Introduction
In 1889, Ollier described a rare disease characterized by multiple, unilateral limb endogenous chondromas associated with limb deformities (1). At present, the etiology of the disease has not been definitively determined, although it has been reported that the occurrence of Ollier disease and Maffucci syndrome (Ollier disease with multiple cavernous hemangiomas) may be related to somatic mosaic mutations of isocitrate dehydrogenase (IDH)I and IDH2(2). The diagnosis of Ollier disease is based on clinical and conventional radiological evaluations, rather than genetic testing.
It mainly manifests as local pain, bone swelling, palpable bone mass and bone deformity (3). The risk of the disease developing into chondrosarcoma is high, and the transformation rates range from 5 to 50%, as previously reported (4). The diagnosis of this disease mainly depends on the clinical description, imaging diagnosis and histological examination, and surgical treatment is the main form of treatment (5). The present study describes a case of Ollier disease with chondrosarcoma and also presents a summary of the relevant literature in an aim to shed further light on this rare disease.
Case report
A 37-year-old man had initially developed an obvious mass on his left index, middle and index fingers at the age of 6 years; however, no special treatment was administered. After 30 years, the masses on the patient's hands had grown in size, acquiring a dendritic shape, and were accompanied by pain, leading to limited function and movement. Moreover, the left hand (Fig. 1A), left hip, left knee, left ankle and left foot (Fig. 1B) were swollen and this condition progressively became aggravated. The mass was hard and exhibited nodular changes. At a later stage, the mass in the left hip became rapidly enlarged and the patient was in obvious pain. He also visited relevant hospitals without receiving any special treatment. The patient came to the Second Affiliated Hospital of Shanxi Medical University for treatment in 2018 due to severe pain in the left hip joint. Since the onset of the disease, the patient did not have any symptoms of a frequent fever, fatigue, emaciation, dizziness, headaches, blurred vision or any other discomfort, In addition, the patient had no family history of any hereditary diseases, no history of exposure to any specific chemicals and radiation, and was a single farmer in occupation and did not use tobacco and consume alcohol. A physical examination upon admission revealed the following: The left lower extremity of the patient was significantly shorter than the right lower extremity, by ~10 cm. Dendritic-shaped masses were observed in the left hand, swelling and deformity were observed on the left hip, left knee and left foot, and the range of motion of the left hip and left knee was markedly restricted compared with that of the normal right hip and knee. An X-ray examination revealed multiple osteopathies of the left hand, left hip, left knee, left ankle and left foot (Fig. 2). A magnetic resonance imaging (MRI) examination of the hip revealed abnormal signals of bone and soft tissue in the proximal left femur (Fig. 3), and malignant lesions were considered. The clinical diagnosis of Ollier disease was thus made. The results of the patient's laboratory tests are presented in Table I. However, due to the clear diagnosis, the patient did not undergo radionuclide bone imaging and a computed tomography (CT) scan.
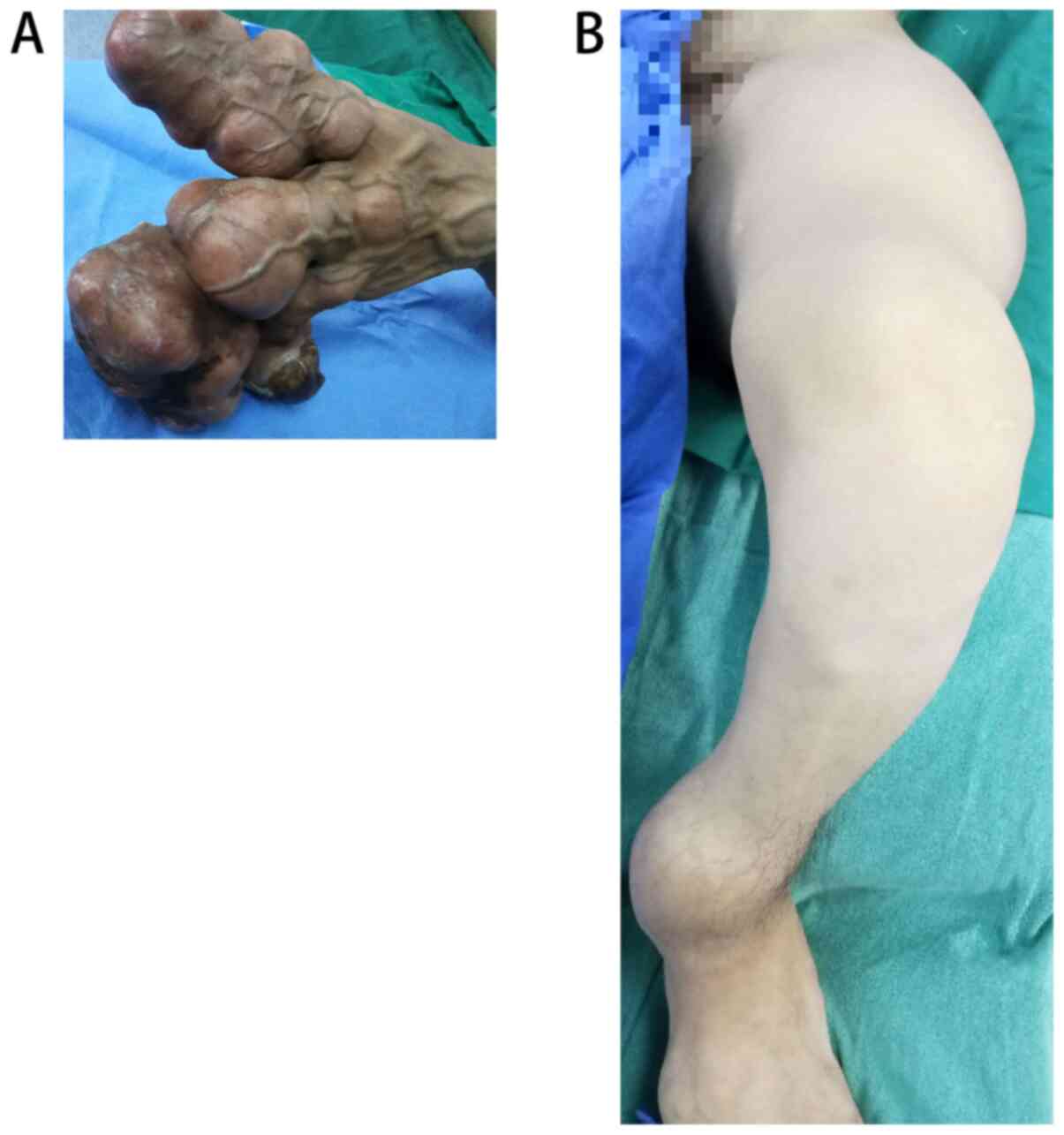 |
Figure 1
General images of (A) the left hand and (B) the left lower extremity.
|
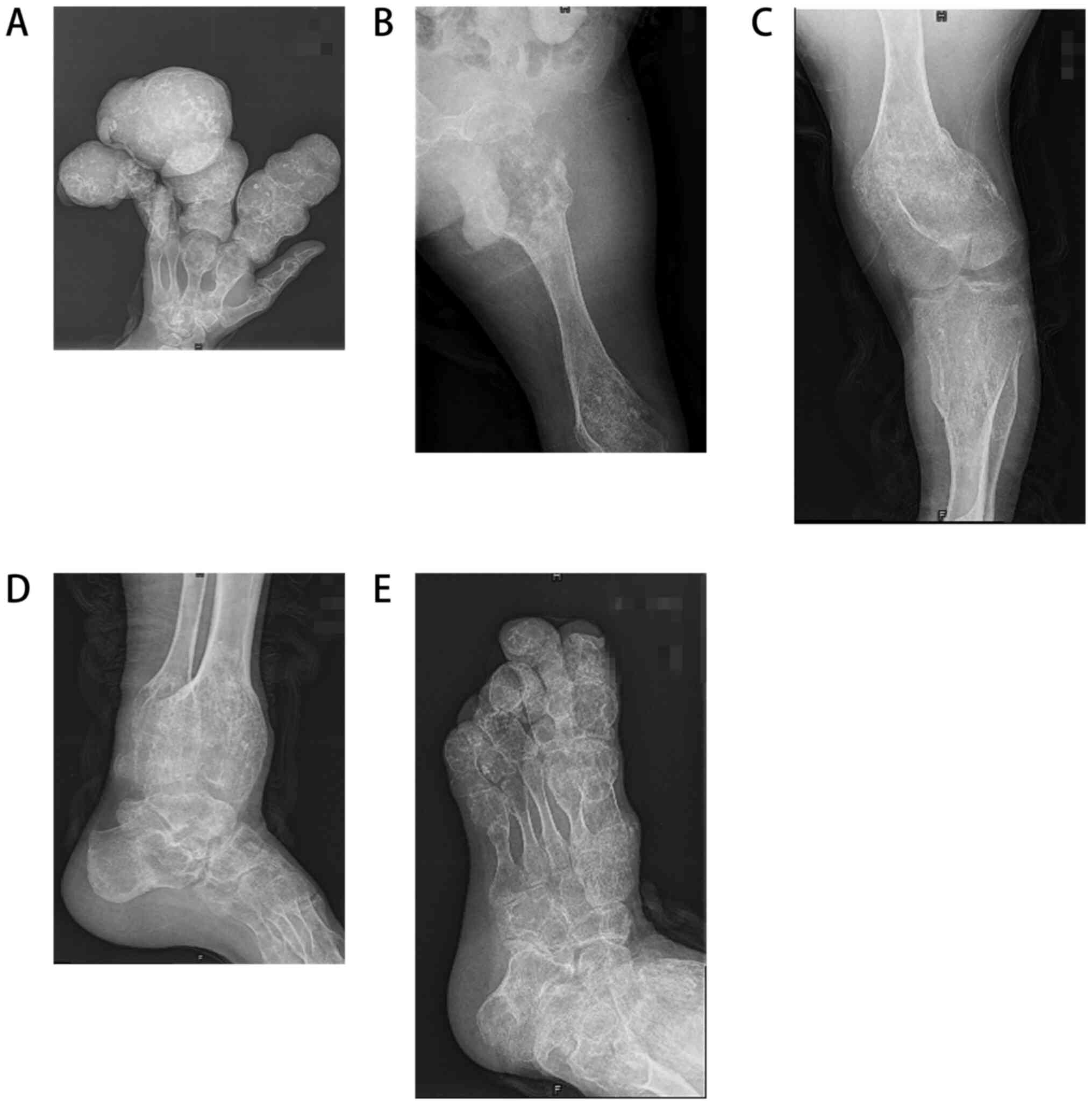 |
Figure 2
Localized bone destruction and bone expansive growth in the (A) left hand, (B) left hip, (C) left knee, (D) left ankle and (E) left foot.
|
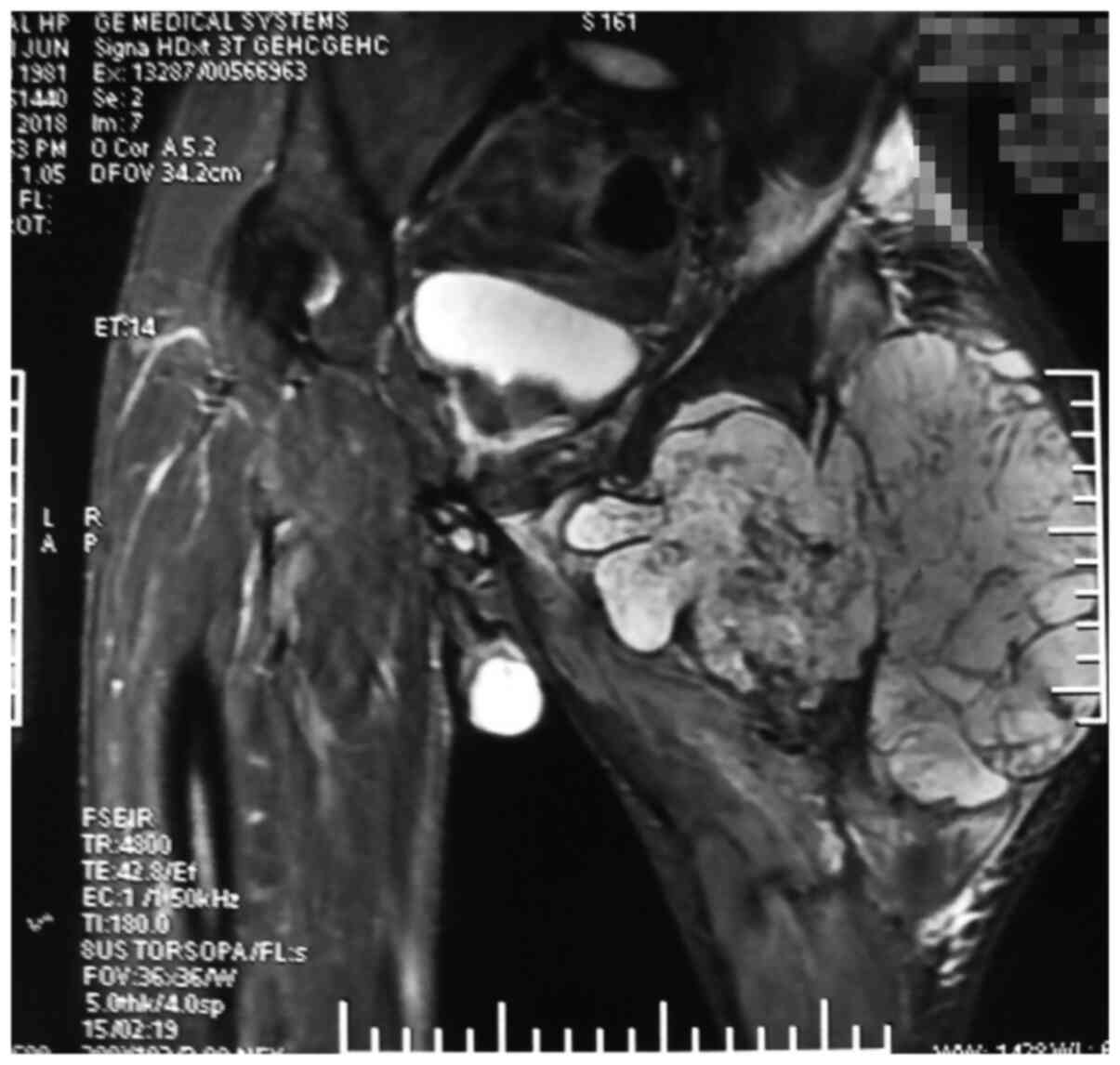 |
Figure 3
Bone destruction of the left hip joint and abnormal signal of the surrounding soft tissue can be seen. Post-operative pathological examination revealed that the lesion in the left hip joint developed into chondrosarcoma.
|
 |
Table I
Results of the laboratory examination of the patient.
|
Table I
Results of the laboratory examination of the patient.
| Parameter |
First admission |
Last follow-up |
Normal reference range |
| Platelet count (109 g/l) |
389 |
462 |
125-350 |
| Alkaline phosphatase (U/l) |
149 |
188 |
45-125 |
| Lactate dehydrogenase (U/l) |
255 |
267 |
120-250 |
| Neuron-specific enolase (ng/ml) |
15.9 |
- |
<13 |
| ESR (mm/h) |
6 |
61 |
0-15 |
| CRP (mg/l) |
7 |
182 |
0-10 |
| D-Dimer (ng/ml) |
175 |
652 |
<255 |
Following a pre-operative preparation, and under brachial plexus anesthesia and local anesthesia, the 2nd, 3rd and 4th metacarpophalangeal joints of the left hand were severed, and the lesions of the proximal left femur were biopsied. The doctor sent the severed finger and the lesion on the proximal left femur to the pathology department for examination. The results revealed that the proximal focus of left femur exhibited a tumor-like hyperplasia of chondrocytes, nuclear atypia, suggesting chondrosarcoma grade II (Fig. 4). Due to the findings that the mass on the left hip joint was malignant, it was recommended that a left hip joint dissection should be performed; however, the patient and his family could not accept this and the patient was thus discharged after his condition was relatively stable. After a year and a half of follow-up, the patient felt that the function of the left hand was good, the lesion of the left thigh was enlarged, the local ulcer ruptured, and the pain had become more severe. The patient passed away three months later.
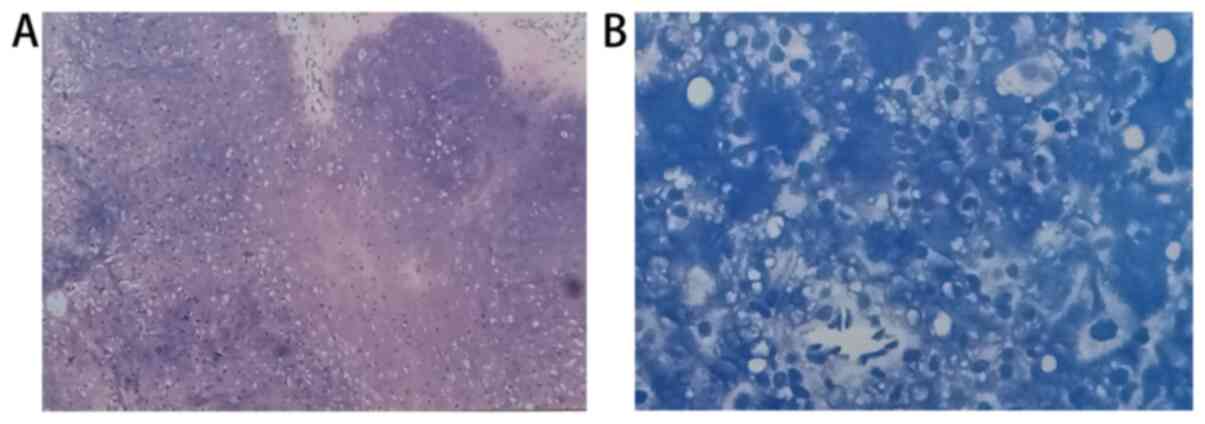 |
Figure 4
(A) Biopsy sample from the end of the finger, revealing a cartilaginous tumor with nodular lobular hyperplasia and calcification, consistent with Ollier disease. (B) Biopsy sample from the left hip joint. The size of the nucleus is different, atypia is evident, and pathological mitosis can be seen, thus suggesting chondrosarcoma grade II.
|
Discussion
Multiple enchondromatosis is a rare heterogeneous skeletal disease, which can be divided into six subtypes as follows: Ollier disease, Maffucci syndrome, metaphyseal chondromatosis, metatarsal chondromatosis, spinal interstitial chondrodysplasia and spinal interstitial chondromatosis (6). Over the years, it has been reported that Ollier disease may be associated with pelvic diseases, hematological diseases, gliomas of the skull base, pancreatic cancer and other diseases (7-14), and this has guided clinicians in the diagnosis of patients who may have Ollier disease. However, attention should be paid to lesions of the nervous system (15), digestive system (16) and blood system. In addition, due to of the high risk of degeneration into chondrosarcoma (4), patients should undergo close and long-term follow-up in order to identify the relevant systemic diseases and malignant lesions as early as possible.
Disease pathogenesis
In terms of etiology, Amary et al (2), in 2011, suggested that the onset of Ollier disease and Maffucci syndrome may be related to somatic mosaic mutations in the IDHI and IDH2 genes. Silve and Jüppner (17), in 2006, also suggested that the onset of Ollier disease may be related to somatic mutations in the parathyroid hormone 1 receptor (PTHR1) (R150C) gene. These two theories suggest that the disease is a non-hereditary disease; this is a non-familial genetic disease that occurs spontaneously and is not inherited. Although the etiology of the disease has been reported, the disease pathogenesis has not yet been fully elucidated.
Diagnosis
The diagnosis of Ollier disease is based on clinical manifestations and a routine radiological evaluation. A histological analysis is mainly used to identify malignant tumors. Other examinations, such as radionuclide scanning, ultrasound and MRI, are not helpful in determining the diagnosis. They are used to evaluate and monitor skin lesions with symptoms (pain and increased volume). However, there is no simple method for the genetic diagnosis in clinical practice (18). The clinical diagnosis of Ollier disease is relatively simple, and there is no obvious trend as regards sex in the majority of patients. The first onset of Ollier disease usually appears within the first 10 years of life, appearing as a single or multiple bone mass. With the increase in age, corresponding limb deformities appear in Ollier disease, deformities, which affects the growth and development of children (5).
Clinical description
The disease mainly manifests in the short tubular bones of the hand, followed by the femur, tibia, fibula, humerus, radius, ulna (18). However, there are also reported cases of the disease in flat bone, such as the scapula (19). In the present study, it was found that this patient exhibited abnormalities and unbalanced growth and development during adolescence. The patient in the present study developed a deformity on the left hand at the age of six, which progressively became more severe. Adult patients often develop arthritis and other skeletal deformities.
Imaging diagnosis
Radionuclide bone imaging is the most effective tool for the diagnosis of multiple lesions and malignant transformation (20). It can guide the clinical and prognostic analysis of Ollier disease. An X-ray is one of the most basic tools for the diagnosis of skeletal diseases. In Ollier disease, the lesions mostly appear in the shaft and metaphysis of short or long bone, and the lesions are mostly oval in shape. In short bone, the lesions can appear as scallop-like impressions due to the limited space, the cortical bone becomes thinner, and thus pathological fractures are common. In the long shaft, due to its large growth space, there is no characteristic scallop-like impression, and the bone cortex does not become thinner (21). It is more important to evaluate the lesion size and soft tissue invasion by a CT scan (16). The lesion is characterized by a round and lobulated density shadow, and the density is equal to or slightly lower than the muscle density shadow (22). A CT scan can also reveal different calcification patterns. It has been indicated that further attention should be paid to calcification patterns when predicting the development of chondrosarcoma (18). In the present study, the MRI revealed a round, quasi-round and striped density inhomogeneous signal in the medullary cavity, particularly soft tissue involvement. For example, in the present study, the MRI revealed the significant destruction of the left femoral cortex and extensive soft tissue involvement, combined with the rapid aggravation of short-term pain in the patient, considering the greater possibility of malignant chondrosarcoma; a biopsy also needs to be performed to determine the nature of the lesion before guiding clinical treatment.
On the whole, an X-ray can diagnose correctly typical cases. A CT scan can reveal calcification foci, which is of great significance in determining whether there a malignant alteration exists. An MRI examination can visualize the cartilage components in the lesions to further clarify the lesions and improve the accuracy of diagnosis.
Histological examination
Lesions sent for examination can reveal the following: The destruction of bone tissue, followed by the extensive proliferation of chondrocytes and visible calcification. The typical changes involved in the development into chondrosarcoma are nuclear polymorphisms and hyperchromaticity, exhibiting pathological mitosis (23).
The clinical diagnosis of Ollier disease is relatively simple, and there is no obvious sex-associated trend in the majority of patients. The first onset of Ollier disease usually appears within the first 10 years of life, appearing as a single or multiple bone mass. With the increase in age, corresponding limb deformities appear in Ollier disease, which subsequently affect the growth and development of children.
Genetic diagnosis
To the best of our knowledge, to date, there is no available method of genetic diagnosis for this disease.
Treatment
It has been reported that there is no specific drug treatment for Ollier disease (15). For patients with Ollier disease without significant deformities or functional impairment, a long-term follow-up can be made (24). The treatment of complications associated with Ollier disease remains predominantly surgical, and is mainly considered in the following cases: i) Patients who have been found to have the disease during childhood, and the disease thus affects the growth and development of the child. Bone lengthening surgery can correct deformities and can maintain balanced limb development as much as possible. Madan et al (25) demonstrated that the Ilizarov technique has a unique advantage in this respect. Of course, when treating children with Ollier disease with bone lengthening, the focus should be on the location of the lesion, the method of lengthening and the expected benefit of lengthening (24), in order to achieve an optimal therapeutic effect. ii) For patients with pathological fractures, the traditional surgical treatment is usually adopted, involving the curettage of lesions and internal fixation with bone grafts, and the patient is followed-up regularly after surgery. iii) Surgical resection remains the primary treatment for chondrosarcoma associated with Ollier disease; however, extensive resection should also be performed, and sometimes partial resection should be performed. Patients may also require amputation in order to reduce the risk of post-operative recurrence and metastasis and to maximize survival (26).
In conclusion, Ollier disease is characterized by multiple endogenous chondromas of one limb with ipsilateral limb deformities. The incidence rate is ~1/100,000. With the development in research methods, the pathological and genetic mutation characteristics of the disease are gradually becoming clearer. However, the follow-up of patients with the disease is crucial, as other systemic diseases and easily malignant changes may accompany the course of the disease (27). In the present study, the pathogenesis, diagnosis and treatment of the disease were systematically described through the analysis of a typical case and a literature review, in order to effectively guide clinical practice.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the Natural Science Foundation of Shanxi Province (no. 201901D111371).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
JW and JL participated in the conception and design of the study. JW, ZW and JL confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from the patient presented in this case report prior to his death.
Patient consent for publication
As the patient passed away, consent for his data to be published was provided by the patient's brother.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Ollier L: De la dyschondroplasie. Bull Soc Chir Lyon. 3:22–24. 1900.
|
|
2
|
Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, et al: Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 43:1262–1265. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
D'Angelo L, Massimi L, Narducci A and Di Rocco C: Ollier disease. Childs Nerv Syst. 25:647–653. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Verdegaal SH, Bovee JV, Pansuriya TC, Grimer RJ, Ozger H, Jutte PC, San Julian M, Biau DJ, van der Geest IC, Leithner A, et al: Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicenter study of 161 patients. Oncologist. 16:1771–1779. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yang H and Lu H: Progress in comprehensive diagnosis and treatment of endogenous chondroma. Zhejiang J Integr Tradit Chin West Med. 26:877–880. 2016.
|
|
6
|
Superti-Furga A, Spranger J and Nishimura G: Enchondromatosis revisited: New classification with molecular basis. Am J Med Genet C Semin Med Genet. 160C:154–164. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fridirici ZC, Petrusek JJ, Thorpe EJ and Leonetti JP: Ollier disease of the lateral skull base. Otol Neurotol. 39:e52–e53. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gajavelli S, Nakhla J, Nasser R, Yassari R, Weidenheim KM and Graber J: Ollier disease with anaplastic astrocytoma: A review of the literature and a unique case. Surg Neurol Int. 7 (Suppl 23):S607–S611. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kenny SL, Patel K, Humphries A, Davis M, Flanagan AM and McCluggage WG: Ovarian cellular fibroma harbouring an isocitrate dehydrogenase 1 (1DH1) mutation in a patient with Ollier disease: Evidence for a causal relationship. Histopathology. 62:667–670. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mandonnet E, Anract P, Martin E, Roujeau T, Spena G, Cormier-Daire V, Duffau H and Baujat G: Collaborators. Brain and skull base MRI findings in patients with Ollier-Maffucci disease: A series of 12 patient-cases. Clin Neurol Neurosurg. 160:147–151. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Moser T, Lin XZ, Bazille G, Fleury M, Dietemann JL and Kremer S: Progressive hemianopsia caused by intracranial enchondroma in Ollier disease. Neurology. 71(2018)2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rietveld L, Nieboer TE, Kluivers KB, Schreuder HW, Bulten J and Massuger LF: First case of juvenile granulosa cell tumor in an adult with Ollier disease. Int J Gynecol Pathol. 28:464–467. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sampagar AA, Jahagirdar RR, Bafna VS and Bartakke SP: Juvenile granulosa cell tumor associated with Ollier disease. Indian J Med Paediatr Oncol. 37:293–295. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
White MS, Martin PL and McLean TW: Acute myelogenous leukemia associated with Ollier disease. Pediatr Blood Cancer. 50:645–646. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ding C, Chen W, Liu F, Xiong M and Chen J: Skull base chondrosarcoma caused by Ollier disease: A case report and literature review. World Neurosurg. 127:103–108. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kumar A, Jain VK, Bharadwaj M and Arya RK: Ollier disease: Pathogenesis, diagnosis, and management. Orthopedics. 38:497–506. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Silve C and Jüppner H: Ollier disease. Orphanet J Rare Dis. 1(37)2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sun TTT, Keung IF, Wong TC, Leung POY and Chan JSY: Enchondroma of the hand: Result of surgery curettage and grafting and possible factors affecting the outcome. J Orthop Trauma Rehabil (Hong Kong). 22:13–17. 2017.
|
|
19
|
Wang JP, Xu ZY, Bao ZQ, Dai XS, Ma L, Yao N, Wang ZP, Tao YS and Chai DM: Ollier disease: Two case reports and a review of the literature. Am J Transl Res. 10:3818–3826. 2018.PubMed/NCBI
|
|
20
|
Vaz RM and Turner C: Ollier disease (enchondromatosis) associated with ovarian juvenile granulosa cell tumor and precocious pseudopuberty. J Pediatr. 108:945–947. 1986.PubMed/NCBI View Article : Google Scholar
|
|
21
|
El Abiad JM, Robbins SM, Cohen B, Levin AS, Valle DL, Morris CD and de Macena Sobreira NL: Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am J Med Genet A. 182:1093–1103. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Miyawaki T, Kinoshita Y and Iizuka T: A case of Ollier's disease of the hand. Ann Plast Surg. 38:77–80. 1997.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jacobi CM, Hiranya ES, Gay A, Holzmann D, Kollias S and Soyka MB: Enchondroma of the nasal septum due to Ollier disease: A case report and review of the literature. Head Neck. 37:E30–E33. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang H: Bone lengthening for children with limb shortening due to Ollier's disease. Chin J Orthop Surg. 5:471–476. 2018.
|
|
25
|
Madan SS, Robinson K, Kasliwal PD, Bell MJ, Saleh M and Fernandes JA: Limb reconstruction in Ollier's disease. Strategies Trauma Limb Reconstr. 10:49–54. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Atalay İB, Yılmaz S, Şimşek MA, Ekşioğlu MF and Güngör BŞ: Chondrosarcomas of the phalanges of the hand. Eklem Hastalik Cerrahisi. 29:34–39. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rexach Fumanya M, Castro Guardiola A and García-Bragado Dalmau F: Ollier's disease. Med Clin (Barc). 139(e5)2012.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|














