1. Introduction
Vasoactive intestinal peptide (VIP) secreting tumour
(VIPoma), also known as Verner-Morrison syndrome, is a
neuroendocrine tumour (NET) secreting VIP in an uncontrolled
manner. Werner and Morrison first described this syndrome in 1958,
reporting two patients with profuse watery diarrhoea leading to
hypokalemia and death from shock and dehydration (1). Other names for this syndrome include
pancreatic cholera (1) and WDHA
syndrome (watery diarrhoea, hypokalemia, achlorhydria) due to the
most common symptoms (2).
2. Epidemiology
VIPomas are rare tumours that comprise <10% of
all pancreatic endocrine tumors (PETs) with an estimated incidence
of 1/10,000,000 individuals per year (3). In total, 95% of VIPomas occur in
solitary forms, although they likewise appear on the grounds of
multiple endocrine neoplasia type 1 (MEN-1) syndrome (4). In adults, they develop most commonly
in the fortieth year of life with a sparse female predominance
(male:female ratio of 1:3) (5).
In children, the diagnosis is generally performed between 2 to 4
years of age (6). The majority of
VIPomas are intrapancreatic and are observed in the body and tail
of the pancreas, while 25% are found in the pancreatic head
(7). Nevertheless, there are
cases of a neoplasm with extra-pancreatic origin, such as the
bronchus, colon, liver, sympathetic nerve chains, pituitary and
thyroid glands (8). In infants,
on the other hand, these tumours commonly arise in the adrenal
glands and sympathetic ganglia (9).
3. Aetiology and pathogenesis
A VIP is a 28-amino-acid polypeptide which was
formerly isolated from the intestine in 1970(10). It is a neurohormone that adheres
to receptors on intestinal epithelial cells and induces the
activation of adenylate cyclase and cAMP production. This pathway
initiates the excretion and suppresses the reabsorption of sodium,
chloride, potassium and water in the intestine, resulting in
profound secretory diarrhoea (6).
VIP also displays vasomotor action on vessels, glucogenolytic
effect on the liver and reduces gastric acid secretion (11).
4. Clinical manifestations
The most notable clinical feature is watery
diarrhoea (54.5%), accompanied by hypokalemia (45.6%) and
achlorhydria (42.4%) (12).
Watery diarrhoea is often excessive, surpassing 3 litres per day.
It occurs without steatorrhea, and, in contrast to osmotic
diarrhoea, it persists while fasting (13). The causes of hypokalemia are
associated with aldosterone synthesis, VIPoma-induced chronic
diarrhoea, or direct potassium excretion by enterocytes (14). Hypokalemia may result in
manifestations, such as muscle weakness, flaccid paralysis,
respiratory distress and changes in the ECG (flattened T-waves).
There is also bicarbonate wasting through stool, leading to
hypokalemic nonanion gap metabolic acidosis (15). Hypochlorhydria or achlorhydria is
typically due to the inhibitory effect on parietal cells of gastric
mucosa, resulting in reduced gastric acid production (16). This usually leads to the
malabsorption of essential electrolytes and vitamins.
Other indications of excessive VIP discharge involve
hyperglycemia (20-50%), hypercalcaemia (25-50%), hypochlorhydria
(20-50%) and flushing (15-30%) (17). Hypercalcemia presents in almost
50% of patients with VIPoma (18). Its causes are unclear; however, it
has been associated with severe dehydration, electrolyte
disarrangements, multiple endocrine neoplasia (MEN) syndrome
followed by hyperparathyroidism, or the excretion of a calcitrophic
peptide by the tumour. Hypomagnesemia is generally secondary to
diarrhoea and leads to tetany in some cases. Almost 8% of patients
exhibit facial flushing, connected with prostaglandin production by
the tumour. The profound glycogenolytic effects of VIP on the liver
lead to diminished glucose intake by tissues and consequent
hyperglycemia (18). Additional
signs of VIPoma include skin rash, bloating, nausea, vomiting,
lethargy and an involuntary decrease in weight (19).
5. Diagnosis
A previous study on 41 cases from the Chinese
literature revealed that the average time from the manifestation of
symptoms to final diagnosis was >15 months, although patients
experience a range of distinguishing signs (20). By definition, VIP plasma levels
are increased in almost all patients with VIPoma (4). The diagnosis of the neoplasm is
confirmed in patients with secretory diarrhoea commonly >700
ml/day with a serum VIP level >200 pg/ml (reference range is
<190 pg/ml) (21). In order to
verify the diagnosis, it is essential to renew the VIP levels'
test, as, during the incidents of diarrhoea, plasma VIP levels
remain within the normal range (22). Amid children, catecholamine
amounts should also be estimated.
Supplementary blood laboratory analyses include
hypochlorhydria, hypokalemia, hypercalcemia, hyperglycemia and
hypomagnesemia. Moreover, high blood urea nitrogen levels are
associated with renal insufficiency (11). Of note, in 66% of patients, the
levels of gastrin and insulin are also elevated (23). In addition, in one case reported
in the literature, a patient with VIPoma had increased dopamine
levels, implying that neuroendocrine cells can secrete both
catecholamines, as well as pancreatic peptides (24).
There is a significant advantage of imaging studies
for the establishment of diagnosis (25,26). CT is essential in determining the
size, the location of the tumour origin, the involvement of nearby
structures, vessels, lymph nodes and the presence of calcification
(6). VIPomas >3 cm in diameter
can be efficiently recognised by CT scans (4). MRI can obtain neoplasms as small as
1 cm in diameter and are useful for the assessment of spinal
tumours (27). More novel imaging
with PET-CT Gallium-68 dotatate is 97% sensitive for the detection
of VIPomas, while the responsiveness of contrast-enhanced CT and
MRI is at 80 and 85%, respectively (28).
There are high amounts of somatostatin receptors in
up to 90% of pancreatic neuroendocrine tumors (PNETs). Thus,
somatostatin receptor scintigraphy applying radiolabeled
somatostatin analogue octreotide or lanreotide is a beneficial
approach for the identification of hidden metastases (4).
Additional methods comprise endoscopic ultrasound,
which helps to define the precise extent of the disease, as well as
to perform the biopsy of the lesion. Immunohistochemically, VIPomas
stain positively for VIP, somatostatin, neuron-specific enolase,
chromogranin A, synaptophysin and cytokeratin (29).
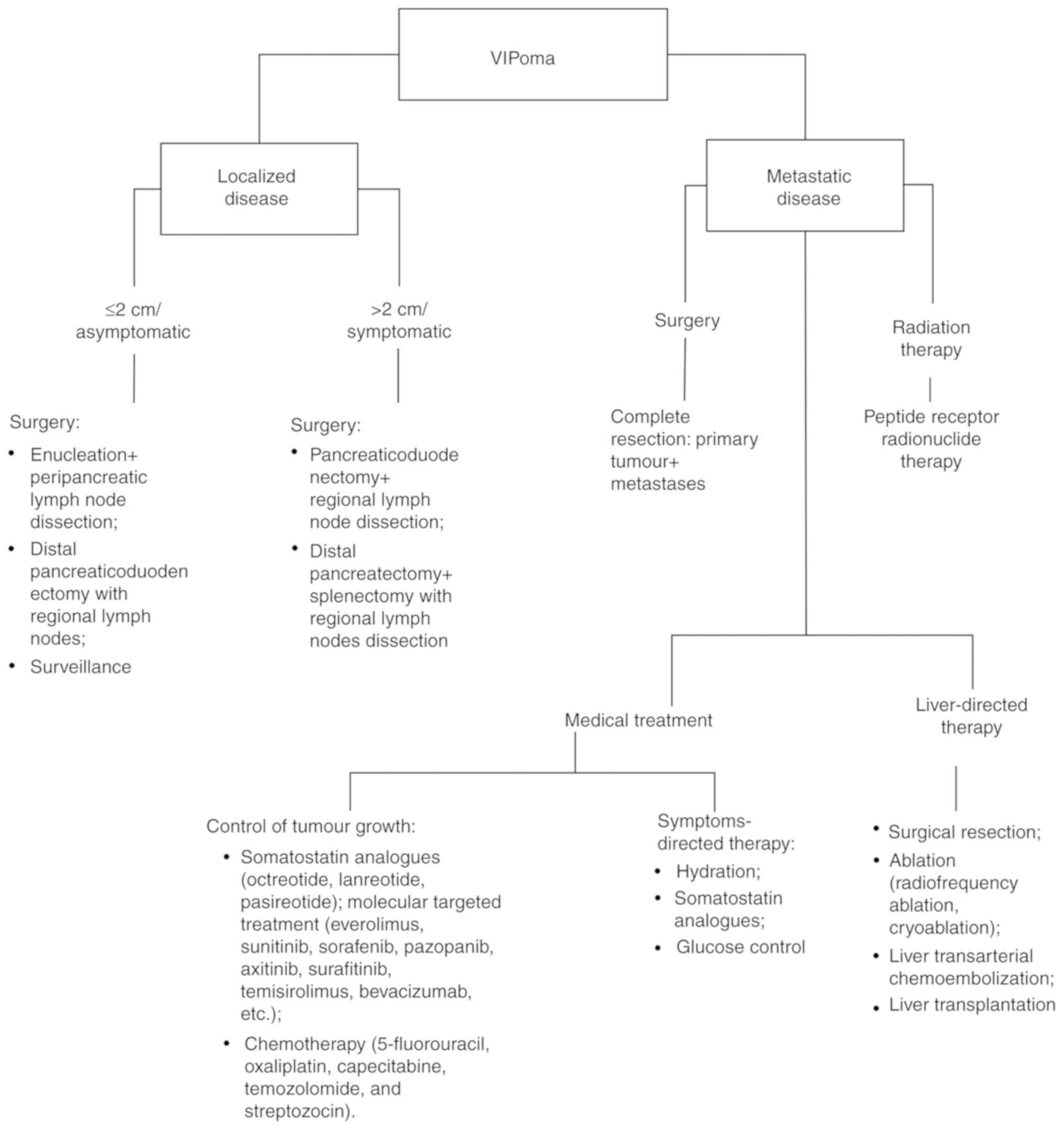
6. Treatment
The treatment of VIPomas comprises medical
supervision and surgery (Fig. 1).
Initial therapeutic control is intended principally for the
suppression of the symptoms of the disease. It includes a rapid
substitution of fluids and electrolytes to prevent dehydration and
electrolyte abnormalities, and to restore the acid-base balance.
The additional administration of glucocorticoids is performed in
patients who are insensitive to somatostatin analogues (30).
Various studies on functional NETs have proven that
managing excessive hormone levels is essential to lowering both the
morbidity and mortality of patients (31,32). Somatostatin is a peptide that
restrains the secretion of a wide range of hormones, and
somatostatin analogues (octreotide, lanreotide and pasireotide) can
reproduce its effect on the cell membrane receptors (7,33).
VIPomas, as well as the majority of NETs, usually
express somatostatin receptors on their surface; somatostatin will
adhere to these receptors, thus inhibiting hormone excretion from
the tumour cells. Somatostatin analogues are competent in both
regulating the symptoms and growth of the neoplasm (34). The CLARINET (Controlled study of
Lanreotide Antiproliferative Response In Neuroendocrine Tumors)
trial published in 2014, demonstrated the anti-proliferative
efficacy of the somatostatin analogue, lanreotide, in patients with
NETs (34). Octreotide is a
synthetic long-acting SST analogue that efficiently hinders VIP
discharge from tumour and has been approved by the FDA for
treatment of VIPomas (35).
However, long-term treatment with octreotide may result in drug
resistance, leading to the administration of extremely high doses
for the achievement of a desirable effect (36,37). In patients who exhibit a reduced
efficacy of somatostatin, interferon-α can be introduced with
octreotide to ameliorate symptoms and promote tumour regression
(38). The general adverse
effects of somatostatin analogue treatment comprise indigestion,
vomiting, bloating, diarrhoea with steatorrhea following fat
malabsorption, as well as mild glucose intolerance; nonetheless,
symptoms tend to fade over time (34).
Surgery is recommended by The World Health
Organization for all localized PNETs, regardless of the size
(39). The type of surgery
depends essentially on tumour localization and size, and leads to
curative results in 40% of patients (40,41). A total surgical resection
comprises the extraction of the primary mass, as well as all distal
metastases to the lymph nodes. The pancreatic body and tail are
resected during distal pancreatectomy, which can be achieved with
or without splenectomy (42). A
pancreaticoduodenectomy is a standard approach for a neoplasm with
a location in the pancreatic head. For tumours which are <2 cm
in size, parenchyma-sparing surgery, such as the enucleation of the
tumour, is also an option. It conserves a considerable amount of
pancreatic tissue and maintains sufficient endocrine and exocrine
function of the pancreas as opposed to conventional surgery
(43). Tumour debulking is not a
curative procedure, although it benefits symptom control and
prolongs patient survival (12).
Norton et al described 20 patients with
advanced NETs to whom aggressive surgery had been done. The
postoperative complication rate was 30% with no operative deaths
(44). Metastases have also been
noted in 60% of cases at the time of neoplasm detection (23) and most commonly arise in the
liver, kidney, lymph nodes and bones (45). In cases of hepatic metastases,
surgical resection of the liver is designated for patients without
diffuse involvement of both lobes, diminished liver functions,
extrahepatic metastases, or advanced neuroendocrine carcinoma
(46). In particular, for
patients with metastasis predominantly in the liver, debulking
surgery can be suggested (46).
In cases of small metastases (<3 cm), radiofrequency ablation
and cryoablation are a common choice of treatment (47). Additionally, ablation can be
applied mutually with surgical resection to bypass hepatectomy. In
patients with inoperable liver metastases, liver transarterial
chemoembolization (TACE) has emerged as a palliative treatment
procedure (40). Still, there is
a great hazard of perihepatic sepsis and liver abscess associated
with liver-directed therapy (48).
For selected patients, for whom medical treatment is
not an option, liver transplantation can be considered. The
selection standards for liver transplantation suggested by
Mazzaferro et al include a recipient age <55 years, no
evidence of disease recurrence for at least 6 months during the
pre-transplantation period, the extraction of all extrahepatic
metastases preceding liver transplantation and an involvement of
liver parenchyma <50% (49).
Gedaly et al in the retrospective report of the UNOS
database revealed that 150 orthotopic liver transplants (OLTs)
(amidst 87,820 ones performed between 1998 and 2008), were
performed for metastatic NETs. The average recipient age was 45
years. The overall survival rate was 81% at 1 year, as opposed to
65% at 3 and 49% at 5 years following transplantation (50). In the meta-analysis by Máthé et
al comprising 89 patients with NETs undergoing OLT, the authors
indicate a 1-year survival rate of 71%, together with 55%, and 44%
at 3 and five years, respectively (51). The comprehensive systematic review
of 64 cases revealed that liver transplantation resembles to
provide a survival benefit amid patients with diffuse liver
metastases; nevertheless, a high incidence of tumour recurrence
rate implies that the strict selection of patients is critical
(52).
Resection of the primary mass in cases of inoperable
metastatic cancer is still controversial. Data from a previous
systematic review determined that the main benefit of primary
tumour resection (PTR) is to alleviate manifestations caused by the
primary tumour, histologically verify the diagnosis and potentially
improve overall survival. Additionally, PTR was safe with a low
perioperative risk of mortality (53). However, due to the scarcity of
randomized controlled trials, the decision to implement PTR,
particularly in asymptomatic patients with the inoperable
metastatic condition, should still be made on an individual basis
(54).
Everolimus (Afinitor) is an oral mammalian target of
rapamycin (mTOR) inhibitor. It is applied as second-line therapy
for patients with advanced neoplasms. In the RAD001 in Advanced
Neuroendocrine Tumors-3 (RADIANT-3) trial, everolimus revealed the
reduction of disease-related hormonal symptoms and exhibited an
extended average progression-free survival (55).
Sunitinib is an inhibitor of the vascurlar
endothelial growth factor (VEGF) pathway, which was accepted as a
therapeutic approach for patients with non-surgical, progressive
metastatic NETs. Even though it does not significantly lengthen
progression-free survival (56),
sunitinib achieves complete, rapid and sustained anti-secretory
effects (57).
Additional molecularly targeted therapies include
sorafenib, pazopanib, axitinib and surafitinib, multi-targeted
kinase inhibitors (58), along
with the combination of temisirolimus, another mTOR inhibitor, with
the VEGF inhibitor, bevacizumab (59).
Cytotoxic chemotherapy includes agents, such as
5-fluorouracil (5-FU), oxaliplatin, capecitabine, temozolomide and
streptozocin. Often, a combination of these will be favoured:
Temozolomide with capecitabine, 5-FU/doxorubicin/streptozocin
(FAS), or streptozocin with doxorubicin or 5-FU (60). Systemic chemotherapy with a
streptozotocin and 5-FU mixture is a standard procedure for
patients with bulky extensive growths together with extrahepatic
metastases (4).
Peptide receptor radionuclide therapy (PRRT) with
the radiolabeled somatostatin analogue,
177Lu-tetra-azacyclodo-decanetetraacetic acid-octreotide
(177Lu-DOTATATE), is a novel treatment approach for
nonfunctioning PNETs (61-64).
A recent trial with 34 subjects with a metastatic functioning PNET,
including 5 cases of VIPoma, demonstrated that treatment with PRRT
with 177Lu-DOTATATE was safe, including PR in 56% of
patients and stable disease in 24% of patients. Moreover, it
resulted in a reduction of syndrome-specific syndromes (71%) with a
considerable improvement of QOL. Notably, there was a reduction in
diarrhoea in 4 (80%) patients with a metastatic VIPoma. However,
hormonal crises should be avoided during treatment (65). Other studies had shown the
excellent outcome followed by a total metabolic response to the
administered PRRT (66), and the
improvement of the quality of life of patients with NETs (67). Ataeinia et al presented
successful treatment with 177Lu-DOTATOC in a case of
pancreatic tumour recurrence with comprehensive nonsurgical hepatic
metastasis and IVC compression. PRRT can be counted as an
advantageous treatment approach in such patients with inoperable
extended metastasis nearby major vessels (68).
Lutathera®
[(177Lu)Lu-DOTA-TATE] is the first approved drug therapy
for PRRT. It is designated for the treatment of SSTR-positive
gastroenteropancreatic NETs. Lutathera® provokes DNA
breaks, leading to cell death of the tumour. The positive outcomes
of the multicenter phase-III clinical trial (69), NETTER-1, led to its approval by
medicines agencies in America and Europe.
7. Prognosis
The average survival rate of patients with VIPoma is
96 months (70). Ghaferi et
al reported that 59% of patients at an average follow-up of 15
months were alive with no indication of disease, 23% had succumbed
to the disease, and 18% were alive with the presence of the
condition (9). Prognosis is
largely dependent on tumour staging, surgical situation and the
severity of the metastases (71).
An age <40 years and >60 years, a tumour size >4 cm in
diameter, the poor management of water, electrolyte and acid-base
profiles, critical metastatic situation and tumour inoperability
are all indicated as unfortunate prognostic circumstances (72). The mortality rate associated with
VIPoma emerges from untreated WDHA syndrome leading to prolonged
dehydration with critical electrolyte and acid-base imbalances, and
subsequently leading to renal failure, cardiac arrest and
eventually death (73).
8. Conclusion
In conclusion, VIPoma is a unique tumour and can be
difficult to diagnose. If diarrhoea perseveres while fasting,
VIP-producing tumours should be considered, and blood plasma
specimens should be analysed for VIP in these patients. If the VIP
level is increased, the diagnosis of VIPoma should be considered.
Before any palliative treatment is commenced, the patient's water
and electrolyte profile should be adjusted. The neoplasm can be
cured adequately by surgical resection. If an operation is
undesirable, surgical debulking, somatostatin analogues can be
applied. Moreover, adjuvant therapy with PRRT is an efficient and
safe addition to surgery or in cases of widespread metastatic
disease or unresectable primary tumour.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
LA performed the literature search, collected the
data from different studies and wrote and edited this review
article. The author has read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that there are no competing
interests.
References
|
1
|
Verner JV and Morrison AB: Islet cell
tumor and a syndrome of refractory watery diarrhea and hypokalemia.
Am J Med. 25:374–380. 1958.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jensen RT, Cadiot G, Brandi ML, de Herder
WW, Kaltsas G, Komminoth P, Scoazec JY, Salazar R, Sauvanet A and
Kianmanesh R: Barcelona Consensus Conference participants: ENETS
consensus guidelines for the management of patients with digestive
neuroendocrine neoplasms: Functional pancreatic endocrine tumor
syndromes. Neuroendocrinology. 95:98–119. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yao JC, Eisner MP, Leary C, Dagohoy C,
Phan A, Rashid A, Hassan M and Evans DB: Population-based study of
islet cell carcinoma. Ann Surg Oncol. 14:3492–3500. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Parbhu SK and Adler DG: Pancreatic
neuroendocrine tumors: Contemporary diagnosis and management. Hosp
Pract 1995. 44:109–119. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Long RG, Bryant MG, Mitchell SJ, Adrian
TE, Polak JM and Bloom SR: Clinicopathological study of pancreatic
and ganglioneuroblastoma tumours secreting vasoactive intestinal
polypeptide (vipomas). Br Med J (Clin Res Ed). 282:1767–1771.
1981.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Belei OA, Heredea ER, Boeriu E, Marcovici
TM, Cerbu S, Mărginean O, Iacob ER, Iacob D, Motoc AGM and Boia ES:
Verner-Morrison syndrome. Literature review. Rom J Morphol Embryol.
58:371–376. 2017.PubMed/NCBI
|
|
7
|
Chen Y, Shi D, Dong F, Han SG, Qian ZH,
Yang LI, Wang Y, Yu RS, Li QH and Fu YB: Multiple-phase spiral CT
findings of pancreatic vasoactive intestinal peptide-secreting
tumor: A case report. Oncol Lett. 10:2351–2354. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Krejs GJ: VIPoma syndrome. Am J Med.
82:37–48. 1987.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ghaferi AA, Chojnacki KA, Long WD, Cameron
JL and Yeo CJ: Pancreatic VIPomas: Subject review and one
institutional experience. J Gastrointest Surg. 12:382–393.
2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Said SI and Mutt V: Polypeptide with broad
biological activity: Isolation from small intestine. Science.
169:1217–1218. 1970.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Friesen SR: Update on the diagnosis and
treatment of rare neuroendocrine tumors. Surg Clin North Am.
67:379–393. 1987.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Schizas D, Mastoraki A, Bagias G, Patras
R, Moris D, Lazaridis II, Arkadopoulos N and Felekouras E:
Clinicopathological data and treatment modalities for pancreatic
vipomas: A systematic review. J BUON. 24:415–423. 2019.PubMed/NCBI
|
|
13
|
Murphy MS, Sibal A and Mann JR: Persistent
diarrhoea and occult vipomas in children. BMJ. 320:1524–1526.
2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Brentjens R and Saltz L: Islet cell tumors
of the pancreas: The medical oncologist's perspective. Surg Clin
North Am. 81:527–542. 2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mekhjian HS and O'Dorisio TM: VIPoma
syndrome. Semin Oncol. 14:282–291. 1987.PubMed/NCBI
|
|
16
|
Remme CA, de Groot GH and Schrijver G:
Diagnosis and treatment of VIPoma in a female patient. Eur J
Gastroenterol Hepatol. 18:93–99. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Metz DC and Jensen RT: Gastrointestinal
neuroendocrine tumors: Pancreatic endocrine tumors.
Gastroenterology. 135:1469–1492. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Piet R, Dunckley H, Lee K and Herbison AE:
Vasoactive intestinal peptide excites GnRH neurons in male and
female mice. Endocrinology. 157:3621–3630. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Abu-Zaid A, Azzam A, Abudan Z, Algouhi A,
Almana H and Amin T: Sporadic pancreatic vasoactive intestinal
peptide-producing tumor (VIPoma) in a 47-year-old male. Hematol
Oncol Stem Cell Ther. 7:109–115. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen C, Zheng Z, Li B, Zhou L, Pang J, Wu
W, Zheng C and Zhao Y: Pancreatic VIPomas from China: Case reports
and literature review. Pancreatology. 19:44–49. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Anderson CW and Bennett JJ: Clinical
presentation and diagnosis of pancreatic neuroendocrine tumors.
Surg Oncol Clin N Am. 25:363–374. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Grozinsky-Glasberg S, Mazeh H and Gross
DJ: Clinical features of pancreatic neuroendocrine tumors. J
Hepatobiliary Pancreat Sci. 22:578–585. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Smith SL, Branton SA, Avino AJ, Martin JK,
Klingler PJ, Thompson GB, Grant CS and van Heerden JA: Vasoactive
intestinal polypeptide secreting islet cell tumors: A 15-year
experience and review of the literature. Surgery. 124:1050–1055.
1998.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nilubol N, Freedman EM, Quezado MM, Patel
D and Kebebew E: Pancreatic neuroendocrine tumor secreting
vasoactive intestinal peptide and dopamine with pulmonary emboli: A
case report. J Clin Endocrinol Metab. 101:3564–3567.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pasricha G, Padhi P, Daboul N and Monga
DK: Management of well-differentiated gastroenteropancreatic
neuroendocrine tumors (GEPNETs): A review. Clin Ther. 39:2146–2157.
2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Debray MP, Geoffroy O, Laissy JP, Lebtahi
R, Silbermann-Hoffman O, Henry-Feugeas MC, Cadiot G, Mignon M and
Schouman-Claeys E: Imaging appearances of metastases from
neuroendocrine tumours of the pancreas. Br J Radiol. 74:1065–1070.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Semelka RC, Custodio CM, Cem Balci N and
Woosley JT: Neuroendocrine tumors of the pancreas: Spectrum of
appearances on MRI. J Magn Reson Imaging. 11:141–148.
2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lam S, Liew H, Khor HT, Dalan R, Kon YC,
Jong M, Chew DE and Leow MK: VIPoma in a 37-year-old man. Lancet.
382(832)2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ram R, Natanzi N, Saadat P, Eliav D and
Vadmal MS: Skin metastasis of pancreatic vasoactive intestinal
polypeptide tumor: Case report and review of the literature. Arch
Dermatol. 142:946–947. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
O'Dorisio TM, Mekhjian HS and Gaginella
TS: Medical therapy of VIPomas. Endocrinol Metab Clin North Am.
18:545–556. 1989.PubMed/NCBI
|
|
31
|
Ito T, Igarashi H, Uehara H and Jensen RT:
Pharmacotherapy of Zollinger-Ellison syndrome. Expert Opin
Pharmacother. 14:307–321. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jensen RT, Niederle B, Mitry E, Ramage JK,
Steinmuller T, Lewington V, Scarpa A, Sundin A, Perren A, Gross D,
et al: Frascati Consensus Conference; European neuroendocrine tumor
society: Gastrinoma (duodenal and pancreatic). Neuroendocrinology.
84:173–182. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang X, Zhou L, Liu Y, Li W, Gao H, Wang
Y, Yao B, Jiang D and Hu P: Surgical resection of vasoactive
intestinal peptideoma with hepatic metastasis aids symptom
palliation: A case report. Exp Ther Med. 11:783–787.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Caplin ME, Pavel M, Ćwikła JB, Phan AT,
Raderer M, Sedláčková E, Cadiot G, Wolin EM, Capdevila J, Wall L,
et al: Lanreotide in metastatic enteropancreatic neuroendocrine
tumors. N Engl J Med. 371:224–233. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Plouin PF, Bertherat J, Chatellier G,
Billaud E, Azizi M, Grouzmann E and Epelbaum J: Short-term effects
of octreotide on blood pressure and plasma catecholamines and
neuropeptide Y levels in patients with phaeochromocytoma: A
placebo-controlled trial. Clin Endocrinol (Oxf). 42:289–294.
1995.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Nguyen HN, Backes B, Lammert F, Wildberger
J, Winograd R, Busch N, Rieband H and Matern S: Long-term survival
after diagnosis of hepatic metastatic VIPoma: Report of two cases
with disparate courses and review of therapeutic options. Dig Dis
Sci. 44:1148–1155. 1999. View Article : Google Scholar
|
|
37
|
Lamberts SW, Pieters GF, Metselaar HJ, Ong
GL, Tan HS and Reubi JC: Development of resistance to a long-acting
somatostatin analogue during treatment of two patients with
metastatic endocrine pancreatic tumours. Acta Endocrinol (Copenh).
119:561–566. 1988.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Faiss S, Pape UF, Böhmig M, Dörffel Y,
Mansmann U, Golder W, Riecken EO and Wiedenmann B: International
Lanreotide and Interferon Alfa Study Group: Prospective,
randomized, multicenter trial on the antiproliferative effect of
lanreotide, interferon alfa, and their combination for therapy of
metastatic neuroendocrine gastroenteropancreatic tumors-the
International Lanreotide and Interferon Alfa Study Group. J Clin
Oncol. 21:2689–2696. 2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lapeña Rodríguez M, Cholvi Calduch R,
Muñoz Forner E, Garcés Albir M and Sabater Ortí L: Life-threating
diarrhea and acute renal failure secondary to pancreatic VIPoma
treated by surgery. Rev Esp Enferm Dig. 111:641–643.
2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Dréanic J, Lepère C, El Hajjam M, Gouya H,
Rougier P and Coriat R: Emergency therapy for liver metastases from
advanced VIPoma: Surgery or transarterial chemoembolization? Ther
Adv Med Oncol. 8:383–387. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kos-Kudła B, Ćwikła J, Ruchała M,
Hubalewska-Dydejczyk A, Jarzab B, Krajewska J and Kamiński G:
Current treatment options for gastroenteropancreatic neuroendocrine
tumors with a focus on the role of lanreotide. Contemp Oncol
(Pozn). 21:115–122. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shah M, Goldner W, Halfdanarson T,
Bergsland E, Berlin J, Halperin D, Chan J, Kulke M, Benson A,
Blaszkowsky L, et al: NCCN clinical practice guidelines in
oncology: Neuroendocrine and adrenal tumors. J Natl Compr Canc
Netw. 16:693–702. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Partelli S, Bartsch DK, Capdevila J, Chen
J, Knigge U, Niederle B, Nieveen van Dijkum EJM, Pape UF, Pascher
A, Ramage J, et al: Antibes consensus conference participants:
ENETS consensus guidelines for standard of care in neuroendocrine
tumours: Surgery for small intestinal and pancreatic neuroendocrine
tumours. Neuroendocrinology. 105:255–265. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Norton JA, Kivlen M, Li M, Schneider D,
Chuter T and Jensen RT: Morbidity and mortality of aggressive
resection in patients with advanced neuroendocrine tumors. Arch
Surg. 138:859–866. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Fernández-Cruz L, Blanco L, Cosa R and
Rendón H: Is laparoscopic resection adequate in patients with
neuroendocrine pancreatic tumors? World J Surg. 32:904–917.
2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Pavel M, O'Toole D, Costa F, Capdevila J,
Gross D, Kianmanesh R, Krenning E, Knigge U, Salazar R, Pape UF, et
al: Vienna Consensus Conference participants: ENETS consensus
guidelines update for the management of distant metastatic disease
of intestinal, pancreatic, bronchial neuroendocrine neoplasms (NEN)
and NEN of unknown primary site. Neuroendocrinology. 103:172–185.
2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Moug SJ, Leen E, Horgan PG and Imrie CW:
Radiofrequency ablation has a valuable therapeutic role in
metastatic VIPoma. Pancreatology. 6:155–159. 2006.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kennedy A, Bester L, Salem R, Sharma RA,
Parks RW and Ruszniewski P: NET-Liver-Metastases Consensus
Conference: Role of hepatic intra-arterial therapies in metastatic
neuroendocrine tumours (NET): Guidelines from the
NET-liver-metastases consensus conference. HPB (Oxford). 17:29–37.
2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Mazzaferro V, Pulvirenti A and Coppa J:
Neuroendocrine tumors metastatic to the liver: How to select
patients for liver transplantation? J Hepatol. 47:460–466.
2007.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gedaly R, Daily MF, Davenport D, McHugh
PP, Koch A, Angulo P and Hundley JC: Liver transplantation for the
treatment of liver metastases from neuroendocrine tumours: An
analysis of the UNOS database. Arch Surg. 146:953–958.
2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Máthé Z, Tagkalos E, Paul A, Molmenti EP,
Kóbori L, Fouzas I, Beckebaum S and Sotiropoulos GC: Liver
transplantation for hepatic metastases of neuroendocrine pancreatic
tumors: A survival-based analysis. Transplantation. 91:575–582.
2011. View Article : Google Scholar
|
|
52
|
Moris D, Tsilimigras DI,
Ntanasis-Stathopoulos I, Beal EW, Felekouras E, Vernadakis S, Fung
JJ and Pawlik TM: Liver transplantation in patients with liver
metastases from neuroendocrine tumors: A systematic review.
Surgery. 162:525–536. 2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Tsilimigras DI, Ntanasis-Stathopoulos I,
Kostakis ID, Moris D, Schizas D, Cloyd JM and Pawlik TM: Is
resection of primary midgut neuroendocrine tumors in patients with
unresectable metastatic liver disease justified? A systematic
review and meta-analysis. J Gastrointest Surg. 23:1044–1054.
2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Guo J, Zhang Q, Bi X, Zhou J, Li Z, Huang
Z, Zhang Y, Li M, Chen X, Hu X, et al: Systematic review of
resecting primary tumor in MNETs patients with unresectable liver
metastases. Oncotarget. 8:17396–17405. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yao JC, Shah MH, Ito T, Bohas CL, Wolin
EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG,
et al: Everolimus for advanced pancreatic neuroendocrine tumors. N
Engl J Med. 364:514–523. 2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Raymond E, Dahan L, Raoul JL, Bang YJ,
Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A,
et al: Sunitinib malate for the treatment of pancreatic
neuroendocrine tumors. N Engl J Med. 364:501–513. 2011.PubMed/NCBI View Article : Google Scholar
|
|
57
|
De Mestier L, Walter T, Brixi H,
Lombard-Bohas C and Cadiot G: Sunitinib achieved fast and sustained
control of VIPoma symptoms. Eur J Endocrinol. 172:K1–K3.
2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Phan AT, Halperin DM, Chan JA, Fogelman
DR, Hess KR, Malinowski P, Regan E, Ng CS, Yao JC and Kulke MH:
Pazopanib and depot octreotide in advanced, well-differentiated
neuroendocrine tumours: A multicentre, single-group, phase 2 study.
Lancet Oncol. 16:695–703. 2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Hobday TJ, Qin R, Reidy-Lagunes D, Moore
MJ, Strosberg J, Kaubisch A, Shah M, Kindler HL, Lenz HJ, Chen H
and Erlichman C: Multicenter phase II trial of temsirolimus and
bevacizumab in pancreatic neuroendocrine tumors. J Clin Oncol.
33:1551–1556. 2015.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Akirov A, Larouche V, Alshehri S, Asa SL
and Ezzat S: Treatment options for pancreatic neuroendocrine
tumors. Cancers (Basel). 11(E828)2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Auernhammer CJ, Spitzweg C, Angele MK,
Boeck S, Grossman A, Nölting S, Ilhan H, Knösel T, Mayerle J,
Reincke M and Bartenstein P: Advanced neuroendocrine tumours of the
small intestine and pancreas: Clinical developments, controversies,
and future strategies. Lancet Diabetes Endocrinol. 6:404–415.
2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Strosberg J, El-Haddad G, Wolin E,
Hendifar A, Yao J, Chasen B, Mittra E, Kunz PL, Kulke MH, Jacene H,
et al: NETTER-1 trial investigators. Phase 3 trial of
177lu-dotatate for midgut neuroendocrine tumours. N Engl J Med.
376:125–135. 2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Hicks RJ, Kwekkeboom DJ, Krenning E, Bodei
L, Grozinsky-Glasberg S, Arnold R, Borbath I, Cwikla J, Toumpanakis
C, Kaltsas G, et al: ENETS consensus guidelines for the standards
of care in neuroendocrine neoplasia: Peptide receptor radionuclide
therapy with radiolabeled somatostatin analogues.
Neuroendocrinology. 105:295–309. 2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Brabander T, van der Zwan WA, Teunissen
JJM, Kam BLR, Feelders RA, de Herder WW, van Eijck CHJ, Franssen
GJH, Krenning EP and Kwekkeboom DJ: Long-term efficacy, survival,
and safety of [177Lu-DOTA0, Tyr3]octreotate in patients with
gastroenteropancreatic and bronchial neuroendocrine tumours. Clin
Cancer Res. 23:4617–4624. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Zandee WT, Brabander T, Blažević A, Kam
BLR, Teunissen JJM, Feelders RA, Hofland J and de Herder WW:
Symptomatic and radiological response to 177Lu-DOTATATE for the
treatment of functioning pancreatic neuroendocrine tumors. J Clin
Endocrinol Metab. 104:1336–1344. 2019.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Adnan A and Basu S: Rare site primary soft
tissue neuroendocrine tumour with metastases and near-complete
resolution with 177Lu-DOTATATE: Documenting a promising
clinical application of peptide receptor radionuclide therapy. J
Nucl Med Technol. 119(227058): Aug 10, 2019 (Epub ahead of print).
View Article : Google Scholar
|
|
67
|
Jin XF, Spampatti MP, Spitzweg C and
Auernhammer CJ: Supportive therapy in gastroenteropancreatic
neuroendocrine tumors: Often forgotten but important. Rev Endocr
Metab Disord. 19:145–158. 2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ataeinia B, Loberg C, Kravets H, Beheshti
M, von Mallek D, Mottaghy FM and Heinzel A: Successful palliative
peptide receptor radionuclide therapy for impending compression of
vena cava due to unresectable liver metastasis of neuroendocrine
tumor. EXCLI J. 18:273–276. 2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Hennrich U and Kopka K:
Lutathera®: The first FDA- and EMA-approved
radiopharmaceutical for peptide receptor radionuclide therapy.
Pharmaceuticals (Basel). 12(E114)2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Roland CL, Bian A, Mansour JC, Yopp AC,
Balch GC, Sharma R, Xie XJ and Schwarz RE: Survival impact of
malignant pancreatic neuroendocrine and islet cell neoplasm
phenotypes. J Surg Oncol. 105:595–600. 2012.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Loh HH and Tan F: Pancreatic vasoactive
intestinal peptide producing tumor (VIPoma): A case report and
literature review. Endokrinolojide Diyalog. 10:32–37. 2013.
|
|
72
|
Karim N, Zarzour A, Daw HA, Palaparty P,
Taftaf R, Shehata M and Taylor HC: Prolonged survival in a patient
with metastatic vasoactive intestinal peptide producing pancreatic
neuroendocrine tumors. J Clin Case Rep. 2(210)2012. View Article : Google Scholar
|
|
73
|
Soga J and Yakuwa Y: Vipoma/diarrheogenic
syndrome: A statistical evaluation of 241 reported cases. J Exp
Clin Cancer Res. 17:389–400. 1998.PubMed/NCBI
|