Introduction
Non-alcoholic fatty liver disease (NAFLD) is
characterized by histological changes similar to those observed in
individuals with alcoholic hepatitis, while alcohol intake is
absent or not significant. NAFLD includes conditions ranging from
bland steatosis to nonalcoholic steatohepatitis (NASH), and it has
also been associated with obesity and metabolic syndromes (1). Pathogenesis of NAFLD is believed to be
a result of a series of liver insults, also known as the
‘multi-hit’ hypothesis (2–4). Insulin resistance-induced hepatic
steatosis constitutes the first hit that leads to elevated serum
levels of non-esterified or free fatty acids (FFAs). Subsequently,
the increased transport of FFAs into hepatocytes leads to enhanced
hepatic de novo lipogenesis with exceeded hepatic FFA
β-oxidation and an extremely low-density lipoprotein (VLDL) export,
resulting in hepatic steatosis. A number of previous studies have
suggested hepatic steatosis to occur due to an increase in
lipogenesis and a decrease in lipid export.
NAFLD treatments have mostly targeted the main
components of the metabolic syndrome, including obesity, diabetes,
hypertension and dyslipidemia. Additional mediations have focused
on insulin resistance, oxidative stress, pro-inflammatory
cytokines, apoptosis, bacterial overgrowth and the angiotensin
pathway, the pathways potentially involved in the pathogenesis of
NAFLD. No pharmacological agent has been approved for the treatment
of NASH, or has been determined for routine use in the clinic
(5).
Quercetin is one of the most studied flavonoids. It
is present in several fruits, vegetables, nuts and seeds mainly as
glycoside and, thus, is a main component of the daily human diet
(6). Quercetin is known to protect
cells from oxidative stress and it has also been shown to modify
eicosanoid biosynthesis, prevent platelet aggregation, protect
low-density lipoprotein from oxidation and promote relaxation of
cardiovascular smooth muscle (6).
The anti-oxidant activity of quercetin has often been suggested to
be associated with a pharmacological function in the cardiovascular
system (7). The aim of this study
was to investigate the effect of quercetin on NAFLD. Consequently,
the effects of quercetin on FFA-induced steatosis and
insulin-induced IR in HepG2 cells as well as the underlying
mechanism of action were examined. The characteristics of the cell
model used were consistent with the pathogenesis characteristics of
NAFLD, a fact facilitating the investigation of potential effective
therapies for NAFLD.
Materials and methods
Cell culture and treatment
HepG2 cells were grown as a monolayer culture in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin, 100 g/ml
streptomycin, in monolayer culture, and were incubated at 37°C in a
humidified atmosphere containing 5% CO2 in air. The
experiments were performed when cells reached ∼80% confluence. All
cell culture components were purchased from Gibco-BRL (Carlsbad,
CA, USA). The HepG2 cell model of FFA-induced steatosis and
insulin-induced IR was developed as previously described (8,9). Cells
(1×104/ml) were plated into a 48-well plate containing
100 μl of cell culture medium in triplicate. When ∼80% confluence
was reached and HepG2 cells were cultured in FBS-free medium for 24
h, the cells were treated with 500 μl of FFA solution (1.0 mM) and
insulin (50 nM) to develop a steatosis model with IR for 48 h.
Control cells were treated with FFA-free medium containing 0.1%
dimethyl sulfoxide (DMSO) and 1% bovine serum albumin (BSA).
Cell proliferation assay
Cell proliferation was determined using an MTT assay
as previously described (10).
Briefly, 1×104 cells/ml were plated in a 96-well plate
containing 100 μl of cell culture medium in triplicate and were
treated with various concentrations of quercetin (0.5, 1,5, 10, 50
and 100 μM; National Institute for the Control of Pharmaceutical
and Biological Products, Beijing, China). The effects of quercetin
on HepG2 cell growth were then determined using optical density
absorbance, as previously described (10).
Oil Red O (ORO) cell staining
To estimate the lipid accumulation, the cellular
lipid droplets were stained using ORO, according to a method
described in a previously published study (11). After removing the induction solution
from each well of the 48-well culture plate, the cells were washed
three times with PBS and incubated with formalin (10% formaldehyde,
90% PBS) for 15 min, and then fixed. After fixing, cells were
washed 3–4 times with tap water. ORO solution (500 μl) was then
added to each well and the cells were incubated at room temperature
for 15 min. After removing the ORO solution from each well, the
cells were washed a number of times with ddH2O until the
solution became clear. After being dried and mounted with glycerin,
the cells were examined under a light microscope, and the red oil
droplet in staining in the cells indicated FFA-induced steatosis.
Isopropanol (200 μl) was added to each well. After shaking and
incubating at room temperature for 15 min, the extract was added to
a 48-well culture plate and the absorbance was determined at 500
nm.
Determination of triglyceride in the
HepG2 cell model
Following removal of the induction solution from
each well of the 48-well culture plate, the cells were washed three
times with PBS. RIPA extract was added to each well on ice for
10–20 min. The cells were then collected and lysed using
ultrasonication methods. Supernatant (50 μl) was used for protein
assay by coomassie blue. Double mixture of chloroform and methanol
(2:1) were added to the other supernatant and the solution was then
mixed. After resting for 30 min, samples were centrifuged (4°C;
12,000 × g; 2 min), the upper solution was removed and the lower
solution was dried at 70°C. PBS (20 μl) was added to dissolve
lipids (12). TG was determined
using a kit, according to the manufacturer’s instructions (Bei Hua
KangTai Clinical Reagent Co., Beijing, China).
Western blot analysis
HepG2 cells were lysed in a buffer containing 50 mM
Tris-HCl (pH 8.0), 150 mM NaCl, 0.02% NaN3, 1% SDS, 1 mM
ethylenediaminetetraacetic acid (EDTA), 0.5% sodium deoxycholate,
100 mg/ml phenylmethylsulfonyl fluoride (PMSF), 1 mg/ml leupeptin
and 1% NP-40. Sixty micrograms of protein were analyzed in western
blot analysis experiments with an anti-SREBP-1c antibody (dilution,
1:200; Santa Cruz Biotechnology, Inc., Carlsbad, CA, USA), FAS
(dilution, 1:200; Santa Cruz Biotechnology, Inc.), IRβ, P-IRβ,
IRS1, P-IRS1 (dilution, 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA) or an anti-β-actin antibody (dilution, 1:1,000).
Images were captured and quantified using ChemiDoc™ XRS (Bio-Rad,
Hercules, CA, USA).
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells using TRIzol
reagent. Complementary DNAs (cDNAs) were synthesized using the
High-Capacity cDNA Reverse Transcription kit (Applied Biosystems,
Inc., Foster City, CA, USA). Semi-quantitative detection of
peroxisome proliferator-activated receptor α (PPARα), SREBP-1c,
FAS, fatty-acid-binding proteins (FABP), carnitine
O-palmitoyltransferase-1 (CPT-1), microsomal TG transfer protein
(MTTP) and β-actin was performed. Primers for each gene (Table I) were synthesized by China Takara
Biomedical Technology (Beijing, China). PCR amplification was
performed in a total volume of 20 μl, containing 1 μl cDNA
solution, 10 μl of 2X PCR Master mix, 0.25 μl of each primer at 10
μM and 8.5 μl of nuclease-free water. PCR was conducted for 28–30
cycles. PCR conditions were: denaturation at 94°C for 2 min, 28–30
cycles of 94°C for 30 sec and annealing at 54°C for 2 min,
extension at 72°C for 1 min and a final extension at 72°C for 5
min. The PCR products (10 μl) were run in a 1.5% agarose gel, and
the DNA was visualized by ethidium bromide, using UV
transilluminator and then photographed. The signal intensities were
estimated using the Gel Doc™ software. β-actin was used to
normalize the expression values of the other genes.
 | Table I.Sequences of the primers used in the
PCR measurements. |
Table I.
Sequences of the primers used in the
PCR measurements.
| Gene | Sequence (5′→3′) | GenBank no./ref. |
|---|
| PPARα | Forward:
GCCATCCCAGGCTTCGCAAACT
Reverse: CAAAATCGTGCTGCTCCCCCGT | NM_001001928 |
| SREBP-1c | Forward:
CTTAGAGCGAGCACTGAACTG
Reverse: TGGCCTCATGTAGGAACACC | NM_004176 |
| FAS | Forward:
TTCGTTTGTGAGCCTGACTGC
Reverse: GCTCCCGGATCACCTTCTTG | NM_004104 |
| FABP | Forward:
TCTTCTTCTGCATGCCTGCGCC
Reverse: TAGCCCACGTTGCTGGAGGTGA | NM_002080 |
| CPT-1 | Forward:
TCTACCATGATGGGCGGCTGCT
Reverse: CGTCTGGGCTCGTGCGACATTT | NM_001031847 |
| MTTP | Forward:
TAATCGCAGCCACCCCTGACGA
Reverse: ACCTCTGCCTGTGGACAGCCTT | NM_000253 |
| β-actin | Forward:
CTGGCCGGGACCTGACTGACTA
Reverse: TGCTCGCTCCAACCGACTGC | NM_001101 |
Statistical analysis
Results were shown as the means ± standard deviation
(SD). Differences were evaluated by one-way analysis of variance
(ANOVA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of quercetin on hepatic lipid
accumulation
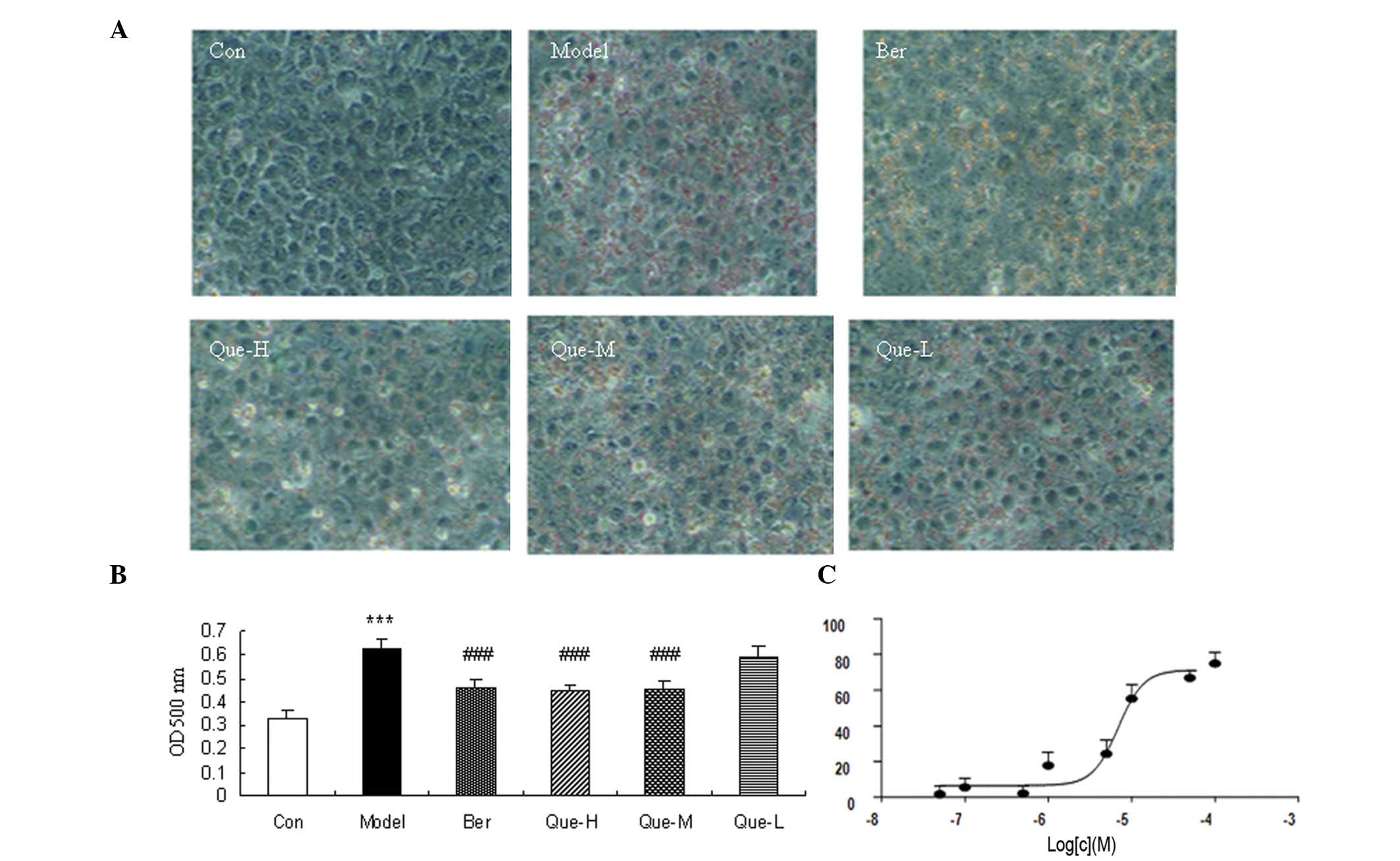
The changes of the cellular lipid accumulation were
detected using ORO staining. The results showed that intracellular
lipids were almost absent in the control group, the FFA- and
insulin-induced intracellular lipid vacuoles were observed under
optical microscopy in model cells. Additionally, intracellular
lipid content was significantly reduced following berberine
treatment (Fig. 1). Quercetin also
strongly affected cellular lipid accumulation in a dose-dependent
manner (Fig. 1).
Effects of quercetin on proliferation and
cell cycle progression of HepG2 cells
We determined whether or not quercetin affected cell
proliferation using MTT assay. Results showed that quercetin
significantly inhibited HepG2 cell proliferation at a concentration
of 100 and 50 μM (P<0.05) (Fig.
2A). No difference was detected in cell proliferation when
additional quercetin concentrations were used.
Effects of quercetin on TG levels in the
HepG2 cell model
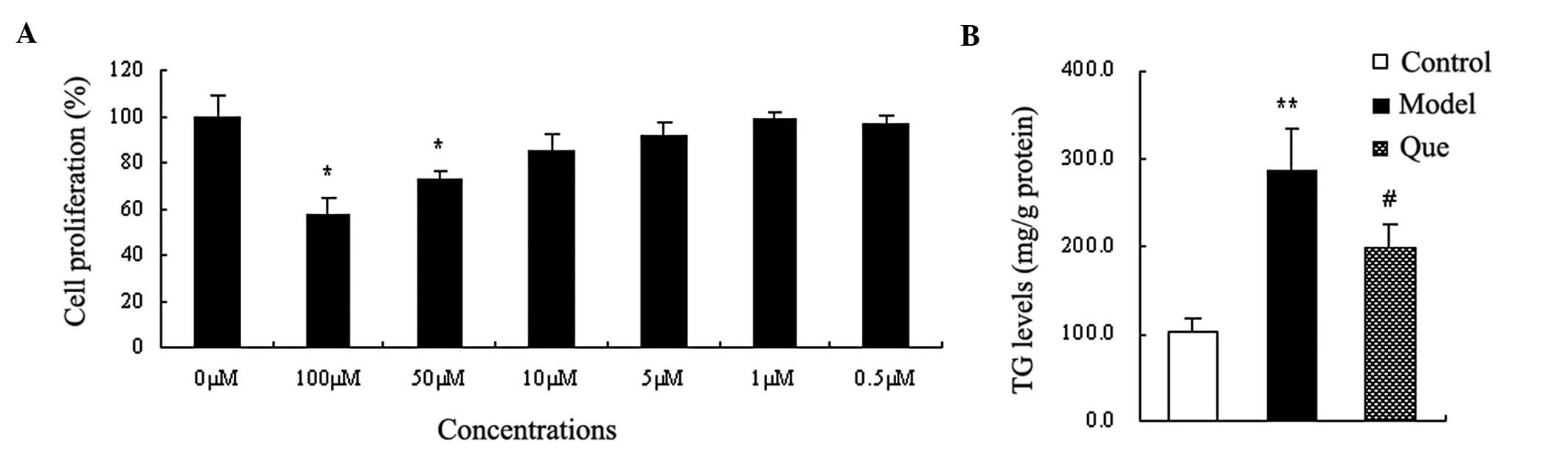
To confirm the effect of quercetin on hepatic fat
accumulation, TG levels were determined. TG levels in the model
group were significantly higher compared to those in the control
group (P<0.01) (Fig. 2B).
Compared to the model group, the levels of TG in the
quercetin-treated groups were decreased (P<0.05) (Fig. 2B).
Quercetin induced the phosphorylation of
IR and IRS1 in the HepG2 cell model
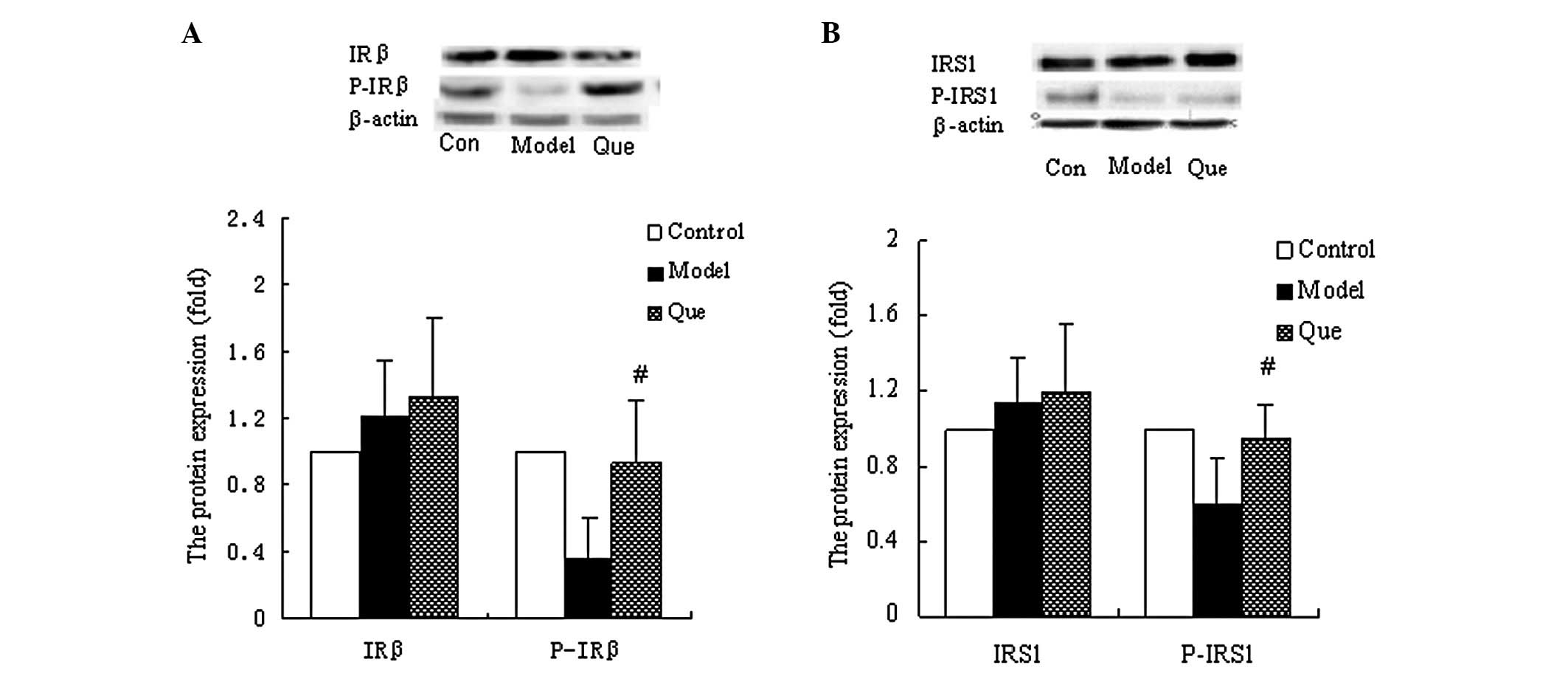
The effects of quercetin on insulin signaling in the
FFA- and insulin-induced cell models were observed using western
blot analysis. There was no difference in the expression of IRβ and
IRS1, in the model and quercetin-treated groups (Fig. 3A), while the phosphorylation of IRβ
and IRS1 was markedly enhanced in the quercetin-treated group
(Fig. 3B).
Changes in levels of PPARα, SREBP-1c,
FABP, CPT-1, MTTP and FAS
In order to investigate the mechanisms of
quercetin-improving hepatic lipid accumulation, we monitored the
mRNA levels of PPARα, SREBP-1c, FABP, CPT-1, MTTP and FAS in
various groups. The results demonstrated that SREBP-1c and FAS
expression was reduced in the quercetin-treated compared to the
model group (P<0.01) (Fig. 4A).
The mRNA expression levels of PPARα, FABP, CPT-1 and MTTP did not
demonstrate a statistically significant change in each group
(Fig. 4A). We confirmed the protein
expression of SREBP-1c and FAS, using western blot analysis. The
results also showed that quercetin downregulated SREBP-1c and FAS
gene expression (Fig. 4B).
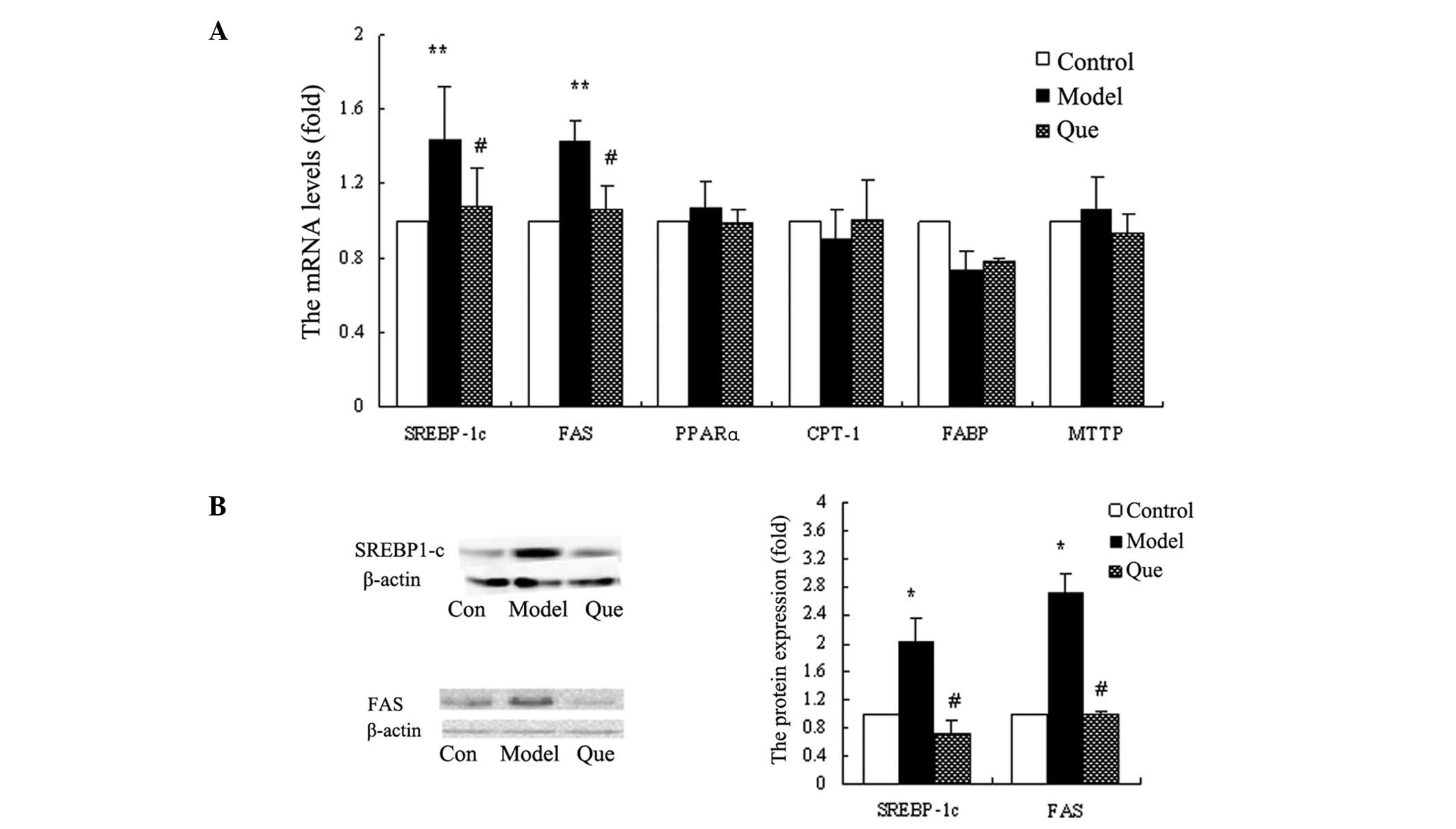 | Figure 4.Effects of quercetin on gene
expression of hepatic genes with regard to fatty acid metabolism in
HepG2 cells. (A) The mRNA levels of SREBP-1c, FAS, PPARα, CPT-1,
FABP and MTTP in each group. Data are the means ± standard
deviation (SD) (n=3). **P<0.01, model vs. control
group; #P<0.01, quercetin-treated vs. model group.
(B) SREBP-1c and FAS protein expression levels in each group.
*P<0.05, model vs. control group;
#P<0.05, quercetin-treated vs. model group. Con,
control group; Model, model group; Que, quercetin-treated
group. |
Discussion
NAFLD includes a spectrum of diseases that have
insulin resistance in common and are associated with metabolic
conditions, such as obesity, type 2 diabetes mellitus and
dyslipidemia. The prevalence of NAFLD has been on the increase in
the last two decades and it affects ∼30% of the adult population in
the USA (13). Currently, no
pharmacological agent has been approved for the treatment of NASH,
or has been included for use in the clinical practice. As a result,
novel drugs are needed for the treatment of NAFLD.
Quercetin, one of the most studied flavonoids, has a
variety of biological functions, including antioxidant,
anti-inflammatory, anti-virus and anticancer effects. In the
present study, we examined the effect of quercetin on insulin
resistance and the hepatic lipid accumulation of NAFLD as well as
the underlying mechanism, in order to identify effective
therapies.
Insulin resistance and steatosis are highly involved
in the development of NAFLD. We used a NAFLD cell model of
FFA-induced steatosis and insulin-induced IR, characteristics that
are consistent with the pathogenesis of NAFLD, in order to
investigate the effect of quercetin on NAFLD. The results of ORO
staining and TG level examination showed markedly accumulated
intracellular lipids and higher TG levels in the HepG2 cell model.
These results might be used for the investigation of disease
mechanisms as well as the development of novel therapy for
NAFLD.
The association between NAFLD and insulin resistance
is known to be almost universal (14). Phosphorylation of IRβ and IRS1 was
found to be improved in the quercetin-treated group, suggesting
that quercetin improved IR by enhancing insulin signal
transduction.
The lipid droplets and TG levels were lower in the
berberine, high- and medium-dose quercetin groups compared to the
model group that used ORO staining and ORO-based colorimetric
quantitative assay. This finding suggested that-quercetin
significantly improved hepatic lipid accumulation. Hepatic lipid
accumulation is involved in various pathways, such as the
synthesis, transport and oxidation of long-chain fatty acids and
TG. When the amount of intake and synthesis of fatty acid surpasses
its outtake and catabolism, the lipid is expected to be deposited
in the liver cells potentially resulting in a fatty liver (15). To further investigate the mechanism
of quercetin on the hepatic lipid accumulation amelioration, key
factors of fatty acid synthesis and metabolism were determined
using semi-quantitative RT-PCR and western blot analysis. A major
cause of steatosis is the increased fatty acid flux to the liver
due to a high availability of plasma FFA with regard to peripheral
oxidative requirements. SREBP-1c and FAS are known to be involved
in fatty acid synthesis (16). The
upregulated SREBP-1c and FAS levels in the model group indicated
that SREBP-1c and FAS are involved in the development and
pathogenesis of NAFLD with steatosis and IR. After treatment with
quercetin, the expression of SREBP-1c and FAS was inhibited,
showing that quercetin improved hepatic lipid accumulation by
affecting fatty acid synthesis mediated by the inhibition of
SREBP-1c and FAS expression.
NAFLD is one of the most common forms of liver
disease worldwide. The exact mechanisms promoting progressive liver
injury are not well-defined, although substrates derived from
adipose tissues, such as FFA, tumor necrosis factor-α (TNF-α),
leptin and adiponectin have been suggested. The NAFLD cell model
induced by FFA and insulin may be used for the investigation of the
mechanism of development, pathogenesis and treatment of diseases.
As a result, quercetin might protect the liver from NAFLD and also
decrease hepatic lipid accumulation by reducing fatty acid
syntheses, as well as improve insulin resistance through the
regulation of the insulin signaling pathway.
Abbreviations:
|
CPT-1
|
carnitine O-palmitoyltransferase-1
|
|
FABP
|
fatty-acid-binding proteins
|
|
FAS
|
fatty acid synthase
|
|
FFA
|
free fatty acid
|
|
MTTP
|
microsomal triglyceride transfer
protein
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
ORO
|
Oil Red O
|
|
PPARα
|
peroxisome proliferator-activated
receptor α
|
|
SREBP-1c
|
sterol regulatory element-binding
protein-1c
|
|
TG
|
triglyceride
|
|
VLDL
|
very low-density lipoprotein
|
Acknowledgements
This study was supported by the
Natural Sciences Fund of the Inner Mongolia Autonomous Region (no.
20080404Zd31).
References
|
1.
|
Ludwig J, Viggiano TR, McGill DB and Oh
BJ: Nonalcoholic steatohepatitis: Mayo Clinic experiences with a
hitherto unnamed disease. Mayo Clin Proc. 55:434–438.
1980.PubMed/NCBI
|
|
2.
|
Malhi H and Gores GJ: Molecular mechanisms
of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver
Dis. 28:360–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Estep JM, Baranova A, Hossain N, et al:
Expression of cytokine signaling genes in morbidly obese patients
with non-alcoholic steatohepatitis and hepatic fibrosis. Obes Surg.
19:617–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Miele L, Grieco A, Armuzzi A, et al:
Hepatic mitochondrial beta-oxidation in patients with nonalcoholic
steatohepatitis assessed by 13C-octanoate breath test. Am J
Gastroenterol. 98:2335–2336. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lam B and Younossi ZM: Treatment options
for nonalcoholic fatty liver disease. Therap Adv Gastroenterol.
3:121–137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Formica JV and Regelson W: Review of the
biology of Quercetin and related bioflavonoids. Food Chem Toxicol.
33:1061–1080. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hayek T, Fuhrman B, Vaya J, et al: Reduced
progression of atherosclerosis in apolipoprotein E-deficient mice
following consumption of red wine, or its polyphenols quercetin or
catechin, is associated with reduced susceptibility of LDL to
oxidation and aggregation. Arterioscler Thromb Vasc Biol.
17:2744–2752. 1997. View Article : Google Scholar
|
|
8.
|
Cui W, Chen SL and Hu KQ: Quantification
and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J
Transl Res. 2:95–104. 2010.PubMed/NCBI
|
|
9.
|
Zhang WY, Lee JJ, Kim Y, Kim IS, Park JS
and Myung CS: Amelioration of insulin resistance by scopoletin in
high-glucose-induced, insulin-resistant HepG2 cells. Horm Metab
Res. 42:930–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Hu KQ, Yu CH, Mineyama Y, McCracken JD,
Hillebrand DJ and Hasan M: Inhibited proliferation of
cyclooxygenase-2 expressing human hepatoma cells by NS-398, a
selective COX-2 inhibitor. Int J Oncol. 22:757–763. 2003.PubMed/NCBI
|
|
11.
|
Hwang JT, Park IJ, Shin JI, et al:
Genistein, EGCG, and capsaicin inhibit adipocyte differentiation
process via activating AMP-activated protein kinase. Biochem
Biophys Res Commun. 338:694–699. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Konno A, Suzuki Y, Ogawa T and Taniuchi T:
UV irradiation promotes the accumulation of triglyceride in
Lipomyces lipofer. Biosci Biotechnol Biochem. 73:2474–2477.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wieckowska A, McCullough AJ and Feldstein
AE: Noninvasive diagnosis and monitoring of nonalcoholic
steatohepatitis: present and future. Hepatology. 46:582–589. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Sanyal AJ, Campbell-Sargent C, Mirshahi F,
et al: Nonalcoholic steatohepatitis: association of insulin
resistance and mitochondrial abnormalities. Gastroenterology.
120:1183–1192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Koteish A and Diehl AM: Animal models of
steatosis. Semin Liver Dis. 21:89–104. 2001. View Article : Google Scholar
|
|
16.
|
Horton JD, Bashmakov Y, Shimomura I and
Shimano H: Regulation of sterol regulatory element binding proteins
in livers of fasted and refed mice. Proc Natl Acad Sci USA.
95:5987–5992. 1998. View Article : Google Scholar : PubMed/NCBI
|