
gefitinib for 24 h, the percentage of HLA-DR positive macrophages
increased slightly. (C1 and C2) When co-cultured with PC-9 cells in
different ratio, the percentage decreased. (D1 and D2) After
macrophages co-cultured with PC-9 for 24 h, then macrophages and
PC-9 were treated with gefitinib together (Mr co-culture way),
HLA-DR-positive percentage remained low at either co-culture ratio.
(E1 and E2) If PC-9 cells were treated with gefitinib first, then
co-cultured with macrophages (Mm co-culture way), HLA-DR-positive
macrophages remained low. There were few CD206-positive macrophages
during the whole process. HLA, human leukocyte antigen; PMA,
phorbol-12-myristate-13-acetate; Mr, resident macrophage; Mm,
migrated macrophage. |
Discussion
Tumor cell activities change the microenvironment,
and the tumor microenvironment also shapes tumor cell
characteristics. The present study treated PC-9 cells with
gefitinib in three different microenvironments. It was revealed
that PC-9 cells always acquired resistance to gefitinib, either
without macrophages co-culture or with macrophages co-culture in
two ways. Similar to patients with lung cancer in gefitinib
treatment who present individual differences, in the present study,
PC-9 cells indicated different features after being exposed to
gefitinib for a period of time with the same dose and frequency in
different environments. PC-9/GR and PC-9/Mr/GR were similar. Both
decreased EGFR activation, which is the binding target of
gefitinib, and lost the expression of other proliferated-related
membrane receptors, such as IGF-1R and TGF-βIIR. It was not strange
that PC-9/GR and PC-9/Mr/GR cells were not sensitive to gefitinib
when the binding target was missing. EGFR, IGF-1R and TGF-βIIR
triggered by corresponding ligands will activate downstream
signaling pathways, including the Ras-MAPK and PI3K signaling
pathways, which in turn promotes cell proliferation and cell
survival (31,32). Due to reduced stimulation of
proliferation signaling, the aforementioned two sublines mostly
accumulated in phases prior to G2 in the cell cycle.
This ‘low proliferation status’ was the result of receptors
closing, which could help the cells to avoid from further gefitinib
treatment.
Notably, all the three resistant sublines showed
different resistance ability, but none of them showed the most
common EGFR exon 20 mutation (T790M). In our previous study, PC-9
cells were induced to exhibit resistance to gefitinib with another
administration regimen, the alteration of high-dose stimulation and
low-dose maintenance, which led to the appearance of the T790M
mutation in the resistant subline (Zhao et al, unpublished
data). While the mechanism is unclear, changes in the environment
modulate tumor cells to develop secondary resistance, which has
been shown in the clinic where the resistance mechanism is
individually characterized, even among patients with lung cancer
who harbored the same sensitive mutation with the same
administration regimen (2,4,33).
Macrophages renewing locally and migrating from the
blood vessels change the microenvironment continuously (6). The present study explored how
macrophage renewal affected PC-9 cells to develop secondary
resistance to gefitinib. Macrophages that renew locally always
share the same environment with tumor cells (6,19);
thus, co-culture was performed, which started before PC-9 treatment
with gefitinib (Mr) to let macrophages always accompany PC-9 cells.
Cell damage is the common signal to attract macrophages, either
monocyte-derived or resident macrophages, to carry out clearance
activity and reparative response (6,17,20,21).
Thus, the co-culture starting after PC-9 cell treatment (Mm)
simulated this renewal mode. Notably, in Mm co-culture, macrophages
markedly assisted PC-9 cell recovery from gefitinib treatment and
developed resistance rapidly.
However, if macrophages appeared before gefitinib
treatment, they hardly protected PC-9 cells from gefitinib
treatment. It seemed that resident (Mr) macrophages could not
recognize PC-9 cells exposure to gefitinib as an abnormal event as
Mm macrophages did. Unlike cytotoxic killing agents, gefitinib
inhibits PC-9 cells proliferation, while not killing directly
(34). Both PC-9 cells and
gefitinib-resistant sublines mainly showed elongation of
G0/G1 phase after gefitinib treatment in the
first 24 h. The gradual proliferation inhibition resembled slow
physiological proliferation, and could mislead macrophages (Mr) in
the same microenvironment with PC-9 cells from turning on
protection. By contrast, if macrophages (Mm) could recognize it as
a damage signal they may help PC-9 cells to survive. It is not
clear how Mm macrophages protected PC-9 cells from gefitinib. TGF-β
secretion was increased in the Mm group, while it was inhibited in
the Mr group. At the same time, PC-9/Mm/GR maintained TGF-βIIR
expression, while PC-9/Mr/GR did not. This indicated that
TGF-β/TGFR signal transduction could be activated in PC-9/Mm/GR.
Normally, TGF-β contributes to tumor initiation, progression,
metastasis and chemotherapy resistance by regulating inflammatory
factors, fibrotic process and epithelial mesenchymal transition,
besides promoting proliferation (35–37).
The present study revealed a relatively strong vimentin expression
in PC-9/Mm/GR cells compared with that in other sublines, which
suggested that the cells exhibited certain mesenchymal
character.
In the future, it would be helpful to detect
inflammatory factors and fibrosis in vivo or co-culture
fibroblasts and lymphocytes in vitro. In the present study,
Mm macrophages not only assisted PC-9 cells to keep a stable
phenotype compared with that of their parental cells, but they
displayed a relative strong vimentin expression, which could be
regulated by TGF-β signaling and may be the potential reason for
the PC-9/Mm/GR acquisition of the fastest and strongest resistance
ability observed. Decreasing TGF-β expression or inhibiting its
binding with receptor to inhibit TGF-β signal transduction are
needed to confirm the function of TGF-β in the process, which is a
limitation of the current study. Renewal modes of macrophages may
lead to different distribution in time and space. Migrated
macrophages, while not resident ones, may play a protective
function, which could affect the development of resistance. If
macrophage infiltration prior to gefitinib treatment and migrated
macrophages to the TME were monitored, development of secondary
resistance could be predicted accurately.
Previous clinical studies aimed to investigate the
association between the efficacy of EGFR-TKIs treatment and the
density of macrophages, but the results are controversial (13,14,22,23,38).
The discrepancy in results is not unexpected, since the majority of
samples are obtained by surgical excision, while operations are
usually performed prior to EGFR-TKIs treatment. The macrophages in
the above tissues are likely to be associated with tumorigenesis
and malignant potential, rather than treatment response (24,25).
High levels of certain inflammatory cytokines and chemokines are
found to be associated with EGFR-TKIs resistance, but the clinical
data on concentration changes before and after treatment are not
enough yet (39,40). In numerous studies, macrophages are
categorized into M1 or M2 polarization (13–17).
The present study did not induce macrophage polarization. Upon
treatment of PC-9 cells with gefitinib in Mr or Mm co-culture,
macrophages did not polarize. This suggested that macrophages may
be activated to protect tumor cells prior to polarization.
Considering that migrated and resident macrophages play their own
roles in individual inflammatory status and are associated with
smoking, infection, carcinogenesis and toxic reaction after
treatment (6,18,21,22,25,41),
the present study attempted to exclude other interference. However,
the role of the renewal type in vivo was not evaluated,
which is a limitation of the present study.
From traditional chemotherapy to target therapy,
cancer treatment strategies always aim to fight tumor cells
directly. Although the latter causes less injury to the host body,
resistance always develops after the interaction of tumor cells and
the TME (2–4,42).
Immunotherapy has been largely developed, but the tactic remains to
avoid or limit harmful effects in the host body. Macrophages may be
a prospective target to be ‘trained tumor killing cells’ by host
immunoreaction (43,44). Despite several cases showing good
responses, it is not clear if novel resistance-like process will
develop or whether adverse events will occur (45,46).
Since cell killing is followed by resistance and host injury, it
should be considered changing the treatment strategy to quarantine
tumor cells to mold them to be regulated by the host body or limit
their harmful activity. Macrophages could be a prospective target
to be ‘trained’ to regulate the TME.
In summary, macrophage renewal modes could interfere
with the development of secondary resistance by altering their
distribution in time and space. The present study may provide a
different perspective to identify novel approaches to monitor
EGFR-TKIs response and design new treatment strategies against lung
cancer.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
We thank Professor Jie Zhu in the Clinical Flow
Cytometry Center of the Second Hospital of Dalian Medical
University (Dalian, China), for his help in the analysis of flow
cytometry results.
Funding
This work was supported by grants from the National Natural
Science Foundation of China (grant nos. 81972022 and 81572919),
Natural Science Foundation of Liaoning Province (CN; grant no.
2019-MS-087), Scientific Research Foundation of Liaoning
Educational Department (CN; grant no. LZ2019036) and Excellent
Young Talents Fund Program of Higher Education Institutions of
Liaoning Province (CN; grant no. LR2019022).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The data generated in the present study may be found in
the Figshare at the following URL: https://figshare.com/articles/online_resource/Sanger_sequencing_results_rar/21680843.
Authors' contributions
BZ and ML designed the outline of the study. BZ and
YZ conducted the experiments and data analysis. ML wrote the
manuscript. BZ, SL and ML supervised the study and contributed to
data interpretation and manuscript revision. BZ and ML confirmed
the authenticity of all raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TKI
|
tyrosine kinase inhibitor
|
|
TME
|
tumor microenvironment
|
|
Mr
|
resident macrophage
|
|
Mm
|
migrated macrophage
|
|
PC-9/Mr/GR
|
gefitinib-resistant sublines induced
with PC-9 cells in Mr co-culture
|
|
PC-9/Mm/GR
|
gefitinib-resistant sublines induced
with PC-9 cells in Mm co-culture
|
|
PC-9/GR
|
gefitinib-resistant sublines induced
with PC-9 cells without macrophage co-culture
|
|
PC-9/Mr
|
PC-9 cells co-cultured with Mr
|
|
PC-9/Mm
|
PC-9 cells co-cultured with Mm
|
|
ELISA
|
Enzyme-linked immunosorbent assay
|
|
PMA
|
phorbol-12-myristate-13-acetate
|
|
GR
|
gefitinib resistant
|
|
p-EGFR
|
phosphorylated-EGFR
|
|
IGF-1R
|
insulin-like growth factor 1
receptor
|
|
TGF-βIIR
|
TGF-βII receptor
|
|
HLA
|
human leukocyte antigen
|
|
HRM
|
high-resolution melting
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohashi K, Maruvka YE, Michor F and Pao W:
Epidermal growth factor receptor tyrosine kinase
inhibitor-resistant disease. J Clin Oncol. 31:1070–1080. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Konig D, Prince SS and Rothschild SI:
Targeted therapy in advanced and metastatic non-small cell lung
cancer. An update on treatment of the most important actionable
oncogenic driver alterations. Cancers (Basel). 13:804–840. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bivona TG and Doebele RC: A framework for
understanding and targeting residual disease in oncogene-driven
solid cancers. Nat Med. 22:472–478. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin A, Wei T, Meng H, Luo P and Zhang J:
Role of the dynamic tumor microenvironment in controversies
regarding immune checkpoint inhibitors for the treatment of
non-small cell lung cancer (NSCLC) with EGFR mutations. Mol Cancer.
18:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Loyher PL, Hamon P, Laviron M,
Meghraoui-Kheddar A, Goncalves E, Deng Z, Torstensson S, Bercovici
N, de Chanville CB, Combadière B, et al: Macrophages of distinct
origins contribute to tumor development in the lung. J Exp Med.
215:2536–2553. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonde AK, Tischler V, Kumar S, Soltermann
A and Schwendener RA: Intratumoral macrophages contribute to
epithelial-mesenchymal transition in solid tumors. BMC Cancer.
12:352012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mantovani A, Marchesi F, Malesci A, Laghi
L and Allavena P: Tumour-associated macrophages as treatment
targets in oncology. Nat Rev Clin Oncol. 14:399–416. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Palma M and Lewis CE: Macrophage
regulation of tumor responses to anticancer therapies. Cancer Cell.
23:277–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mantovani A and Allavena P: The
interaction of anticancer therapies with tumor-associated
macrophages. J Exp Med. 212:435–445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jia Y, Li X, Jiang T, Zhao S, Zhao C,
Zhang L, Liu X, Shi J, Qiao M, Luo J, et al: EGFR-targeted therapy
alters the tumor microenvironment in EGFR-driven lung tumors:
Implications for combination therapies. Int J Cancer.
145:1432–1444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang DH, Lee HS, Yoon D, Berry G, Wheeler
TM, Sugarbaker DJ, Kheradmand F, Engleman E and Burt BM:
Progression of EGFR-mutant lung adenocarcinoma is driven by
alveolar macrophages. Clin Cancer Res. 23:778–788. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garrido-Martin EM, Mellows TWP, Clarke J,
Ganesan AP, Wood O, Cazaly A, Seumois G, Chee SJ, Alzetani A, King
EV, et al: M1(hot) tumor-associated macrophages boost
tissue-resident memory T cells infiltration and survival in human
lung cancer. J Immunother Cancer. 8:e0007782020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao F, Liu N, Ma X, Qin J, Liu Y and Wang
X: M2 macrophages reduce the effect of gefitinib by activating
AKT/mTOR in gefitinib-resistant cell lines HCC827/GR. Thorac
Cancer. 11:3289–3298. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang B, Zhang Y, Zhao J, Wang Z, Wu T, Ou
W, Wang J, Yang B, Zhao Y, Rao Z and Gao J: M2-polarized
macrophages contribute to the decreased sensitivity of EGFR-TKIs
treatment in patients with advanced lung adenocarcinoma. Med Oncol.
31:1272014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung FT, Lee KY, Wang CW, Heh CC, Chan
YF, Chen HW, Kuo CH, Feng PH, Lin TY, Wang CH, et al:
Tumor-associated macrophages correlate with response to epidermal
growth factor receptor-tyrosine kinase inhibitors in advanced
non-small cell lung cancer. Int J Cancer. 131:E227–E235. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gentek R, Molawi K and Sieweke MH: Tissue
macrophage identity and self-renewal. Immunol Rev. 262:56–73. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mascia F, Lam G, Keith C, Garber C,
Steinberg SM, Kohn E and Yuspa SH: Genetic ablation of epidermal
EGFR reveals the dynamic origin of adverse effects of anti-EGFR
therapy. Sci Transl Med. 5:199ra1102013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ginhoux F and Guilliams M: Tissue-resident
macrophage ontogeny and homeostasis. Immunity. 44:439–449. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu-Vanpala S, Deerhake ME, Wheaton JD,
Parker ME, Juvvadi PR, MacIver N, Ciofani M and Shinohara ML:
Functional heterogeneity of alveolar macrophage population based on
expression of CXCL2. Sci Immunol. 5:eaba73502020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du J, Li G, Jiang L, Zhang X, Xu Z, Yan H,
Zhou Z, He Q, Yang X and Luo P: Crosstalk between alveolar
macrophages and alveolar epithelial cells/fibroblasts contributes
to the pulmonary toxicity of gefitinib. Toxicol Lett. 338:1–9.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mukaida N, Nosaka T, Nakamoto Y and Baba
T: Lung macrophages: Multifunctional regulator cells for metastatic
cells. Int J Mol Sci. 20:1162018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Z, Maeda D, Yoshida M, Umakoshi M,
Nanjo H, Shiraishi K, Saito M, Kohno T, Konno H, Saito H, et al:
The intratumoral distribution influences the prognostic impact of
CD68- and CD204-positive macrophages in non-small cell lung cancer.
Lung Cancer. 123:127–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Casanova-Acebes M, Dalla E, Leader AM,
LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L,
Chen ST, et al: Tissue-resident macrophages provide a
pro-tumorigenic niche to early NSCLC cells. Nature. 595:578–584.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cotechini T, Atallah A and Grossman A:
Tissue-resident and recruited macrophages in primary tumor and
metastatic microenvironments: Potential targets in cancer therapy.
Cells. 10:9602021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Strober W: Trypan blue exclusion test of
cell viability. Curr Protoc Immunol. 111:A3.B.1–A3.B.3. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao BX, Wang J, Song B, Wei H, Lv WP,
Tian LM, Li M and Lv S: Establishment and biological
characteristics of acquired gefitinib resistance in cell line
NCI-H1975/gefinitib-resistant with epidermal growth factor receptor
T790M mutation. Mol Med Rep. 11:2767–2774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li M, Zhang Q, Liu L, Liu Z, Zhou L, Wang
Z, Yue S, Xiong H, Feng L and Lu S: The different clinical
significance of EGFR mutations in exon 19 and 21 in non-small cell
lung cancer patients of China. Neoplasma. 58:74–81. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun Xuerui WY, Jialin G and Li Mei:
Difference between LCGreen PLUS and EvaGreen in detecting EGFR
mutation with HRM. J Dalian Med Univ. 42:391–394. 2020.
|
|
30
|
R Core Team. R, . A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna, Austria: ISBN 3-900051-07-0, URL. http://www.R–project.org/
|
|
31
|
Hakuno F and Takahashi SI: IGF1 receptor
signaling pathways. J Mol Endocrinol. 61:T69–T86. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ark AV, Cao J and Li X: TGF-β receptors:
In and beyond TGF-beta signaling. Cell Signal. 52:112–120. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rotow J and Bivona TG: Understanding and
targeting resistance mechanisms in NSCLC. Nat Rev Cancer.
17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Paez JG, Janne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Batlle E and Massague J: Transforming
growth factor-β signaling in immunity and cancer. Immunity.
50:924–940. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Caja L, Dituri F, Mancarella S,
Caballero-Diaz D, Moustakas A, Giannelli G and Fabregat I: TGF-β
and the tissue microenvironment: Relevance in fibrosis and cancer.
Int J Mol Sci. 19:12942018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu X, Buttgereit A, Lelios I, Utz SG,
Cansever D, Becher B and Greter M: The cytokine TGF-beta promotes
the development and homeostasis of alveolar macrophages. Immunity.
47:903–912. e9042017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang H, Wang X, Li X, Fan Y, Li G, Guo C,
Zhu F, Zhang L and Shi Y: CD68(+)HLA-DR(+) M1-like macrophages
promote motility of HCC cells via NF-κB/FAK pathway. Cancer Lett.
345:91–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-beta IL-6 axis mediates selective and adaptive
mechanisms of resistance to molecular targeted therapy in lung
cancer. Proc Natl Acad Sci USA. 107:15535–15540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fukuoka M, Yoshioka K and Hohjoh H: NF-κB
activation is an early event of changes in gene regulation for
acquiring drug resistance in human adenocarcinoma PC-9 cells. PLoS
One. 13:e02017962018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wan L, Wang Y, Tang Y, Tan Y, He F, Zhang
Y, Yang K, Chen Z, Song C, Gu R, et al: Gefitinib-induced cutaneous
toxicities in brown norway rats are associated with macrophage
infiltration. Inflammation. 43:2137–2146. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bukowski K, Kciuk M and Kontek R:
Mechanisms of multidrug resistance in cancer chemotherapy. Int J
Mol Sci. 21:32332020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anderson NR, Minutolo NG, Gill S and
Klichinsky M: Macrophage-based approaches for cancer immunotherapy.
Cancer Res. 81:1201–1208. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kennedy LB and Salama AKS: A review of
cancer immunotherapy toxicity. CA Cancer J Clin. 70:86–104. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Im JS, Herrmann AC, Bernatchez C, Haymaker
C, Molldrem JJ, Hong WK and Perez-Soler R: Immune-modulation by
epidermal growth factor receptor inhibitors: Implication on
anti-tumor immunity in lung cancer. PLoS One. 11:e01600042016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hussaini S, Chehade R, Boldt RG, Raphael
J, Blanchette P, Vareki SM and Fernandes R: Association between
immune-related side effects and efficacy and benefit of immune
checkpoint inhibitors-A systematic review and meta-analysis. Cancer
Treat Rev. 92:1021342021. View Article : Google Scholar : PubMed/NCBI
|