1. Introduction
Periodontitis is a chronic multifactorial inflammatory disease associated with dysbiotic plaque biofilms and is characterized by the progressive destruction of the tooth supporting apparatus (1). This tissue destruction is initiated by an excessive inflammatory host response to periodontal pathogenic bacteria. The production of the key pro-inflammatory cytokine, interleukin (IL)-1β, by various host-immune cells such as macrophages, oral fibroblasts, oral epithelial cells and osteoblasts induces the production of inflammatory mediators, matrix metalloproteinases and osteoclasts in periodontal tissues, leading to the destruction of connective tissue and alveolar bone (2).
The role of IL-1, particularly IL-1β, in the pathogenesis of periodontal diseases was initially documented by Jandinski et al (3) in 1991. They demonstrated that the number of cells stained with anti-human IL-1β antibody was almost 3-fold higher in periodontally diseased tissue compared with normal tissue, suggesting that IL-1β produced by cells in periodontal tissues was related to the pathological processes associated with periodontal disease. Kinane et al (4) measured the amount of IL-1 in gingival crevicular fluid during human experimental gingivitis; an increase in IL-1 levels was observed with subsequent gingival inflammation, suggestive of IL-1, an early biomarker of gingival inflammation. In addition, it has been well-established that an increased IL-1 production is a strong indicator of the susceptibility to severe periodontitis and peri-implantitis (5). This IL-1β remains in an inactive form as pro-IL-1β. A series of processes results in the formation of a multiprotein complex termed the ‘inflammasome’ in the cytosol, which is responsible for converting pro-IL-1β into its activated form, IL-1β (6). The inhibition of inflammasome activation by periodontal pathogens may allow other bacteria in the subgingival ecosystem to survive for longer period of time, thus contributing to persistent chronic inflammation and further periodontal damage (7). It has also been reported that titanium ions also stimulate inflammasome activation, which may be responsible for inflammation around implants in peri-implantitis (8). Current evolving evidence suggests that the inflammasome plays an essential role in the pathogenesis and progression of periodontal and peri-implant disease. Thus, the main objective of the present review was to provide a short overview of the inflammasome and to discuss various strategies that may target inflammasomes that may be used as novel approaches for periodontal treatment.
2. Background, role and function of inflammasomes
Inflammation is a double-edged sword. Inadequate inflammation leads to persistent pathogenic infection whereas excessive inflammation leads to the development of chronic inflammatory diseases. Therefore, it is essential for the host to balance the extent of inflammation by proper inflammasome activation.
As regards the characterization of the inflammasome, according to Martinon et al (9), the inflammasome is defined as ‘the set of intracellular protein complexes that enable autocatalytic activation of inflammatory caspases, which drives host immune responses by releasing mature cytokines and alarmins into circulation and by inducing pyroptosis’. The role of the inflammasome in the innate immunity was first documented by Martinon et al (9) in 2002, followed by Bostanci et al (10) in 2009, who reported its involvement in the pathogenesis of periodontal disease. Since then, studies focusing on the inflammasome in periodontal diseases have been published (7,10-15).
The cytosolic multimeric protein structures of inflammasomes mainly comprise of three components: i) a sensor molecule; ii) an adaptor, and iii) a zymogen procaspase-1. The sensor molecule has a ‘nucleotide-binding domain leucine-rich repeat-containing receptor (NLR)’ and the adaptor has ‘apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC)’ (16).
To date, >10 different inflammasomes have been reported in the literature, which include NLR family pyrin domain (PYD) containing protein (NLRP)1, NLRP3, NLRP6, NLRP12, pyrin, NLR family apoptosis inhibitory protein (NAIP)/NLR family caspase activation and recruitment domain (CARD) domain containing (NLRC)4, retinoic acid inducible gene 1 (RIG-1), absent in melanoma-2 (AIM2), interferon (IFN), gamma-inducible protein 16 (IFI16), NLRC3 and NLP6 (15,17,18). NLRP1 is recognized as the first protein forming an inflammasome along with its potential role in the host innate immunity and inflammatory diseases (7). A well-documented inflammasome is NLRP3. NLRP3 also known as cryopyrin, is predominantly expressed in macrophages, and is encoded by the NLRP3 gene on human chromosome 1(19).
Inflammasome triggering is associated with high levels of inflammation; thus, it should be strictly regulated to prevent its aberrant activation. Under normal healthy conditions, the host balances the extent of inflammation by the proper activation of inflammasomes. Inflammasomes function as intracellular sensors for various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns/danger-associated molecular patterns (DAMPs). Although myeloid cells (dendritic cells, macrophages and monocytes) and neutrophils are usually responsible for inflammasome activation, inflammasomes can also be triggered in gingival fibroblasts, keratinocytes and mucosal epithelial cells (20). The main function of this inflammasome is to convert pro-active forms of inflammation-associated cytokines into their biologically active forms. Inflammasome activation leads to the maturation of the pro-inflammatory cytokines, IL-1β and IL-18 through their cleavage by an enzyme known as caspase-1(7). Various pleiotropic actions, such as the activation of B cells, the recruitment of neutrophils and other innate immune cells, antibody production and the differentiation of T-cells occurs once mature IL-1β and IL-18 are recognized by their receptors. Moreover, inflammasome activation induces pyroptosis in host immune cells. Pyroptosis occurs through the cleavage of caspase-1, leading to the oligomerization of the N-terminal gasdermin D fragment of gasdermin D protein (cytoplasmic protein) and insertion into the plasma cell membrane, resulting in pore formation. This pore formation by gasdermin D induces cell lysis and the release of intracellular components (DAMPS) into the extracellular environment, accentuating inflammation.
3. Inflammasome activation
Inflammasome activation involves a two-step signaling process, i.e., cell priming along with cell recognition and formation. Cell priming process serves two purposes: i) The transcriptional and translational upregulation of inflammasome components (comprising of the sensor molecule, caspase-1 and pro-IL-1β); and ii) the post-translational alteration of the sensor molecule and adaptor molecule (ASC).
The transcriptional upregulation of inflammasome components occurs through the activation of the transcription factor, NF-κB, caused by the recognition of various PAMPs or DAMPs that engage multiple Toll-like receptors or by the cytokines, TNF and IL-1β. These host cell receptors and molecules are recognized by bacterial components, which initiate different signaling pathways. The second step is recognizing a PAMP or DAMP specific to each inflammasome, which then induces inflammasome formation and activation (7). Once PAMPs or DAMPs specific for certain sensor molecules, such as pattern recognition receptors (PRRs) are recognized, the oligomerization of the PRR is induced.
4. Inflammasomes in periodontal disease
Various in vitro, animal and clinical studies have demonstrated the role of inflammasomes (mainly NLRP3) in periodontal inflammation (10,11,21,22). Higher levels of inflammasome expression have been detected in the gingival and salivary samples of patients with periodontal disease (13,23). Mature cytokines processed by caspase-1, mainly IL-1β, control osteoclastic differentiation and activity by exerting direct effects on osteoclasts and indirectly by modulating the expression of receptor activator of nuclear factor-κB ligand (RANKL) in other immune cells. It has been demonstrated that osteoblasts express inflammasome components and NALP3, which is a core protein of NLRP3. The interaction of osteoblastic cells with Aggregatibacter actinomycetemcomitans induces the production of IL-1β, IL-18 and pyroptosis mediated by NLRP3 activation. The activation of caspase-1 also induces cell death by the pyroptosis of osteoprogenitor cells (7). These events may be responsible for bone turnover and inflammatory bone resorption in vivo. Higuchi et al (24) recently reported that conventional periodontal treatment modifies the inflammasome priming status of peripheral blood mononuclear cells in patients with chronic periodontitis. Yamaguchi et al (25) further suggested that the NLRP3 inflammasome plays a critical role in periodontal disease and atherosclerosis induced by Porphyromonas gingivalis through sustained inflammation. Isaza-Guzmán et al (23) also revealed that salivary levels of NLRP3, ASC and IL-1β function as strong and independent indicators of the extent of periodontal breakdown (potential biomarkers) in both chronic periodontitis and aggressive periodontitis; thus they may have potential for use for the prevention and treatment of periodontitis (23).
5. Therapeutics targeting the inflammasome
Host modulation in the treatment of periodontal disease constitutes an important approach. Sudden and abrupt inflammasome activation leads to the continuous production of pro-inflammatory cytokines, which results in periodontitis. Therefore, regulators or inhibitors/antagonists aiming at inflammasome components and their improper activation can be considered as an effective approach for the early intervention and management of periodontal disease. The release of various PAMPs and DAMPs accelerates the priming events by binding to their associated receptors; the overexpression of these proteins (priming) in diseased tissues can be used as an alert signal for therapeutics (18,26). Drugs such as lipoxins and resolvins, targeting TNF and IL-1, are already used as periodontal therapeutic agents, which affect inflammasomes at their priming stage (27). Recently, three approaches have been advocated for the therapeutic inhibition/downregulation of inflammasomes. These include the following (7): i) the inhibition of the upstreaming intracellular signaling mechanism; ii) the blocking of inflammasome components; and iii) the inhibition of inflammasome-mediated cytokines (IL-1β and IL-18).
Inhibition of the upstreaming intracellular signaling mechanism
Periodontitis is associated with decreased salivary and gingival crevicular fluid (GCF) antioxidants, and increased oxidative stress. Trigger factors that lead to NLRP3 inflammasome activation are the upregulation of ion flux (K+, Ca2+ and Cl-), mitochondrial damage, lysosomal disruption and the release of reactive oxygen species. Reactive oxygen species produced by mitochondria, directly or indirectly lead to inflammasome activation. As the role of oxidative parameters in the pathogenesis of periodontal disease is well documented (28,29), drugs that effectively decrease reactive oxygen species production, further leading to decreased inflammasome activation maybe considered as novel approaches. These drugs include (Fig. 1): Allopurinol (Zyloprim/Aloprim®), cardiolipin SS-31 (Elamipretide, Bendavia, MTP-131) and nicotinamide riboside (NR).
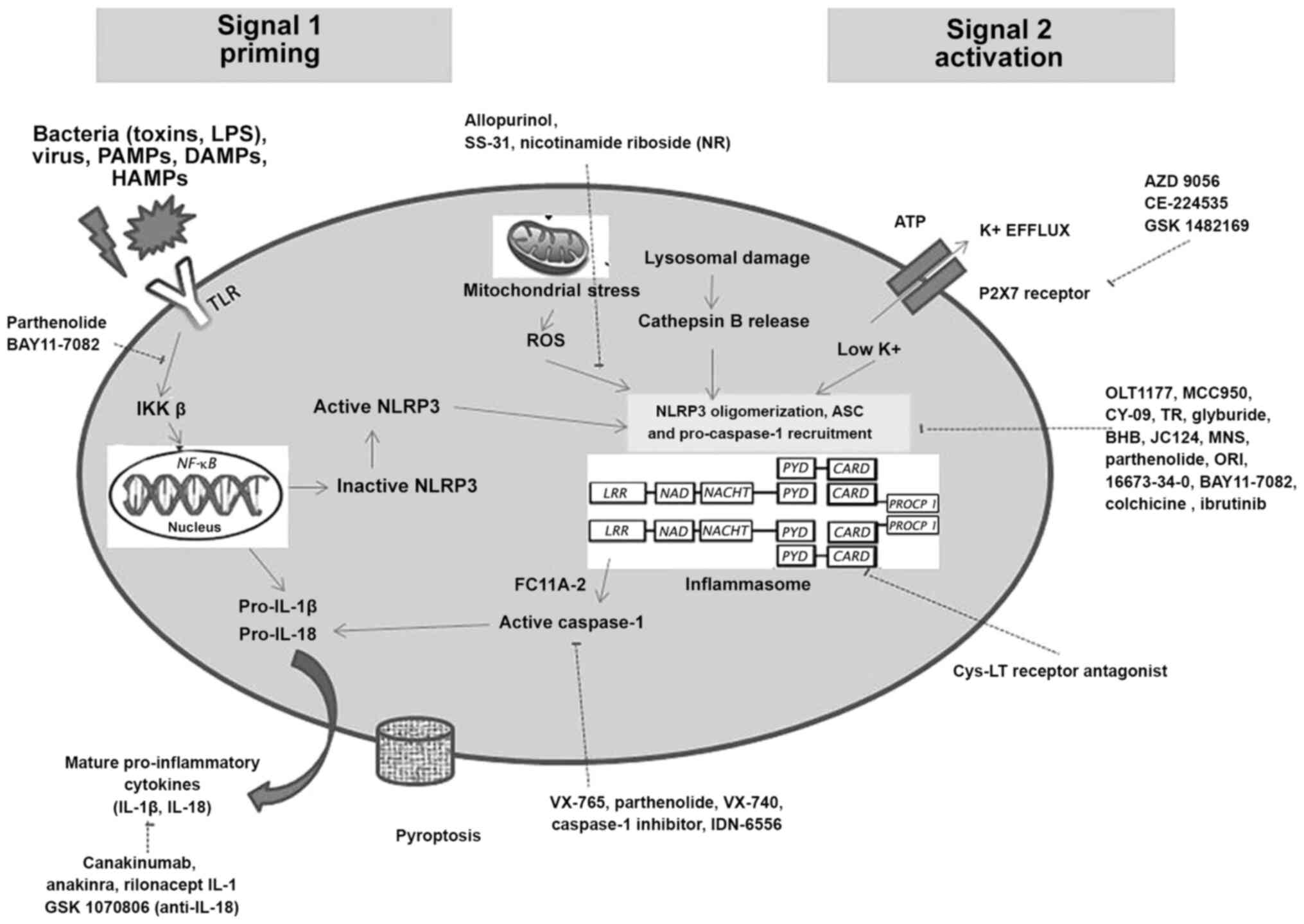 |
Figure 1
Schematic illustration of the inflammasome pathway and potentially novel therapeutic approaches targeting inflammasomes at different blockade sites during steps of its priming and activation.
|
Allopurinol is a prototypical xanthine oxidase inhibitor that is already being used for the treatment of diseases, such as gout (a form of arthritis which is NLRP3 inflammasome-dependent) (30), kidney stones (31) and type 2 diabetic neuropathy (32). Another antioxidant drug being considered to play a potential role in stabilizing a cardiolipin, a phospholipid solely expressed in the inner membrane of mitochondria is Szeto-Schiller peptide (SS-31). This therapeutic drug (SS-31) selectively targets and stabilizes cardiolipin. This drug prevents the excessive production of reactive oxygen species and conserves the electron carrying action of cytochrome c (33). The local application of these drugs can decrease reactive oxygen species produced during periodontitis, thus preventing periodontal tissue and bone damage. SS-31 has already been approved for the treatment of gout and kidney stones and is under research for its application in heart failure (NCT01572909) and primary mitochondrial disease (34). NR is currently available as a dietary supplement. Its therapeutic usage has been clinically tested for various diseases (35), such as atherosclerosis, diabetes and coronary artery disease (36). However, to the best of our knowledge, no significant studies related to these drugs in the periodontal field have been reported to date. As periodontal disease has a two-way association with various systemic diseases, it can be hypothesized that these drugs may aid in the treatment of periodontal diseases associated with these conditions. Therefore, further research on these drugs is encouraged.
Blocking of inflammasome components
The activation of caspase-1 leads to the conversion of the inactive form/precursor form of pro-inflammatory cytokines (pro-IL-1β and pro-IL-18) into IL-1β and IL-18. This activation of caspase-1 can be regulated directly through the PYD and CARD interactions. Thus, targeting PYD only proteins (POPs) and CARD only proteins (COPs) as modifiers of inflammasomes may gain importance. POPs and COPs are key components due to their ability to regulate NF-κB (involved in the priming stage), as well as inflammasome activation. Recent studies have demonstrated that the downregulation of various COPs and POPs in periodontal disease may stimulate the abrupt activation of the inflammasome followed by the activation of IL-1β expression, leading to periodontal tissue breakdown (12,14). Two POPs, which are well known are POP1 (PYDC1) and POP2 (PYDC2). POP1 exhibits ~88% similarity to the PYD domain of ASC and also targets NF-κB activation by inhibiting IkappaB kinase (IKK), thus indirectly inhibiting NLRP3 activation, whereas POP2 directly binds to NLRs and blocks TNF-α-mediated NF-κB activation at the level of p65, thus preventing inflammasome activation. Various drugs included under this approach are the following: Caspase-1 inhibitors [Emricasan (IDN-6556), VX-740 (Pralnacasan) and VX-765], Bruton's tyrosine kinase (BTK) inhibitors [ibrutinib (PCI-32765, Imbruvica)], Acalabrutinib (ACP-196) and NLRP3 tubulin assembly blockers [colchicine (Colcrys) and OLT-1177 (Dapansutrile)].
Drugs targeting caspase-1 are VX-765, VX-740 (Pralnacasan) and Emricasan (IDN-6556), which have been successfully tested in humans (26,37-39). A commonly used caspase-1 inhibitor is VX-765, a reversible inhibitor that becomes active when metabolized by plasma esterase enzyme (40). VX-765 is already undergoing clinical trials (NCT01501383) for the treatment of epilepsy and psoriasis (41). IDN-6556 is already tested for its potential use in diabetes and nonalcoholic steatohepatitis cirrhosis. Marchesan et al documented that the use of VX-765 in male murine models of experimental periodontitis, led to the inhibition of alveolar bone destruction (7). The results of micro-computed tomography revealed that the inhibition of caspase-1 via VX-765 significantly prevented ~50% of alveolar bone loss (7).
BTK inhibitors may also represent novel therapeutic agents for the management of periodontal disease. The interaction of BTK with adaptor proteins is essential for ASC oligomerization during the activation of the NLRP3 inflammasome (42). The inhibition of BTK by pharmacological means, impairs NLRP3 activation, which is responsible for periodontal inflammation. Recently used BTK inhibitors are ibrutinib (PCI-32765, Imbruvica) and Acalabrutinib (ACP-196) (42,43). Ibrutinib is an FDA-approved drug for the treatment of various carcinogenic tumors and chronic graft vs. host disease. BTK inhibitors has also been shown to inhibit alveolar bone destruction in experimental periodontitis models by inhibiting ASC phosphorylation (43). Another drug, ACP-196, has also exhibited similar effects on alveolar bone (43). Pokhrel et al (43) suggested that ACP-196 may be used as a potential therapeutic agent, as it suppresses the differentiation of osteoclastic cells and Porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption by ~50%. Their study suggested that that BTK inhibitors maybe an acceptable treatment for periodontitis (43).
During activation, the spatial arrangement of NLRP3 components is governed by microtubulin dynamics (44). NLRP3 components are one of the main targets currently being evaluated for the treatment of osteoarthritis, cardiac diseases, psoriasis, leukemia, diabetes and non-alcoholic steatohepatitis (26). Colchicine (Colcrys) and OLT-1177 are FDA-approved therapeutic agents used in the treatment of these diseases. Colchicine has been advocated for its antioxidant, anti-mitotic, anti-inflammatory and bone-protective effects. The principal mechanism of colchicine is tubulin disruption by blocking microtubule assembly, thus influencing the innate immune pathway and the activation of the NLRP3 inflammasome (44,45). Aral et al (45) assessed colchicine as a therapeutic measure in experimental periodontitis and concluded that colchicine treatment significantly reduced IL-1β, IL-8, RANKL, RANKL/osteoprotegerin (OPG), total oxidative stress and bone volume ratio levels, and increased total antioxidant status levels in periodontitis cases compared with the controls. Gingival IL-1β levels were also shown to decrease significantly by half during colchicine treatment (45). These results suggested that the administration of colchicine reduced NLRP3-induced periodontal inflammation and destruction.
Inhibition of inflammasome-mediated cytokines
This approach targets IL-1β, IL-18 and type-1 IFN. Drugs targeting inflammasome mediated cytokines include: IL-1β [canakinumab, ACZ885 (Ilaris), anakinra (Kineret) and rilonacept (Arcalyst)], IL-18 (GSK1070806) and type-1 IFN.
IL-1β is the most potent pro-inflammatory cytokine released by inflammasome activation and is involved as an effector molecule in a number of inflammasome-driven diseases. Globally used inhibitors of IL-1β are canakinumab, anakinra and rilonacept (46). Canakinumab is a monoclonal antibody targeting IL-1β, anakinra is a modified IL-1β receptor antagonist and rilonacept is a soluble decoy receptor. Promising results of these approved drugs have been demonstrated in rheumatoid arthritis, atherosclerotic disease and diabetes. Oates et al (2), in an experimental model of periodontitis, concluded that IL-1β antagonist significantly reduced radiographic bone loss by 50% compared with the control sites. Another study by Yoshinari et al (47) demonstrated that at 1 month following conventional non-surgical therapy, IL-1β levels in GCF were still elevated in patients with periodontitis (47). Thus, targeting inflammasome components prior to IL-1β secretion may provide an effective alternate measure for modulating the periodontal host response.
As only a limited number of studies on the role of IL-18 in the pathogenesis of periodontal disease have been documented, fewer clinical trials considering IL-18 antagonist as a newer therapy have been reported. GSK1070806 (an anti-IL-18 monoclonal antibody) may be used as one of the therapeutic agents in the future, although its exact mechanisms of action require further investigation (48). Type 1 IFNs stimulate anti-inflammatory cytokines (such as IL-10) that further utilize STAT3 for inhibiting pro-IL-1β and pro-IL-1α, thus regulating inflammasome activation (49). Type 1 IFNs also use transcription factor (STAT1) to decrease caspase-1 expression, although the exact underlying mechanisms require further validation (49). Notably, to date, to the best of our knowledge, there are no studies available evaluating type-1 IFN and IFN-γ as inflammasome regulators in periodontal disease, which may provide direction for future research.
Other inflammasome modulators include CD40L, Bcl-2 and Bcl-xL (14). Additional inflammasome inhibitors targeting NLRP3, reactive oxygen species and caspase-1 for the treatment of various autoimmune and systemic inflammatory diseases investigated to date are parthenolide, FC11A-2, glyburide, AZD 9056, CE-224535, GSK 1482169, JC124, Bay 11-7082, tranilast (TR), β-hydroxybutyrate (BHB), 16673-34-0, MCC950, 3,4-methylenedioxy-β-nitrostyrene (MNS), oridonin (ORI) and CY-09(16) (Fig. 1). Another approach is TRIM (tripartite motif) proteins which are associated with several physiological processes, including cell proliferation, signal transduction, transcription, DNA repair and pluripotency. TRIM16, which is generally expressed in keratinocytes, is a unique pro-IL-1β binding protein. It enhances IL-1β production by interacting with pro-caspase-1 to enhance innate immunity, whereas its knockdown results in the reduction of IL-1β secretion (50). Another protein TRIM30 which is a negative regulator of TLR signaling, also suppresses NLRP3 activation by hindering reactive oxygen species production in macrophages (51). Although limited data are available, these modulatory mechanisms of inflammasomes may play a potential role in periodontal disease and its treatment.
6. Conclusion and future perspectives
Inflammasomes are intracellular sensors that play an essential role in maintaining host homeostasis by monitoring the inflammation produced. The inappropriate activation of inflammasomes leads to the development of inflammatory diseases, such as periodontitis and peri-implantitis. Various clinical regenerative treatment options are available once the disease has occurred and progressed, causing inflammatory damage to the periodontium; however, to date, there is no treatment available to prevent inflammation at its initial/early stage, at least to the best of our knowledge. Therefore, targeting the disease at the molecular and cellular level may diminish cytokine production and may prevent disease progression to a stage where inflammatory damage has occurred. Targeting inflammasomes directly may provide a novel potential therapeutic approach for the treatment of inflammasome-related periodontal and peri-implant disease. This may modulate the excessive inflammatory response observed in periodontal disease by the use of inflammasome regulators that target abrupt intracellular signaling pathways, inflammasomes components, cytokines and various others proteins (COPs, POPs and TRIM family proteins) at various levels during inflammasome activation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors (AR, AK, FF and ST) contributed in the conception and design of the study. AR and AK confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, Flemmig TF, Garcia R, Giannobile WV, Graziani F, et al: Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. 89 (Suppl 1):S173–S182. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Oates TW, Graves DT and Cochran DL: Clinical, radiographic and biochemical assessment of IL-1/TNF-alpha antagonist inhibition of bone loss in experimental periodontitis. J Clin Periodontol. 29:137–143. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jandinski JJ, Stashenko P, Feder LS, Leung CC, Peros WJ, Rynar JE and Deasy MJ: Localization of interleukin-1 beta in human periodontal tissue. J Periodontol. 62:36–43. 1991.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kinane DF, Winstanley FP, Adonogianaki E and Moughal NA: Bioassay of interleukin 1 (IL-1) in human gingival crevicular fluid during experimental gingivitis. Arch Oral Biol. 37:153–156. 1992.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kornman KS, Crane A, Wang HY, di Giovine FS, Newman MG, Pirk FW, Wilson TG Jr, Higginbottom FL and Duff GW: The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol. 24:72–77. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lopez-Castejon G and Brough D: Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 22:189–195. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Marchesan JT, Girnary MS, Moss K, Monaghan ET, Egnatz GJ, Jiao Y, Zhang S, Beck J and Swanson KV: Role of inflammasomes in the pathogenesis of periodontal disease and therapeutics. Periodontol 2000. 82:93–114. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pettersson M, Kelk P, Belibasakis GN, Bylund D, Molin Thorén M and Johansson A: Titanium ions form particles that activate and execute interleukin-1β release from lipopolysaccharide-primed macrophages. J Periodontal Res. 52:21–32. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Martinon F, Burns K and Tschopp J: The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 10:417–426. 2002.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bostanci N, Emingil G, Saygan B, Turkoglu O, Atilla G, Curtis MA and Belibasakis GN: Expression and regulation of the NALP3 inflammasome complex in periodontal diseases. Clin Exp Immunol. 157:415–422. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Park E, Na HS, Song YR, Shin SY, Kim YM and Chung J: Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect Immun. 82:112–123. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Aral K, Berdeli E, Cooper PR, Milward MR, Kapila Y, Karadede Ünal B, Aral CA and Berdeli A: Differential expression of inflammasome regulatory transcripts in periodontal disease. J Periodontol. 91:606–616. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shibata K: Historical aspects of studies on roles of the inflammasome in the pathogenesis of periodontal diseases. Mol Oral Microbiol. 33:203–211. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aral K, Milward MR, Kapila Y, Berdeli A and Cooper PR: Inflammasomes and their regulation in periodontal disease: A review. J Periodontal Res. 55:473–487. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Strowig T, Henao-Mejia J, Elinav E and Flavell R: Inflammasomes in health and disease. Nature. 481:278–286. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zahid A, Li B, Kombe AJK, Jin T and Tao J: Pharmacological Inhibitors of the NLRP3 Inflammasome. Front Immunol. 10(2538)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rathinam VA and Fitzgerald KA: Inflammasome complexes: Emerging mechanisms and effector functions. Cell. 165:792–800. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo H, Callaway JB and Ting JP: Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 21:677–687. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang Y, Dong Z and Song W: NLRP3 inflammasome as a novel therapeutic target for Alzheimer's disease. Signal Transduct Target Ther. 5(37)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Delaleu N and Bickel M: Interleukin-1 beta and interleukin-18: Regulation and activity in local inflammation. Periodontol 2000. 35:42–52. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kim S, Park MH, Song YR, Na HS and Chung J: Aggregatibacter actinomycetemcomitans-induced AIM2 inflammasome activation is suppressed by xylitol in differentiated THP-1 macrophages. J Periodontol. 87:e116–e126. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T and Ando T: Regulation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated periodontal disease. Inflamm Res. 66:59–65. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Isaza-Guzmán DM, Medina-Piedrahíta VM, Gutiérrez-Henao C and Tobón-Arroyave SI: Salivary Levels of NLRP3 Inflammasome-Related Proteins as Potential Biomarkers of Periodontal Clinical Status. J Periodontol. 88:1329–1338. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Higuchi K, Ziamuddin SM, Yamashita Y, Ozaki Y and Yoshimura A: Initial periodontal treatment affects nucleotide-binding domain leucine-rich repeat-containing protein 3 inflammasome priming in peripheral blood mononuclear cells. Arch Oral Biol. 110(104625)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T and Ando T: Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis. 73(ftv011)2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD and Latz E: Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 17(688)2018.PubMed/NCBI View Article : Google Scholar : Erratum for: Nat Rev Drug Discov 17: 588-606, 2018.
|
|
27
|
Van Dyke TE: Pro-resolving mediators in the regulation of periodontal disease. Mol Aspects Med. 58:21–36. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chapple IL: Reactive oxygen species and antioxidants in inflammatory diseases. J Clin Periodontol. 24:287–296. 1997.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Brock GR, Butterworth CJ, Matthews JB and Chapple IL: Local and systemic total antioxidant capacity in periodontitis and health. J Clin Periodontol. 31:515–521. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
So AK and Martinon F: Inflammation in gout: Mechanisms and therapeutic targets. Nat Rev Rheumatol. 13:639–647. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Favus MJ and Coe FL: The effects of allopurinol treatment on stone formation on hyperuricosuric calcium oxalate stone-formers. Scand J Urol Nephrol Suppl. 53:265–271. 1980.PubMed/NCBI
|
|
32
|
Becker MA, MacDonald PA, Hunt BJ and Jackson RL: Diabetes and gout: Efficacy and safety of febuxostat and allopurinol. Diabetes Obes Metab. 15:1049–1055. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Szeto HH: First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 171:2029–2050. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lynch DR and Farmer G: Mitochondrial and metabolic dysfunction in Friedreich ataxia: Update on pathophysiological relevance and clinical interventions. Neuronal Signal. 5(NS20200093)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mehmel M, Jovanović N and Spitz U: Nicotinamide Riboside-The Current State of Research and Therapeutic Uses. Nutrients. 12(1616)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS, O'Connor C, Goldstein S, Udelson JE and Sabbah HN: Novel mitochondria-targeting peptide in heart failure treatment: A randomized, placebo-controlled trial of elamipretide. Circ Heart Fail. 10(e004389)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cornelis S, Kersse K, Festjens N, Lamkanfi M and Vandenabeele P: Inflammatory caspases: Targets for novel therapies. Curr Pharm Des. 13:367–385. 2007.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, Carreras MC, Poderoso JJ and Gores GJ: The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 35:953–966. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, Decker C, Charifson P, Weber P, Germann UA, et al: (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 321:509–516. 2007.PubMed/NCBI View Article : Google Scholar
|
|
40
|
de Torre-Minguela C, Mesa Del Castillo P and Pelegrín P: The NLRP3 and pyrin inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front Immunol. 8(43)2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
MacKenzie SH, Schipper JL and Clark AC: The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. 13:568–576. 2010.PubMed/NCBI
|
|
42
|
Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A and Morita R: Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 6(7360)2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pokhrel NK, Kim YG, Kim HJ, Kim HJ, Lee JH, Choi SY, Kwon TG, Lee HJ, Kim JY and Lee Y: A novel Bruton's tyrosine kinase inhibitor, acalabrutinib, suppresses osteoclast differentiation and Porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption. J Periodontol. 90:546–554. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T and Akira S: Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 14:454–460. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Aral CA, Aral K, Yay A, Özçoban Ö, Berdeli A and Saraymen R: Effects of colchicine on gingival inflammation, apoptosis, and alveolar bone loss in experimental periodontitis. J Periodontol. 89:577–585. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Cheng R, Wu Z, Li M, Shao M and Hu T: Interleukin-1β is a potential therapeutic target for periodontitis: A narrative review. Int J Oral Sci. 12(2)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yoshinari N, Kawase H, Mitani A, Ito M, Sugiishi S, Matsuoka M, Shirozu N, Ishihara Y, Bito B, Hiraga M, et al: Effects of scaling and root planing on the amounts of interleukin-1 and interleukin-1 receptor antagonist and the mRNA expression of interleukin-1beta in gingival crevicular fluid and gingival tissues. J Periodontal Res. 39:158–167. 2004.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Mistry P, Reid J, Pouliquen I, McHugh S, Abberley L, DeWall S, Taylor A, Tong X, Rocha Del Cura M and McKie E: Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin-18 mAb GSK1070806 in healthy and obese subjects. Int J Clin Pharmacol Ther. 52:867–879. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, Farlik M, Decker T, Du Pasquier RA, Romero P, et al: Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 34:213–223. 2011.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Rathinam VA, Vanaja SK and Fitzgerald KA: Regulation of inflammasome signaling. Nat Immunol. 13:333–342. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hu Y, Mao K, Zeng Y, Chen S, Tao Z, Yang C, Sun S, Wu X, Meng G and Sun B: Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J Immunol. 185:7699–7705. 2010.PubMed/NCBI View Article : Google Scholar
|














