Introduction
Pancreatic cancer (PC) is one of the deadliest
cancer types, due in part to a high incidence of early local
invasion or distant metastasis (1).
During dissemination, cancer cells must adapt to the tumor
microenvironment (TME) for successful migration, invasion, and
formation of a secondary tumor (2).
Various components in the TME act in concert with cancer cells to
create a supportive environment during tumor progression (3), making TME-cancer crosstalk an
attractive therapeutic target.
Obesity is one of the few known risk factors for PC
and correlates with a worse prognosis (4). In accordance with these
epidemiological observations, high-fat diets were shown to
contribute to tumorigenesis and metastasis in mouse models of PC
(5,6). In addition, fatty infiltration in the
pancreas is positively associated with the incidence of PC, even
after adjusting for confounding factors such as body mass index,
indicating the underlying role of obesity in the tumor TME
(7). Obesity reportedly can induce
an inflammatory and fibrotic microenvironment in PC, resulting in a
reduced response to chemotherapy (8). However, among stromal cells in the PC
microenvironment, relatively little attention has been given to
mature adipocytes, which are closely correlated with obesity.
Adipocytes are the major component of adipose tissue
and are a reservoir for energy storage. Adipocytes adjacent to
cancer cells show profound phenotypic and functional alterations.
(9,10). Using an in vitro coculture
system, we previously found that adipocytes cocultured with PC
cells presented with a delipidation and dedifferentiation
phenotype, and these activated cancer-associated adipocytes
participated in tumor progression (11). Adipocytes are rich in lipids, the
loss of which in cocultured adipocytes may be due to lipid transfer
from adipocytes to cancer cells. In breast (12), ovarian (13), and other types of cancer (14,15),
adipocyte-derived lipids are a potent energy source that supports
cancer growth and progression, suggesting that they may influence
cancer metabolism. In the present study, an in vitro
coculture model was utilized to further interrogate how adipocytes
promoted PC progression and to uncover the metabolic interaction
between adipocytes and PC cells. The metabolic competitive and
energy-plundering relationships between PC cells and adipocytes
were identified, where adipocytes showed impaired insulin
sensitivity and decreased lipid storage, and PC cells exhibited
enhanced glycolytic capacity and increased the store of lipids.
Additionally, the increased levels of lipids in cocultured PC cells
contributed to the enhanced metastatic capacity.
Materials and methods
Cells and reagents
The human pancreatic ductal adenocarcinoma cell
lines, Panc-1 and Mia PaCa2, were obtained from The Cell Bank of
Type Culture Collection of The Chinese Academy of Sciences and were
routinely tested for mycoplasma before the experiments. The Panc-1
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific)
supplemented with 10% (v/v) FBS (Gibco; Thermo Fisher Scientific).
Mia PaCa2 cells were maintained in DMEM supplemented with 10% (v/v)
FBS and 2.5% (v/v) horse serum (Gibco; Thermo Fisher Scientific).
The murine 3T3-L1 cell line is a well-established cell line that
can be stably differentiated into mature adipocytes, and due to its
good reproducibility, it is widely used for studies focusing on
obesity, diabetes as well as the tumor microenvironment (14,16).
Additionally, to the best of our knowledge, there are no stable
human preadipocyte or adipocyte cell lines that can be used in such
experiments. Thus, murine 3T3-L1 cells were chosen for the present
study to ensure the stability of the coculture system, which has
been widely used for studies on the crosstalk between cancer cells
and adipocytes (14,17). Murine 3T3-L1 preadipocytes were
obtained from the American Type Culture Collection and were
maintained in DMEM supplemented with 10% newborn calf serum (Gibco;
Thermo Fisher Scientific). All cells were cultured in a humidified
incubator at 37°C supplied with 5% CO2 air. A total of 2
days after reaching confluence, 3T3-L1 cell differentiation was
induced by changing the medium to DMEM supplemented with 10% FBS
(v/v), 1 µg/ml insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 1
µM dexamethasone for 2 days. The cells were then incubated in DMEM
plus 10% (v/v) FBS and 1 µg/ml insulin for a further 2 days. Next,
the differentiated mature adipocytes were cultured in DMEM
supplemented with 10% (v/v) FBS. To study the crosstalk process
between PC cells and adipocytes, the coculture model was
constructed as previously described (11). Briefly, 8 days after induction, the
mature adipocytes were cocultured with PC cells in a Transwell
indirect coculture system (0.4 µm pore size; Corning, Inc.).
Etomoxir (HY-50202, MedChemExpress) was added as a carnitine
palmitoyl transferase 1 (CPT1) inhibitor, and CAY10499 (cat. no.
10007875, Cayman Chemical Company) was added as a nonselective
lipase inhibitor.
Immunohistochemical staining
The human tissues used in the present study were
collected from a 78-year-old female patient with pancreatic ductal
adenocarcinoma who underwent radical surgery in Huadong Hospital
(Shanghai, China) in March 2019. Consent of the patient and
approval from the Institutional Research Ethics Committee of
Huadong Hospital, Fudan University (Shanghai, China; approval no.
2018K098) were obtained. Immunohistochemistry was performed as
described previously (11). The
paraffin-embedded tissues were stained with rabbit anti-FABP4
polyclonal antibody (pAb; cat. no. 12802-1-AP; ProteinTech Group,
Inc.) overnight at 4°C. For each slide, representative images of
adipocytes surrounding the normal tissue and adipocytes in the
vicinity of the PC cells were obtained. Adipocyte cell sizes were
assessed using ImageJ (version 1.8.0; National Institutes of
Health).
BODIPY staining of lipid droplets
(LDs)
To detect LDs in the 3T3-L1 adipocytes, the cells
were cultured alone or with cancer cells for 5 days and then
incubated in DMEM containing the BODIPY-493/503 lipid probe (0.1
µg/ml, cat. no. D3922, Invitrogen) for 15 min at room temperature.
To detect LDs in PC cells treated with 200 µM oleic acid (OA:
Beyotime, China), the cells were washed with PBS and fixed with
3.7% paraformaldehyde for 15 min at room temperature. The cells
were then incubated with 0.1 µg/ml BODIPY and 5 µg/ml DAPI
(Beyotime Institute of Biotechnology) for 15 min at room
temperature. The stained cells were visualized using a confocal
laser scanning microscope (Olympus) (magnification, ×200 or
×1,000).
Triglyceride (TG) content
analysis
Adipocytes were harvested after being cultured with
or without PC cells for 5 days and then lysed with lysis buffer
(Beyotime Institute of Biotechnology) for 15 min. The TG content of
adipocytes was quantified using a TG assay kit (Applygen
Technologies, Inc.). The total protein concentration was measured
using a BCA assay (Thermo Fisher Scientific, Inc.), and the results
are expressed as milligrams of TG per milligram of protein. The
average of the control group was set as one, and all results are
presented as the relative TG content.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
The cells were harvested and lysed using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. PrimeScript RT
Master Mix (Takara Bio, Inc., cat. no. RR036A) was used to
synthesize cDNA according to the manufacturer's protocol. Gene
expression was determined using qPCR with a SYBR-Green PCR
MasterMix Reagent (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling protocol consisted of an initial
denaturation step at 95°C for 10 min, followed by 40 cycles of 95°C
for 15 sec and 60°C for 1 min. All amplifications and detections
were performed using an ABI 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Relative gene
expression was calculated using the 2−ΔΔCq method with
18S rRNA as an endogenous control (18). The average of the control group was
set to one, and all results are presented as the relative mRNA
expression. All assays were performed in triplicate. The primer
sequences used in the present study are listed in Table I.
 | Table I.Sequences of the primers used in the
present study. |
Table I.
Sequences of the primers used in the
present study.
| Gene name | Forward primer
sequence, 5′-3′ | Reverse primer
sequence, 5′-3′ |
|---|
| 18S |
CGCCGCTAGAGGTGAAATTCT |
CATTCTTGGCAAATGCTTTCG |
| hANGPTL4 |
GACCAAGGGGCATGGAGCTT |
CAGGGGACCTACACACAACAG |
| hGLUT1 |
CTTTGTGGCCTTCTTTGAAGT |
CCACACAGTTGCTCCACAT |
| hHK2 |
GATTGTCCGTAACATTCTCATCGA |
CTTGCAGCAGGGCCAGGCAGTCAC |
| hLDHA |
TGGAGATTCCAGTGTGCCTGTATGG |
CACCTCATAAGCACTCTCAACCACC |
| hFATP1 |
TGACAGTCGTCCTCCGCAAGAA |
CTTCAGCAGGTAGCGGCAGATC |
| hDGAT1 |
ACCTCATCTGGCTCATCTTCTTCTA |
CCCGGTCTCCAAACTGCAT |
| hDGAT2 |
GCTACACTGGCAGGCAACTT |
CATTGCCACTCCCATTCTTT |
| hGPAT4 |
CCCGTATTTGCTGCTGTTCC |
CATACTGCGAGTGCTGAGTGT |
| hPLIN2 |
CCTGCTCTTCGCCTTTCG |
TGCAACGGATGCCATTTTT |
| mGLUT4 |
CCGGATTCCATCCCACAAG |
CATGCCACCCACAGAGAAGA |
| mIRS1 |
CCAGCCTGGCTATTTAGCTG |
CCCAACTCAACTCCACCACT |
| mSOCS3 |
GGACCAAGAACCTACGCATCCA |
CACCAGCTTGAGTACACAGTCG |
| mPPARG |
CCGAAGAACCATCCGATTGA |
TTTGTGGATCCGGCAGTTAAG |
| mSREBF1 |
GATGTGCGAACTGGACACAG |
GCATGTCTTCGATGTCGTTCAAA |
| mChREBP |
CACTCAGGGAATACACGCCTAC |
ATCTTGGTCTTAGGGTCTTCAGG |
Western blotting
Total lysates were extracted using 2% SDS lysis
buffer containing phosphatase and protease inhibitors (Roche
Diagnostics GmbH). A BCA assay (Thermo Fisher Scientific, Inc.) was
used to determine the protein concentration. A total of 20 µg
protein was loaded per lane onto 12% SDS-gels, resolved using
SDS-PAGE, transferred to 0.22 µm PVDF membranes (MilliporeSigma),
and blocked for 1 h at room temperature with 5% skim milk. Each
membrane was immunoblotted with the indicated primary antibodies at
4°C overnight and then incubated with a secondary antibody at 37°C
for 1 h. Immunoreactive bands were visualized using an enhanced
chemiluminescence detection kit (Thermo Fisher Scientific, Inc.)
using an ImageQuant LAS 4000 System (GE Healthcare). The following
primary and secondary antibodies were used in the present study:
AKT (1:1,000; ProteinTech Group, Inc.; cat. no. 10176-2-AP),
p-AKTSer473 (1:1,000, Cell Signaling Technology, Inc.;
cat. no. CST4060S), HIF-1α (1:1,000; ProteinTech Group, Inc.; cat.
no. 20960-1-AP), ANGPTL4 (1:1,000; ProteinTech Group, Inc.; cat.
no. 18374-1-AP), GLUT1 (1:1,000; ProteinTech Group, Inc.; cat. no.
66290-1-lg), HK2 (1:1,000; ProteinTech Group, Inc.; cat. no.
22029-1-AP), LDHA (1:1,000; ProteinTech Group, Inc.; cat. no.
19987-1-AP), α-tubulin (1:1,000; ProteinTech Group, Inc.; cat. no.
66031-1-lg), p-AMPKαThr172 (1:1,000; Cell Signaling
Technology, Inc.; cat. no. CST2535), AMPKα (1:1,000; ProteinTech
Group, Inc.; cat. no. 10929-2-AP), p-STAT3Tyr705
(1:1,000; Cell Signaling Technology, Inc.; cat. no. CST9145), STAT3
(1:1,000; Cell Signaling Technology, Inc.; CST12640), Snail
(1:1,000; CST; CST9782), horseradish peroxidase-conjugated
anti-rabbit IgG antibody (1:5,000; ProteinTech Group, Inc.; cat.
no. SA00001-2) and goat anti-mouse IgG secondary antibody HRP
conjugated (1:5,000; Signalway Antibody LLC; cat. no. L3032).
Migration and invasion assays
For the Transwell migration assay, 5×104
Panc-1 or Mia PaCa2 cells in 200 µl DMEM were seeded into the upper
chamber of a Transwell chamber with an 8-µm pore membrane (24-well
insert; Corning, Inc.). The lower chamber was filled with media
supplemented with 10% FBS, while the upper chamber contained
serum-free media. Cells were allowed to adhere for 2 h prior to
drug treatment and then incubated for 24 h for the migration
assays. The cells that had not migrated through the pores were
removed using cotton swabs, and the cells on the bottom of the
membrane were fixed with 100% methanol and stained with 0.1%
crystal violet at room temperature for 30 min. For the invasion
assays, the Transwell chambers were coated in advance with 1 mg/ml
Matrigel (cat. no. 356231, Corning, Inc.). Cancer cells that
invaded through the Matrigel to the underside of the filter were
stained with crystal violet at room temperature for 30 min. The
number of migrated or invaded cells was counted in three randomly
selected fields of view under a brightfield microscope (IX71;
Olympus Corporation) (magnification, ×100). ImageJ was used to
count the number of cells that had migrated or invaded in each
field of view.
Lactate assays
After PC cells were either cultured alone or
cocultured with adipocytes for 5 days, the conditional medium was
collected and centrifuged at 1,000 × g for 5 min at room
temperature to remove debris. The supernatants were then stored at
−80°C until required. Lactate released into the medium was measured
using the Amplite™ Colorimetric L-Lactate Assay Kit (cat. no.
13815, AAT Bioquest) according to the manufacturer's instructions.
Briefly, 50 µl L-Lactate standards and test samples were placed in
a white, clear, bottom 96-well microplate. 50 µl L-Lactate working
solution was then added to each well of the L-Lactate standard,
blank control, and test samples to a final volume of 100 µl/well.
The reaction was incubated at room temperature for 2 h in the dark.
The absorbance was measured at 575/605 nm. A standard curve based
on the absorbance of the L-lactate standards was drawn, and the
lactate concentrations in the supernatants were then
calculated.
Transmission electron microscopy
PC cells were cultured with or without adipocytes
for 5 days. The cells were then collected by centrifugation at
1,000 × g for 10 min at room temperature and immediately fixed in
2.5% glutaraldehyde, 4% paraformaldehyde, and 0.002% picric acid in
a 0.1 M (pH 7.3) cacodylate buffer at 4°C for 3 h. Tissue slices
were then postfixed in 1% OsO4 in the same buffer at 4°C
for 3 h, dehydrated in a graded acetone series, and embedded in
Epon resin. For electron microscopy, 70-nm thick sections were cut
from tissue resin blocks. The sections were then transferred to
formvar-coated copper mesh grids and double-stained with saturated
uranyl acetate for 30 min, followed by lead citrate for 15 min at
room temperature. The ultra-thin sections on the grids were
examined in a JEOL JEM-1400 plus transmission electron microscope
at 80 kV. ImageJ (version 1.8.0; National Institutes of Health) was
used to count the number of LDs.
Oxygen consumption rate (OCR) and
extracellular acidification rate (ECAR)
After culturing with or without adipocytes for 5
days, PC cells were seeded into 96-well plates at a density of
40,000 cells/well and incubated overnight. Mitochondrial function
and cellular glycolytic capacity were determined using the Seahorse
Bioscience XF96 Extracellular Flux Analyzer (Seahorse Bioscience)
and a Seahorse XF Glycolysis Stress Test Kit and a Cell Mito Stress
Test Kit, according to the manufacturer's protocol. For ECAR
assessment, cells were incubated under basal conditions with
non-buffered RPMI 1640 followed by sequential injection of 10 mM
glucose and 1 mM mitochondrial poison (oligomycin; MilliporeSigma).
OCR was evaluated under basal conditions, followed by a sequential
injection of 1 µM oligomycin, 1 µM fluoro-carbonyl cyanide
phenylhydrazone (FCCP; MilliporeSigma), and 2 mM antimycin A and
rotenone (MilliporeSigma). Both ECAR and OCR measurements were
standardized to total protein content.
RNA sequencing (RNA-seq) and gene set
enrichment analysis (GSEA)
RNA-seq was performed on 3T3-L1 cells cultured with
or without Panc-1 PC cells for 5 days as previously described
(11). The sequencing data have
been deposited in the Gene Expression Omnibus under accession
number GSE123939. GSEA was performed using GSEA software
(http://www.broadinstitute.org/gsea/index.jsp). A gene
set enrichment score (ES) estimating genes from a predefined gene
set was calculated using GSEA. The thresholds for significance were
set by permutation analysis with 1000 gene-set permutations.
Liquid chromatography coupled with
tandem mass spectrometry
Panc-1 PC cells were cultured with or without mature
adipocytes for 5 days and then resuspended in an ~8-fold volume of
lysis buffer (4% SDS, 100 mM HEPES, pH=7.6) containing a protease
inhibitor cocktail and PMSF. The homogenate was sonicated on ice
for 30 min. The sample was centrifuged at 25,000 × g for 30 min at
4°C, and the supernatant was stored at −80°C. Proteins were reduced
in 10 mM DTT for 1 h at 37°C. Protein samples were then cooled to
room temperature (RT). The cysteines were blocked in darkness with
30 mM IAA at 37°C for 30 min. The extracted protein was mixed in
equal amounts according to the groups and then precipitated
overnight in acetone. The protein samples were then resuspended in
1 M urea buffer and digested with trypsin overnight. Next, peptides
were isotopically labeled with iTRAQ reagents (Applied Biosystems;
Thermo Fisher Scientific, Inc.) for 2 h at RT. The labeling
reaction was then stopped using water. The samples were then
separated and identified using a TripleTOF 4600 mass spectrometer
(Applied Biosystems; Thermo Fisher Scientific, Inc.).
Statistical analysis
All statistical analysis was performed using
GraphPad Prism version 5.0 (GraphPad Software, Inc.). All results
are presented as the mean ± standard deviation of at least three
repeats. The differences between two groups were determined using
an unpaired two-tailed Student's t-test. Multiple-group comparisons
were conducted using one-way analysis of variance (ANOVA) followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Adipocytes contribute to tumor
progression
Although obesity adversely affects the long-term
outcomes of patients with PC, the crosstalk between adipocytes and
PC cells has not been fully elucidated (19). Immunohistochemical staining of the
adipocyte marker FABP4 in human PC tissue demonstrated that
adipocytes were present in the TME (Fig. 1A). It was also found that adipocytes
directly adjacent to the tumor were smaller in size compared to
those further away (P<0.0001; Fig.
1B). To assess the role of adipocytes in PC progression, an
adipocyte-PC cell coculture system was established (Fig. 1C). Our previous study revealed the
significant phenotypic alterations in mature adipocytes induced by
pancreatic Panc-1 and MIA PaCa2 cells (11); in the present study, the
relationship between adipocytes and PC cells was further explored.
3T3-L1 preadipocytes were differentiated into mature adipocytes and
then cocultured with human Panc-1 or MIA PaCa2 cells. It was found
that cocultured Panc-1 cells exhibited an elongated mesenchymal
morphology compared to monocultured Panc-1 cells, which showed a
characteristic epithelial morphology and formed compacted colonies
(Fig. 1D). It was also found that
PC cells cocultured for 5 days with mature adipocytes had a
significantly increased migratory and invasive capacity (P<0.05;
Fig. 1E and F). Taken together,
these data indicated that adipocytes surrounding tumors may have
promoted PC progression.
Adipocytes enhance hypoxic signaling
in cocultured PC cells
To decipher the potential mechanisms responsible for
the increased aggressiveness of cocultured cancer cells, mass
spectrometry was used to analyze the protein contents of Panc-1
monocultures and those cocultured with adipocytes for 5 days. GSEA
revealed a striking overrepresentation of hallmark database-defined
pathways involved in hypoxic signaling in Panc-1 cells cocultured
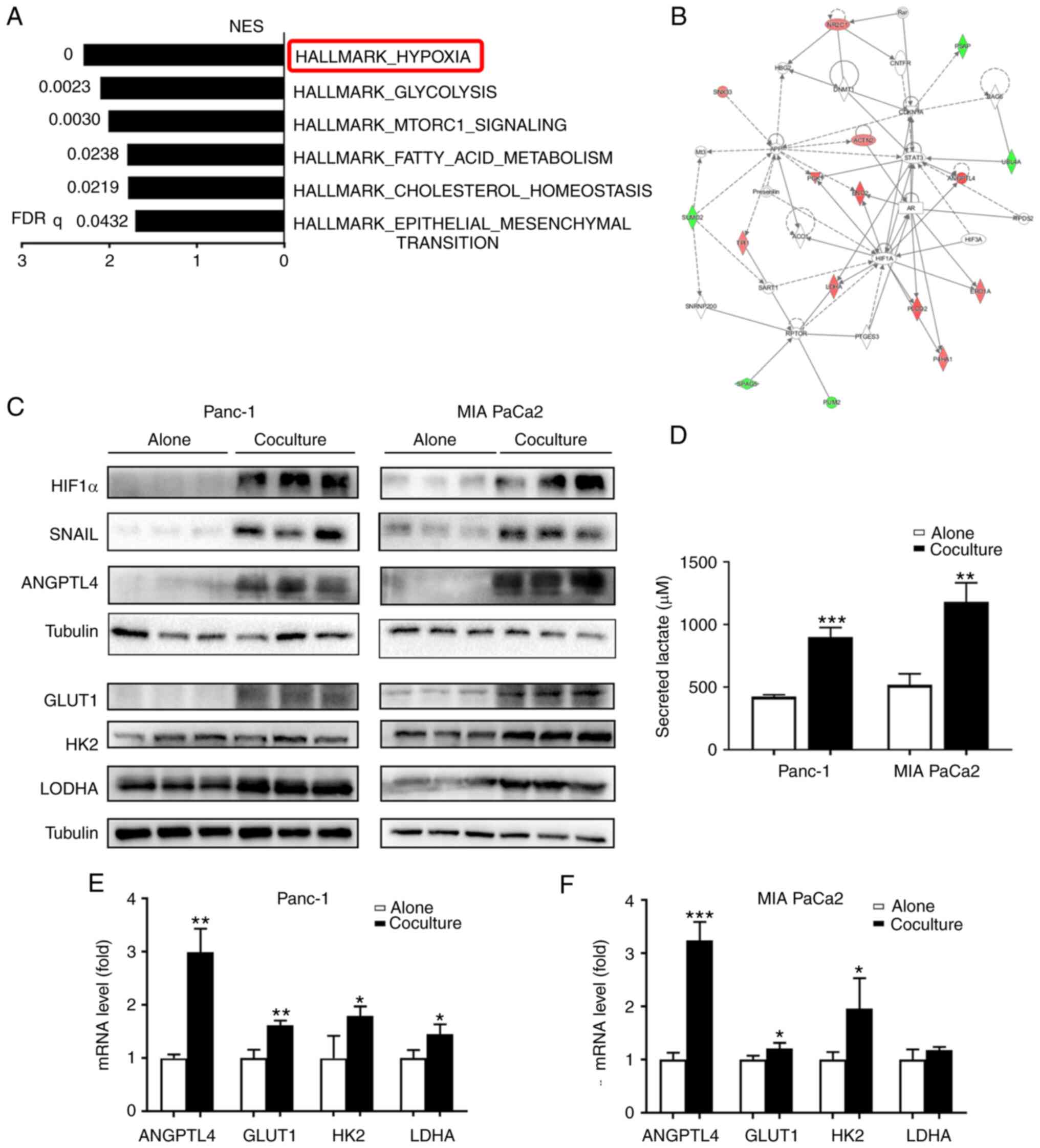
with adipocytes compared to those that were monocultured (Fig. 2A). The majority of the top ten
upregulated proteins (Table II)
are well-established downstream factors of hypoxic signaling, such
as ANGPTL4, ENOG, and LDHA (20–22).
Whole proteome bioinformatics analysis also revealed that certain
metabolic processes and the epithelial-mesenchymal transition were
activated in cocultured tumor cells (Fig. 2A). Based on the protein-protein
interaction network associated with the enriched functional
pathways of carbohydrate metabolism, tissue morphology, and cancer,
it was further confirmed that HIF-1α was activated in the
cocultured Panc-1 cells (Fig. 2B).
Congruently, there was a robust increase in HIF-1α and its related
downstream proteins (SNAIL, ANGPTL4, and glycolytic-associated
proteins) in the cocultured tumor cells (Fig. 2C), which was further confirmed by
RT-qPCR analysis (Fig. 2E and F).
It was also found that lactate production was significantly
increased in the cocultured cells (P<0.01; Fig. 2D). Together, these data suggest that
adipocytes enhance HIF-1α signaling and can reprogram the tumor
metabolic pattern to induce a shift towards anaerobic glycolysis in
PC cells under in vitro coculture conditions.
| Table II.Top ten upregulated proteins in
Panc-1 cells cocultured with adipocytes compared with the
monocultured cells. |
Table II.
Top ten upregulated proteins in
Panc-1 cells cocultured with adipocytes compared with the
monocultured cells.
| Protein name | Gene name | Fold change | P-value |
|---|
| Solute carrier
family 2, facilitated glucose transporter member 1 | SLC2A1 | 2 | <0.0001 |
|
Angiopoietin-related protein 4 | ANGPTL4 | 1.75 | 0.00012 |
|
Procollagen-lysine,2-oxoglutarate
5-dioxygenase 2 | PLOD2 | 1.655172 | 0.000685 |
| γ-enolase | ENOG | 1.642857 | 0.00022 |
| Phosphoglycerate
kinase 1 | PGK1 | 1.6 | 0.003857 |
| Vitronectin | VTNC | 1.586207 | 0.003858 |
| Dynein heavy chain
5, axonemal | DYH5 | 1.560976 | 0.028439 |
| NFX1-type zinc
finger-containing protein 1 | ZNFX1 | 1.56 | 0.024896 |
| Hemoglobin subunit
alpha | HBA | 1.535714 | 0.00257 |
| L-lactate
dehydrogenase A | LDHA | 1.5 | 0.584963 |
PC induces an insulin-resistant
phenotype in adipocytes
Given the enhanced ability of glucose utilization in
cancer cells, whether coculturing of cells resulted in altered
glucose metabolism in adipocytes was next assessed. To address
this, RNA-sequencing on mature 3T3-L1 adipocytes cultured alone or
with Panc-1 PC cells for 5 days was performed. GSEA of
differentially expressed transcripts in cocultured adipocytes
compared with those cultured alone revealed a marked enrichment of
gene sets corresponding to the insulin signaling pathway and the
JAK-STAT3 pathway (Fig. 3A). In
addition, there was a robust decrease in genes associated with the
insulin signaling pathway in cocultured adipocytes (Fig. 3B), indicating insulin resistance in
the adipocytes. It was also found that coculturing the adipocytes
with cancer cells increased STAT3 phosphorylation (Fig. 3C). Additionally, the increased
phosphorylation of STAT3 in adipocytes treated with 20 ng/ml IL-6
resulted in the downregulation of genes related to the insulin
signaling pathway, such as GLUT4 and IRS1 (P<0.05; Fig. 3D and E). In humans, SOCS3 expression
has been shown to be associated with the JAK-STAT3 pathway and
insulin resistance (23). In line
with this, significantly increased SOCS3 expression in adipocytes
treated with IL-6 or cocultured with Panc-1 or MIA PaCa2 PC cells
was observed (P<0.001; Fig. 3F).
Given the close relationship between the JAK-STAT3-SOCS3 axis and
the development of obesity-associated disorders, such as insulin
resistance (24), it was
hypothesized that PC cells could induce an insulin-resistant
phenotype in adipocytes through the JAK-STAT3-SOCS3 axis.
Adipocytes alter PC cell fatty acid
metabolism
Next, both the ECAR and OCR were measured in PC
cells using a Seahorse assay. After monoculture or coculture with
3T3-L1 adipocytes for 5 days, the PC cells were digested and seeded
into 96-well plates for further tests. It was found that the ECAR
did not differ significantly between the two conditions, indicating
no alteration in the rate of glycolysis in PC cells after coculture
with adipocytes (Fig. 4A and B). In
contrast, the cocultured cancer cells underwent significantly
increased OCR in both basal and maximal-uncoupled states compared
with monocultured cancer cells (Fig. 4C
and D), suggesting enhanced fatty acid β-oxidation (FAO) in the
cocultured cancer cells. In cancer, the JAK-STAT3 pathway regulates
lipid metabolism through FAO (25),
and AMPK favors energy-producing processes by activating
β-oxidation (26). Congruently, the
presence of mature adipocytes increased STAT3 and AMPK
phosphorylation and decreased AKT phosphorylation (Fig. 4E). As adipocytes store LDs, it was
hypothesized that adipocytes provide lipids to cancer cells to
enhance their FAO ability. To explore the changes in lipids in
cocultured PC cells, electron microscopy was performed. The results
showed there was a substantial increase in the number of LDs in the
cocultured Panc-1 and MIA PaCa2 cancer cells compared with that in
the monocultured cells (P<0.05; Fig.
4F and G). Substantial changes in the mitochondrial
ultrastructure in the cocultured MIA PaCa2 cells were also observed
(Fig. 4G), suggesting enhanced
respiratory chain activity in cancer cells after coculture with
adipocytes. Taken together, these results showed that adipocytes
resulted in increased lipid content and metabolic reprogramming in
PC cells.
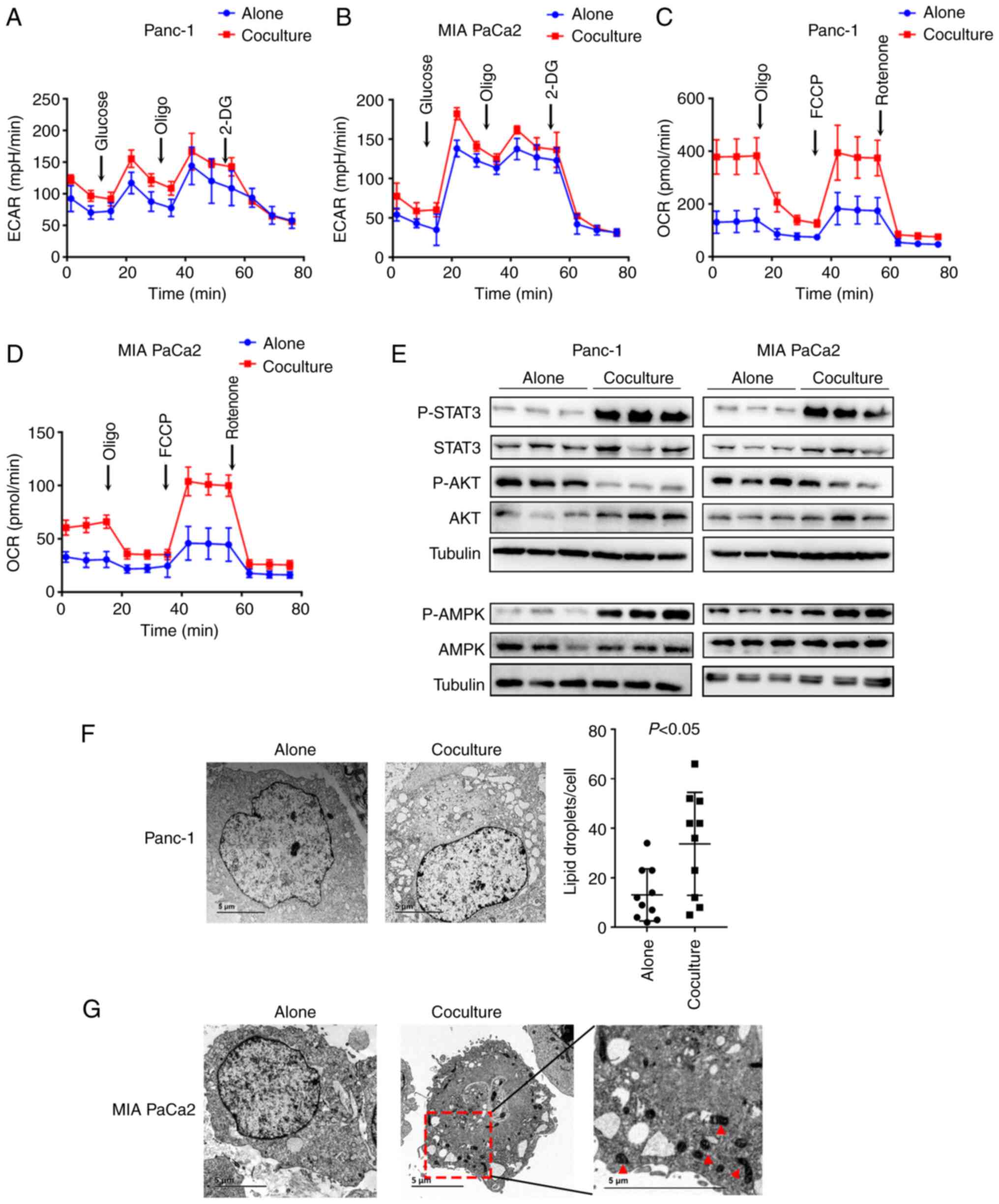 | Figure 4.Adipocytes alter pancreatic cancer
cell fatty acid metabolism. (A and B) ECAR did not differ between
Panc-1 and MIA PaCa2 cancer cells cultured with or without mature
adipocytes for 5 days. ECAR was measured under basal conditions
followed by the sequential addition of 10 mmol/l glucose, 1 mmol/l
oligomycin, and 100 mmol/l 2-deoxy-glucose. (C and D) OCR differed
between Panc-1 and MIA PaCa2 cancer cells cultured with or without
mature adipocytes for 5 days. OCR was measured under basal
conditions followed by the sequential addition of oligomycin (1
µM), FCCP (2 µM), and rotenone (1 µM). (E) Immunoblots of total and
p-STAT3, AKT, and AMPK in Panc-1 and MIA PaCa2 cancer cells
cultured with or without mature adipocytes for 5 days. (F) TEM of
Panc-1 cancer cells cocultured with mature adipocytes for 5 days
compared to cells cultured alone as the control (left).
Quantification of total lipid droplets per cell is shown (right).
n=10 cells/condition. (G) TEM of MIA PaCa2 cells cultured with or
without mature adipocytes for 5 days. The red arrows highlight the
ultrastructural changes of the mitochondrial in the cocultured
cancer cells. ECAR, extracellular acidification rate; OCR, oxygen
consumption rate; TEM, transmission electron microscopy; Oligo,
oligomycin. |
Increased levels of LDs promote the
invasion of cocultured PC cells
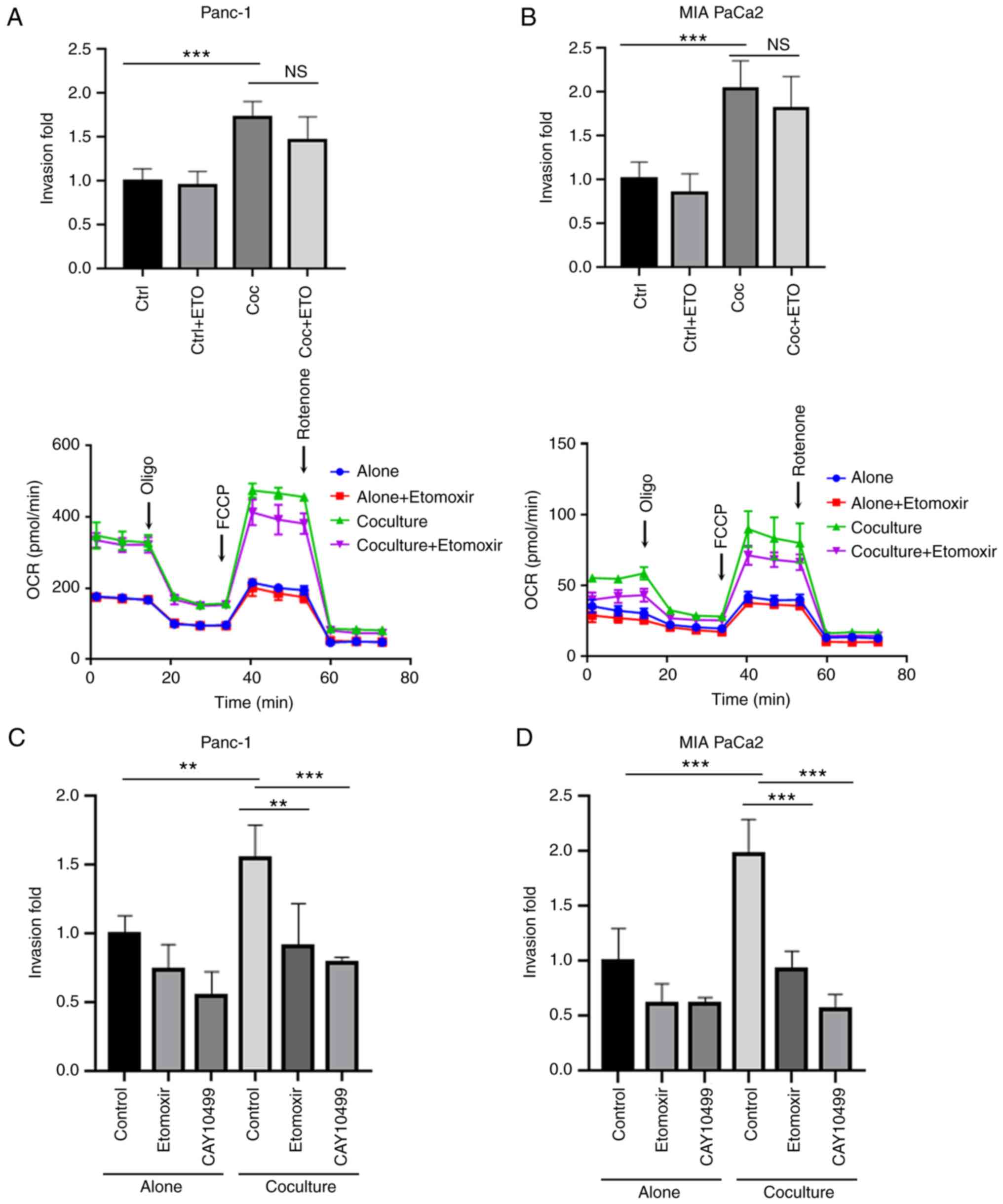
The above data showed that during coculture,
adipocytes stimulated increased invasiveness and enhanced FAO in PC
cells. Thus, whether the utilization of stored lipids in cancer
cells was associated with tumor malignancy was further examined.
Etomoxir can inhibit CPT1, which serves as the primary
rate-limiting factor in the transport of fatty acids to the
mitochondria.
First, cancer cells were cocultured with or without
adipocytes for 5 days with the addition of etomoxir and then the
cancer cells' invasive ability was assessed. It was found that the
inhibition of FAO by etomoxir during coculture did not hamper the
invasive ability of the cocultured cancer cells, in agreement with
the unchanged OCR (Fig. 5A and B).
As catabolism of stored LDs promotes cancer invasion and migration
(27), whether excess stored lipids
contributed to the increased invasive ability of cocultured PC
cells compared with monocultures was assessed. Treating the cancer
cells with etomoxir to inhibit FAO or with CAY10499 to inhibit
lipolysis during the invasion assay significantly reduced the
invasion of the cocultured PC cells (P<0.01; Fig. 5C and D). These data indicate that
the accumulation of LDs during tumor cell coculture with adipocytes
and the utilization of excess lipids during the process of tumor
metastasis together resulted in the increased invasive ability of
the cocultured PC cells. In agreement with the increased levels of
LDs in cocultured cancer cells (Fig. 4F
and G), it was also shown that the fatty acid
transporter-related gene (FATP1) and lipid storage-related genes
(DGAT1, DGAT2, GPAT4, and PLIN2) were upregulated in the cocultured
PC cells (Fig. 6A and B),
suggesting that coculture with adipocytes promoted fatty acid
uptake and storage in the cancer cells. To further confirm this,
Panc-1 and MIA PaCa2 cancer cells were first pretreated with
exogenous OA, which resulted in lipid accumulation (Fig. 6C and D). Preloading exogenous LDs
increased cancer cell aggressiveness, and this effect was abrogated
by etomoxir or CAY10499 treatment (P<0.01; Fig. 6E and F). Taken together, these data
demonstrated that the presence of adipocytes increased the
metastatic capacity of the PC cells, and this was largely due to LD
accumulation in the cocultured tumor cells.
PC cells induce downregulated lipid
metabolism in cocultured adipocytes
The above data indicated that adipocytes altered PC
cell lipid metabolism. Next, whether coculturing with cancer cells
also influenced the lipid metabolism of adipocytes was assessed.
First, a decrease in the size of LDs was found in cocultured
adipocytes, which was consistent with the reduced TG content
(P<0.01; Fig. 7A and B). The
GSEA of transcripts that were downregulated in the cocultured
adipocytes showed enrichment in gene sets corresponding to
oxidative phosphorylation, adipogenesis, and fatty acid metabolism.
Further analysis using Gene Ontology and biological process
compilation confirmed that both fatty acid catabolic and anabolic
processes were suppressed in cocultured adipocytes (Fig. 7C). The GSEA of transcripts that were
downregulated in the cocultured adipocytes also revealed marked
enrichment of gene sets associated with adipogenesis (Fig. 7D). In agreement with this, RT-qPCR
analysis of the adipocytes revealed the reduced expression of key
upstream genes of adipogenesis when cocultured with PC cells
(Fig. 7E). Taken together, these
results indicate that PC cells downregulated lipid metabolism and
adipogenesis in the cocultured adipocytes.
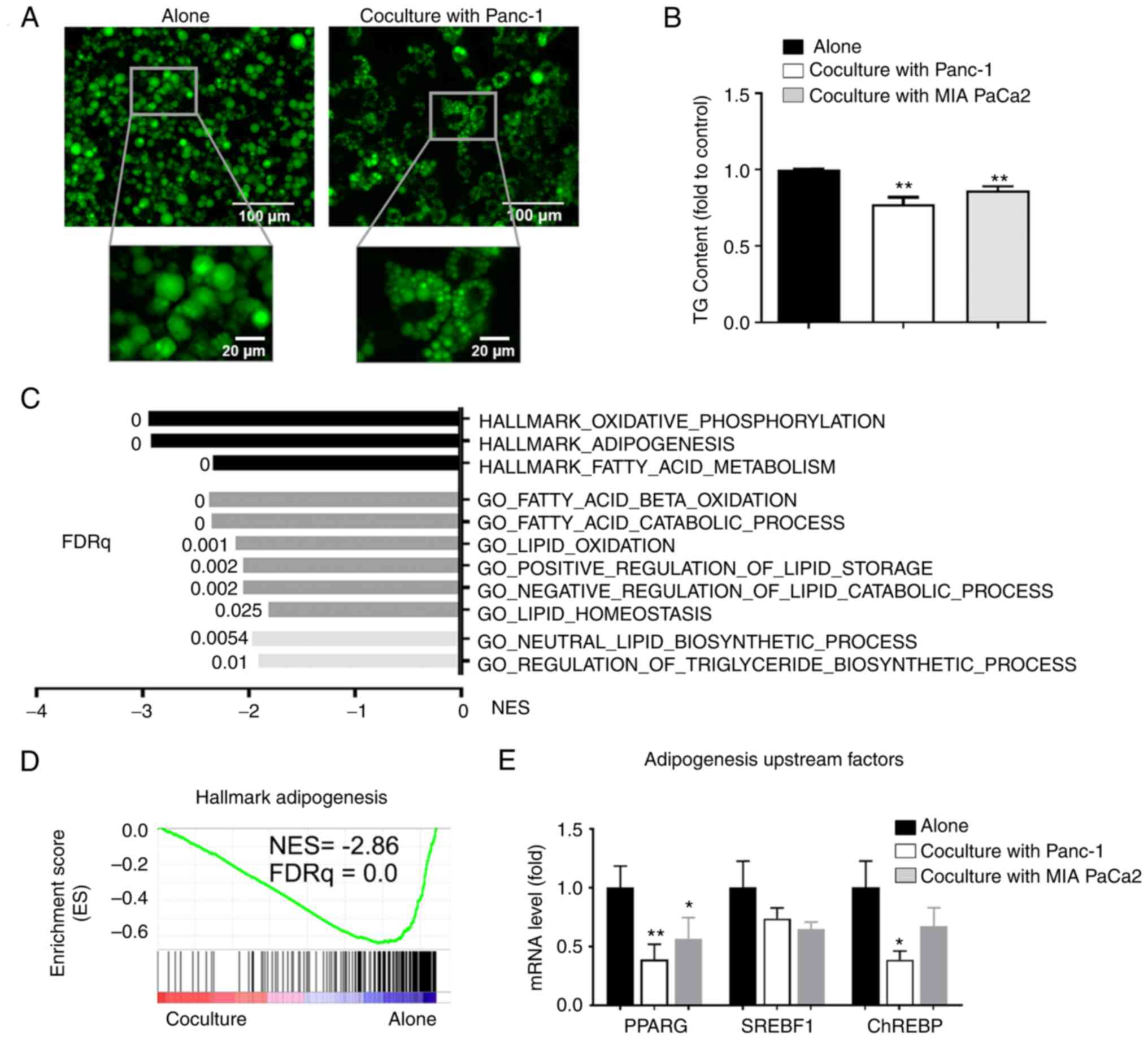 | Figure 7.PC cells downregulate lipid
metabolism in cocultured adipocytes. LD levels in adipocytes
cultured alone or cocultured with PC cells, (A) shown after
staining with BODIPY, the size of LDs in cocultured adipocytes
decreased, or (B) by measure of TG content, the content of TG in
cocultured adipocytes decreased. (C) Top metabolic pathways from
GSEA of downregulated genes in adipocytes cocultured with Panc-1
cancer cells (n=3) using GSEA Hallmark and GO biological process
MSigDB database. (D) Gene Set Enrichment Analysis plot of
enrichment in ‘Hallkmark_Adipogenesis’ signature in adipocytes
cocultured/alone as described in (C). (E) Reverse
transcription-quantitative PCR analysis of adipogenesis upstream
genes in adipocytes under the indicated conditions. Data are
presented as the mean ± standard deviation. *P<0.05,
**P<0.01. PC, pancreatic cancer; LD, lipid droplet; TG,
triglyceride; FDRq, false discovery rate, q value. |
Discussion
Clinical epidemiological observations and
mechanistic research are increasingly establishing the importance
of obesity in PC (4,5). However, the underlying mechanisms by
which excessive adiposity contributes to tumor progression remain
unclear. Adipose tissue as a reservoir for energy storage is
closely associated with the pathophysiological process of obesity.
An increasing number of studies have indicated that crosstalk
exists between adipocytes and cancer cells in the TME. This
crosstalk involves a vicious cycle in which adipocytes are
activated by cancer cells, and in turn, cancer-associated
adipocytes promote tumor progression (28). Altered cellular metabolism is an
important feature of cancer cells that enables unrestricted growth
and motility. Malignant cells tend to rewire their metabolic
properties according to the altered challenges encountered in the
TME (29). In breast (12), ovarian (30), and other types of cancer (14,15,31),
an increasing number of studies have revealed that tumor metabolic
crosstalk with adipocytes contributes to tumor metastasis. In this
study, an intricate metabolic network that contributes to the
anabolic reprogramming of cancer cells and favors tumor
aggressiveness was revealed between PC cells and adipocytes.
First, a metabolic competitive relationship between
PC cells and adipocytes was revealed, where PC cells subverted
adipocyte glucose utilization by desensitizing adipocytes to
glucose. Diabetes is a well-known risk factor for PC, and PC also
seems to cause glucose intolerance (32). Certain clinical studies have shown
that PC-related diabetes is improved following tumor resection
(33,34). In line with this, intraperitoneal
injection of PC cell-conditioned media into immunodeficient mice
resulted in significantly diminished glucose tolerance compared
with controls injected with saline (35). These studies together suggest that
PC can cause glucose desensitization of normal tissues. The ability
of tumors to impair adipocyte insulin sensitivity could serve to
divert insufficient nutrients in the TME. Thus, it was hypothesized
that in the pancreatic TME with limited glucose, PC cells may
induce a diabetic state to obtain a competitive advantage for the
acquisition of glucose. Indeed, it was found that coculturing with
adipocytes induced an increase in glycolytic capacity in PC cells
through the upregulation of glycolytic enzymes.
Most tumors utilize enhanced glucose metabolism to
sustain anabolic processes, which is often related to a more
hypoxic tumor signature (36). The
pancreatic TME is often hypoxic owing to its desmoplastic stroma
(37). Here, it was found that
coculture with adipocytes induced HIF-1α activity in PC cells. A
previous study revealed that adipocytes could induce a glycolytic
phenotype in prostate cancer via HIF-1α activation (38). Moreover, a recent study showed that
HIF-1α signaling was activated by adipocyte-derived extracellular
vesicles, and this functionally augmented the metastatic potential
of breast cancer (39). The
mechanisms by which adipocytes regulate HIF-1α expression in tumors
remain elusive. One possible explanation is related to the fatty
acids released by adipocytes. In liver cancer, OA treatment
activated the FABP5/HIF-1α axis to promote cancer cell
proliferation (40). Taken
together, the present and previous studies highlight the potential
role of adipocytes in the hypoxic TME and imply an intricate
relationship between metabolic reprogramming and hypoxia in the
adipocyte-PC cell coculture system.
Another finding of the present study was the
energy-plundering relationship between PC cells and adipocytes. An
increasing number of studies have confirmed the presence of lipid
transfer from adipocytes to cancer cells (12–15).
In the present study, coculturing with adipocytes led to increased
mRNA levels of the fatty acid transport protein FATP1 and the LD
protein PLIN-2 in cancer cells, suggesting a process of lipid
transportation and storage in cocultured cancer cells. In line with
this, the PC cells induced a decrease in LDs in adipocytes. Another
key concern arising from the present study was that the increased
amount of stored lipids in the cocultured PC cells contributed to
cell invasion. The pharmacological inhibition of lipolysis or lipid
transport into the mitochondria effectively hampered the invasive
ability. Previous studies have shown that the demand for oxidative
phosphorylation and ATP is increased in invasive and metastatic
cancer cells (41–44); however, the source of this required
energy has not been well defined. Intriguingly, recent work has
demonstrated that in PC cells, excess lipids are required for ATP
production to fuel the process of metastasis (27); in an elegant study, the oncogene
KRAS was shown to facilitate the storage of LDs by suppressing
hormone-sensitive lipase (HSL) expression, and stored lipids were
then shown to be catabolized and utilized for tumor progression
(27). The results of the present
study indicated that adipocyte-derived lipids could be stored in
cancer cells and utilized during invasion.
There remain some limitations to the present study.
First, the findings were only confirmed at the cellular level, and
need to be further verified using in vivo experiments.
Second, in this study, the focus was primarily on the migration and
invasion of PC cells. Certain other phenotypes of cancer cells,
such as proliferation, chemoresistance, and immunoregulation, need
to be further investigated. Third, the specific mechanisms of lipid
transfer from adipocytes to PC cells were not revealed. Fourth, the
characteristics of PC cells close to adipocytes in clinical
specimens were not determined, for which, further research in
combination with technologies such as space transcriptome
sequencing is required to assess this. Fifth, only metabolic
crosstalk has been observed in this study. Certain well-known
adipocyte-secreting factors that may drive PC progression should be
further elucidated. The types of cytokines mediating this process
and their specific mechanisms need to be further studied.
In conclusion, these findings reveal a previously
unidentified metabolic interaction between PC cells and adipocytes,
leading to excess lipid storage and the priming of cancer cells for
progression. Based on these results, interrupting the mechanisms of
lipid uptake from adipocytes in the microenvironment may offer a
potential strategy for attenuating PC metastasis.
Acknowledgements
We would like to thank Dr Abousalam Abdoulkader
Ahmed from The Huadong Hospital Affiliated to Fudan University for
his guidance in the development of this manuscript.
Funding
This study was supported by the Shanghai Science and Technology
Commission of Shanghai Municipality (grant no. 20Y11908600), the
Shanghai Shenkang Hospital Development Center (grant no.
SHDC2020CR5008), the Shanghai Municipal Health Commission (grant
no. 20194Y0195), and the Project of Huadong Hospital Affiliated to
Fudan University (grant no. 2019H1285).
Availability of data and materials
The sequencing data have been deposited in the Gene
Expression Omnibus with the assigned accession number GSE123939
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123939).
The datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
ZC and CJ participated in the design of the study.
ZC and YL performed data analysis and prepared the figures. MM, LW,
HW, and ML participated in the analysis of the figures and data. ZC
and YL prepared and revised the manuscript. CJ reviewed the results
and revised the manuscript. ZC, YL and CJ confirm the authenticity
of all the raw data. All authors have read and approved the final
version of this manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional
Research Ethics Committee of Huadong Hospital, Fudan University
(approval no. 2018K098). Written informed consent for the use of
the tissue for scientific research was obtained from all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mizrahi JD, Surana R, Valle JW and Shroff
RT: Pancreatic cancer. Lancet. 395:2008–2020. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sahai E: Illuminating the metastatic
process. Nat Rev Cancer. 7:737–749. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feig C, Gopinathan A, Neesse A, Chan DS,
Cook N and Tuveson DA: The pancreas cancer microenvironment. Clin
Cancer Res. 18:4266–4276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klein AP: Pancreatic cancer epidemiology:
Understanding the role of lifestyle and inherited risk factors. Nat
Rev Gastroenterol Hepatol. 18:493–502. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung KM, Singh J, Lawres L, Dorans KJ,
Garcia C, Burkhardt DB, Robbins R, Bhutkar A, Cardone R, Zhao X, et
al: Endocrine-Exocrine signaling drives obesity-associated
pancreatic ductal adenocarcinoma. Cell. 181:832–847.e18. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okumura T, Ohuchida K, Sada M, Abe T, Endo
S, Koikawa K, Iwamoto C, Miura D, Mizuuchi Y, Moriyama T, et al:
Extra-pancreatic invasion induces lipolytic and fibrotic changes in
the adipose microenvironment, with released fatty acids enhancing
the invasiveness of pancreatic cancer cells. Oncotarget.
8:18280–18295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hori M, Takahashi M, Hiraoka N, Yamaji T,
Mutoh M, Ishigamori R, Furuta K, Okusaka T, Shimada K, Kosuge T, et
al: Association of pancreatic Fatty infiltration with pancreatic
ductal adenocarcinoma. Clin Transl Gastroenterol. 5:e532014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Incio J, Liu H, Suboj P, Chin SM, Chen IX,
Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, et al: Obesity-induced
inflammation and desmoplasia promote pancreatic cancer progression
and resistance to chemotherapy. Cancer Discov. 6:852–869. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quail DF and Dannenberg AJ: The obese
adipose tissue microenvironment in cancer development and
progression. Nat Rev Endocrinol. 15:139–154. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Sullivan J, Lysaght J, Donohoe CL and
Reynolds JV: Obesity and gastrointestinal cancer: The
interrelationship of adipose and tumour microenvironments. Nat Rev
Gastroenterol Hepatol. 15:699–714. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai Z, Liang Y, Xing C, Wang H, Hu P, Li
J, Huang H, Wang W and Jiang C: Cancer-associated adipocytes
exhibit distinct phenotypes and facilitate tumor progression in
pancreatic cancer. Oncol Rep. 42:2537–2549. 2019.PubMed/NCBI
|
|
12
|
Wang YY, Attané C, Milhas D, Dirat B,
Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, et
al: Mammary adipocytes stimulate breast cancer invasion through
metabolic remodeling of tumor cells. JCI Insight. 2:e874892017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang M, Di Martino JS, Bowman RL,
Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal
C, Yong-Gonzales V, et al: Adipocyte-derived lipids mediate
melanoma progression via FATP proteins. Cancer Discov. 8:1006–1025.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wen YA, Xing X, Harris JW, Zaytseva YY,
Mitov MI, Napier DL, Weiss HL, Mark Evers B and Gao T: Adipocytes
activate mitochondrial fatty acid oxidation and autophagy to
promote tumor growth in colon cancer. Cell Death Dis. 8:e25932017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qian SW, Tang Y, Li X, Liu Y, Zhang YY,
Huang HY, Xue RD, Yu HY, Guo L, Gao HD, et al: BMP4-mediated brown
fat-like changes in white adipose tissue alter glucose and energy
homeostasis. Proc Natl Acad Sci USA. 110:E798–E807. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takehara M, Sato Y, Kimura T, Noda K,
Miyamoto H, Fujino Y, Miyoshi J, Nakamura F, Wada H, Bando Y, et
al: Cancer-associated adipocytes promote pancreatic cancer
progression through SAA1 expression. Cancer Sci. 111:2883–2894.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou B, Wu D, Liu H, Du LT, Wang YS, Xu
JW, Qiu FB, Hu SY and Zhan HX: Obesity and pancreatic cancer: An
update of epidemiological evidence and molecular mechanisms.
Pancreatology. 19:941–950. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shuff S, Oyama Y, Walker L and Eckle T:
Circadian Angiopoietin-Like-4 as a Novel Therapy in Cardiovascular
Disease. Trends Mol Med. 27:627–629. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qiao G, Wu A, Chen X, Tian Y and Lin X:
Enolase 1, a moonlighting protein, as a potential target for cancer
treatment. Int J Biol Sci. 17:3981–3992. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharma D, Singh M and Rani R: Role of LDH
in tumor glycolysis: Regulation of LDHA by small molecules for
cancer therapeutics. Semin Cancer Biol. 87:184–195. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pedroso JAB, Ramos-Lobo AM and Donato J
Jr: SOCS3 as a future target to treat metabolic disorders. Hormones
(Athens). 18:127–136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wunderlich CM, Hövelmeyer N and Wunderlich
FT: Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity.
Jakstat. 2:e238782013.PubMed/NCBI
|
|
25
|
Wang T, Fahrmann JF, Lee H, Li YJ,
Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al:
JAK/STAT3-regulated Fatty acid β-oxidation is critical for breast
cancer stem cell self-renewal and chemoresistance. Cell Metab.
27:136–150.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garcia D and Shaw RJ: AMPK: Mechanisms of
cellular energy sensing and restoration of metabolic balance. Mol
Cell. 66:789–800. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rozeveld CN, Johnson KM, Zhang L and
Razidlo GL: KRAS controls pancreatic cancer cell lipid metabolism
and invasive potential through the lipase HSL. Cancer Res.
80:4932–4945. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dumas JF and Brisson L: Interaction
between adipose tissue and cancer cells: Role for cancer
progression. Cancer Metastasis Rev. 40:31–46. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lyssiotis CA and Kimmelman AC: Metabolic
Interactions in the tumor microenvironment. Trends Cell Biol.
27:863–875. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mukherjee A, Chiang CY, Daifotis HA,
Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O and Lengyel
E: Adipocyte-induced FABP4 expression in ovarian cancer cells
promotes metastasis and mediates carboplatin resistance. Cancer
Res. 80:1748–1761. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ye H, Adane B, Khan N, Sullivan T,
Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM,
et al: Leukemic stem cells evade chemotherapy by metabolic
adaptation to an adipose tissue niche. Cell Stem Cell. 19:23–37.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Andersen DK, Korc M, Petersen GM, Eibl G,
Li D, Rickels MR, Chari ST and Abbruzzese JL: Diabetes,
pancreatogenic diabetes, and pancreatic cancer. Diabetes.
66:1103–1110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Permert J, Ihse I, Jorfeldt L, von Schenck
H, Arnquist HJ and Larsson J: Improved glucose metabolism after
subtotal pancreatectomy for pancreatic cancer. Br J Surg.
80:1047–1050. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pannala R, Basu A, Petersen GM and Chari
ST: New-onset diabetes: A potential clue to the early diagnosis of
pancreatic cancer. Lancet Oncol. 10:88–95. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Basso D, Brigato L, Veronesi A, Panozzo
MP, Amadori A and Plebani M: The pancreatic cancer cell line MIA
PaCa2 produces one or more factors able to induce hyperglycemia in
SCID mice. Anticancer Res. 15:2585–2588. 1995.PubMed/NCBI
|
|
36
|
Moldogazieva NT, Mokhosoev IM and
Terentiev AA: Metabolic heterogeneity of cancer cells: An Interplay
between HIF-1, GLUTs, and AMPK. Cancers (Basel). 12:8622020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fuentes NR, Phan J, Huang Y, Lin D and
Taniguchi CM: Resolving the HIF paradox in pancreatic cancer.
Cancer Lett. 489:50–55. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diedrich JD, Rajagurubandara E, Herroon
MK, Mahapatra G, Hüttemann M and Podgorski I: Bone marrow
adipocytes promote the Warburg phenotype in metastatic prostate
tumors via HIF-1α activation. Oncotarget. 7:64854–64877. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
La Camera G, Gelsomino L, Malivindi R,
Barone I, Panza S, De Rose D, Giordano F, D'Esposito V, Formisano
P, Bonofiglio D, et al: Adipocyte-derived extracellular vesicles
promote breast cancer cell malignancy through HIF-1α activity.
Cancer Lett. 521:155–168. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Seo J, Jeong DW, Park JW, Lee KW, Fukuda J
and Chun YS: Fatty-acid-induced FABP5/HIF-1 reprograms lipid
metabolism and enhances the proliferation of liver cancer cells.
Commun Biol. 3:6382020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cunniff B, McKenzie AJ, Heintz NH and Howe
AK: AMPK activity regulates trafficking of mitochondria to the
leading edge during cell migration and matrix invasion. Mol Biol
Cell. 27:2662–2674. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin S, Huang C, Gunda V, Sun J, Chellappan
SP, Li Z, Izumi V, Fang B, Koomen J, Singh PK, et al: Fascin
controls metastatic colonization and mitochondrial oxidative
phosphorylation by remodeling mitochondrial actin filaments. Cell
Rep. 28:2824–2836.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
LeBleu VS, O'Connell JT, Gonzalez Herrera
KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos Chinen LT, Rocha RM, et al: PGC-1α mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16:992–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kelley LC, Chi Q, Cáceres R, Hastie E,
Schindler AJ, Jiang Y, Matus DQ, Plastino J and Sherwood DR:
Adaptive F-actin polymerization and localized ATP production drive
basement membrane invasion in the absence of MMPs. Dev Cell.
48:313–328.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|