Introduction
Pantothenate kinase-associated neurodegeneration
(PKAN), also termed Hallervorden-Spatz syndrome (HSS), is a rare
autosomal recessive neurodegenerative disorder resulting from
mutations of the pantothenate kinase 2 (PANK2) gene
(1). It is the most common form of
neurodegeneration with brain iron accumulation (NBIA) (2). Clinically, it is characterized by
childhood onset of dystonia, dysarthria, rigidity, and
choreoathetosis, with or without pigmentary retinopathy, optic
atrophy, and acanthocytosis (3).
Approximately one-third of the PKAN patients showed cognitive
decline or dementia (3). In typical
PKAN, symptoms present within the first decade of life and usually
rapidly progress, culminating in early mortality. However, in
atypical PKAN, the onset of extrapyramidal symptoms is later, and
the progression is slower and more variable. The characteristic
imaging pattern of PKAN is eye-of-the-tiger sign, which consists of
bilateral hyper-intensity within surrounding hypo-intensity in the
globus pallidus in T2-weighted images (4).
The prevalence of PKAN is low, particularly in the
Chinese population. To the best of our knowledge, no >30 PKAN
cases of Chinese ancestry have been documented so far (5–10). The
current study describes a PKAN patient of a consanguineous Chinese
pedigree who exhibited the novel homozygous PANK2
mutation.
Materials and methods
Subjects
The current study was approved by the ethics
committee of the Second Affiliated Hospital, Zhejiang University
School of Medicine (Hangzhou, China). A single PKAN patient from a
consanguineous Chinese pedigree was recruited. Written informed
consent was obtained from the participants prior to blood sample
collection. In addition, 200 healthy individuals of Chinese
ancestry were recruited as control subjects. The neurological
examinations (including consciousness, recognition, gait, cranial
nerve, motor, sensory, reflex, pathological signs and coordination)
and clinical evaluations (including age at onset, symptoms and
signs, disease course and treatment) were performed. Brain magnetic
resonance imaging (MRI) was conducted in the PKAN patient. Blood
samples (3 ml) were collected for laboratory inspection and gene
sequencing.
Mutation analysis
After obtaining informed consent, genomic DNA was
extracted from 3 ml peripheral blood using a QIAamp DNA Blood
Minikit (Qiagen GmbH, Hilden, Germany). Mutational investigation of
PANK2 gene was performed by polymerase chain reaction (PCR)
amplification and direct DNA sequencing, according to a previous
study (1). Briefly, forward and
reverse primers were designed to amplify each of the seven exons of
PANK2 and all exons of PANK2 and the respective intron-exon
boundaries were amplified by PCR according to a previous study
(1). The purified PCR products were
sequenced using an ABI 3730 Automated DNA Sequencer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The sequencing results were
aligned to the NCBI human reference DNA sequence of PANK2 (Ensembl
gene ID: ENSG00000125779; http://asia.ensembl.org/index.html). Amino acid and
nucleotide changes were identified and numbered corresponding to
their position within PANK2 mRNA. SIFT (http://sift.jcvi.org), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)
and Mutation Taster (http://www.mutationtaster.org) were used to predict
the pathogenicity of the identified mutation.
Results
Clinical features of the patient
The proband was a 20-year-old female who had an
8-year history of gait difficulties and involuntary movements. Her
delivery and postnatal period was unremarkable. The patient had
normal acquisition of developmental milestones until the age of 12
years when unstable walk and involuntary movements began to
develop. Her upper limbs suffered moderate chorea and ballismus,
which became accentuated by anxiety and diminished when she was
calm. Clonazepam at dose of 0.5 mg was prescribed and the symptoms
were significantly relieved. Two years later, difficulties with
walking and the involuntary movements were aggravated and the
patient began to exhibit dysarthria. The patient experienced did
not completely alleviate the symptoms. Oral administration of
benzhexol (2 mg, three times per day) was effective in the first
two months. When the patient sought medical care in the Department
of Neurology, Second Affiliated Hospital, Zhejiang University
School of Medicine (Hangzhou, China) in December 2015, she
presented dysarthria, involuntary movements and unsteady gait.
On cranial nerve examination, the extra-ocular
movements of the patient were normal without nystagmus and visual
field deficits. Choreiform movement was observed in the face.
Pronunciation was slurred with mild palilalia, although her tongue
and soft palate were at midline. The patient's muscle strength was
normal, although muscle tension was decreased in the extremities
and trunk. Chorea was obvious in the patient's upper limbs. No
sensory abnormalities were observed and tendon reflexes were brisk,
while the Babinski sign was bilaterally negative. Finger-nose and
heel-knee-shin test results were normal. The patient's gait was
unstable although the Romberg's test result was negative. The
patient's recognition was not impaired, with a mini-mental state
examination score of 29/30. Laboratory findings included normal
serum ferritin, ceruloplasmin, albumin and lipoprotein levels. The
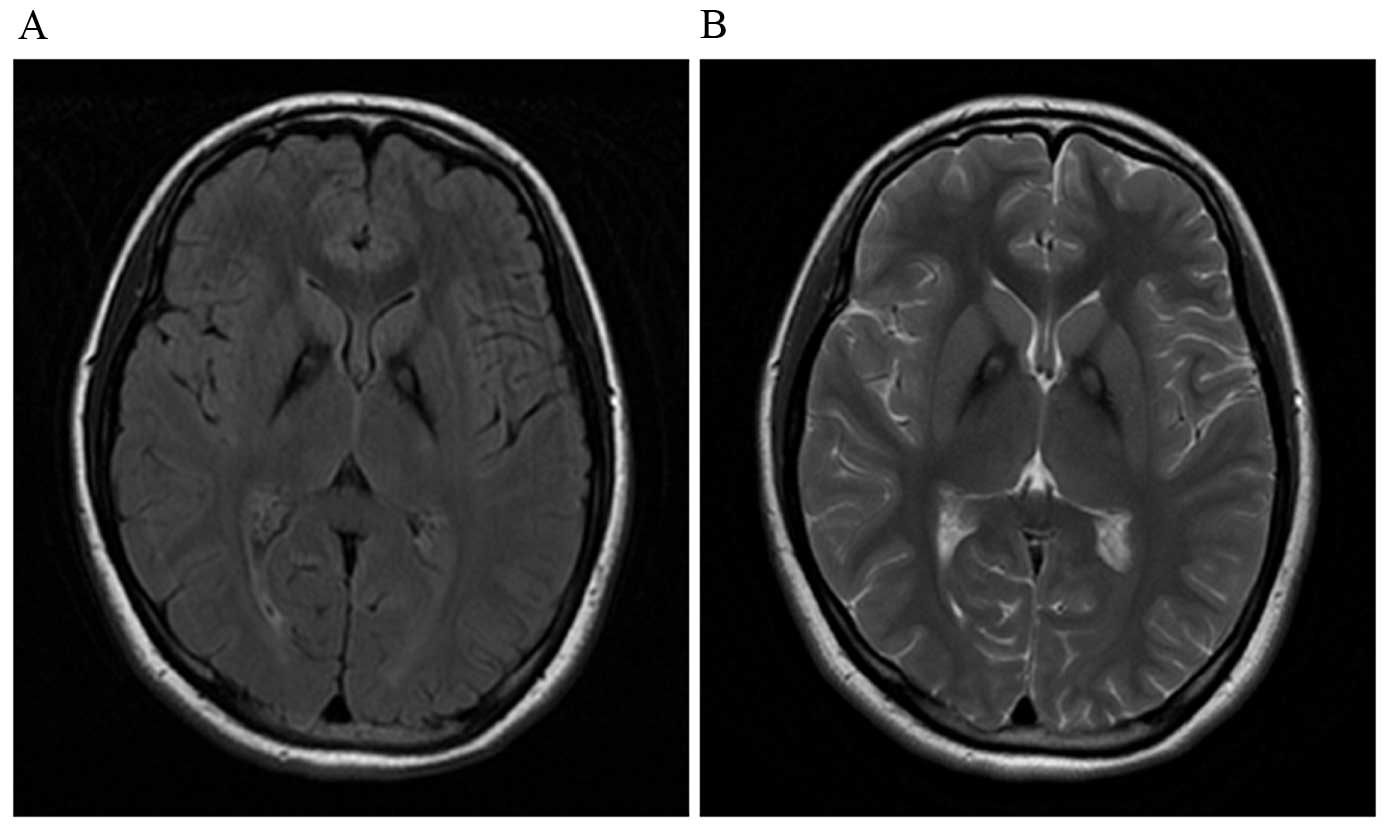
blood smear was negative for acanthocytes. A brain MRI revealed
iron accumulation in the bilateral globus pallidus and putamen
(Fig. 1).
The patient's father and mother were consanguineous,
healthy and did not exhibit gait difficulty or dysarthria.
Identification of the novel PANK2
mutation
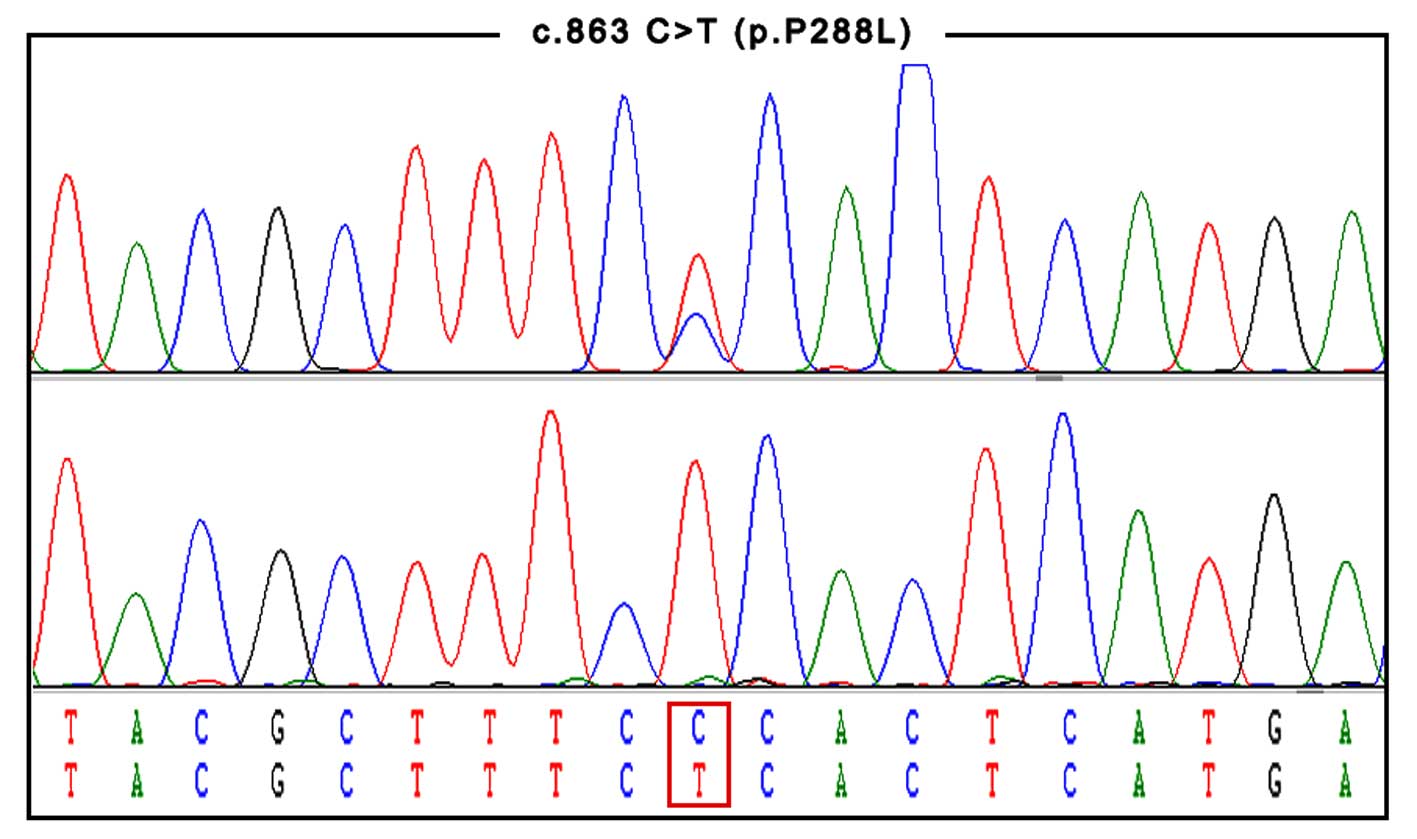
After sequencing of the PANK2 gene, a homozygous
c.863C>T mutation was identified in the proband (Fig. 2). This mutation was located in exon 2
of PANK2, causing a proline to leucine substitution at position 288
(p.P288L) of the enzyme. Her consanguineous parents were
heterozygous carriers of c.863C>T. The homozygous c.863C>T
mutation was absent in the 1000 Genomes database (http://www.1000genomes.org/), The Exome Aggregation
Consortium (ExAc; http://exac.broadinstitute.org/) and all 200 control
individuals. The homozygous c.863C>T mutation of PANK2 was
predicted to be deleterious by SIFT, disease causing by Mutation
Taster and probably damaging by PolyPhen2.
Discussion
PKAN is a neurodegenerative disorder associated with
excessive iron deposition in the globus pallidus (11). It was first described by German
neuropathologists Hallervorden and Spatz in 1922, therefore is also
referred to as HSS (12); however, was
subsequently renamed PKAN (13). In
the present study, a Chinese 20-year-old female with an 8-year
history of unsteady walking and involuntary movements is described.
A brain MRI revealed typical eye-of-the-tiger sign. After
sequencing the PANK2, a homozygous c.863C>T (p.P288L)
mutation was identified in her genetic DNA. Further genetic
investigations revealed a heterozygous c.863C>T mutation in her
consanguineous parents. To the best of our knowledge, this mutation
has not previously been reported; therefore it is considered to be
a novel mutation. The absence of this mutation in the 1000 Genome
database, ExAC, and 200 control individuals demonstrated that this
mutation was potentially pathogenic for PKAN in this particular
family. In addition, it was predicted to be deleterious by SIFT and
Mutation Taster.
The patient described in the current study
experienced early onset and presented with an unsteady walk,
choreic movements, and dysarthria as primary symptoms. The
cognitive impairment was not obvious, although the patient had
experienced the disease for 8 years. These observations were
consistent with two existing reports of Chinese PKAN patients
(5,6).
Notably, cognitive impairment was not prominent in Asian PKAN
patients (2,14,15). By
contrast, the prevalence of pyramidal signs, mental impairment, and
Parkinsonism was higher in Caucasian patients (16). This discrepancy is potentially due to
the ethnic differences.
To date, >100 mutations within PANK2 have been
reported in PKAN cases worldwide (http://www.hgmd.org). The majority of PANK2
mutations are missense mutations, while duplication, deletion,
splice-site mutation and exon deletion have also been reported
(16). The majority of previously
reported Chinese PKAN patients were detected to be carrying
compound heterozygous mutations. In the present study, the patient
carried a homozygous PANK2 mutation. Due to the Chinese
single-child policy that commenced in 1980, this patient did not
have any siblings. Therefore, co-segregation could not be conducted
in this family. However, the early onset of extrapyramidal
symptoms, characteristic eye-of-the-tiger sign in MRI, and
identification of the homozygous PANK2 mutation revealed
that this was a case of PKAN.
The mechanism by which mutations in the PANK2
gene lead to iron accumulation in the brain remains to be
elucidated. PANK2 is an essential regulatory enzyme in
coenzyme A biosynthesis (13). In
addition, the protein encoded by PANK2 is crucial in the
metabolism of pantothenate. A deficiency of PANK2 is
reported to result in the accumulation of cysteine-containing
substrates, which may undergo rapid auto-oxidation in the presence
of iron, leading to free radical generation and cell damage
(1). A limitation of the current study
was that functional experiments were not performed to verify the
pathogenicity of the identified mutation. Thus, further
investigations are required.
Acknowledgements
The authors would like to thank the participants for
their assistance and willingness to take part in the study. Funding
for the present study was granted by the National Natural Science
Foundation of China (grant no. 81500973) and Natural Science
Foundation of Zhejiang Province (grant no. LY16H090006) to Hong-Fu
Li.
References
|
1
|
Zhou B, Westaway SK, Levinson B, Johnson
MA, Gitschier J and Hayflick SJ: A novel pantothenate kinase gene
(PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet.
28:345–349. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hogarth P: Neurodegeneration with brain
iron accumulation: Diagnosis and management. J Mov Disord. 8:1–13.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tonekaboni SH and Mollamohammadi M:
Neurodegeneration with brain iron accumulation: An overview. Iran J
Child Neurol. 8:1–8. 2014.PubMed/NCBI
|
|
4
|
Hayflick SJ, Westaway SK, Levinson B, Zhou
B, Johnson MA, Ching KH and Gitschier J: Genetic, clinical, and
radiographic delineation of Hallervorden-Spatz syndrome. N Engl J
Med. 348:33–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Song XW, Wang YL, Shi YW, Deng WY, Chen
SQ, Lin H, Yi YH and Liao WP: Clinical manifestations and detection
of pantothenate kinase 2 gene mutation in a patient with
Hallervorden-Spatz syndrome. Zhonghua Yi Xue Za Zhi. 89:3320–3323.
2009.(In Chinese). PubMed/NCBI
|
|
6
|
Zhang Y, Tang B, Guo J, Long Z, Xia K, Pan
Q, Hu Z, Wu D, Tang J, Chen T and Yan X: Studies on PANK2 gene
mutations in Chinese patients with Hallervorden-Spatz syndrome.
Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 22:189–191. 2005.(In Chinese).
PubMed/NCBI
|
|
7
|
Fung GP and Chan KY: Cervical myelopathy
in an adolescent with Hallervorden-Spatz disease. Pediatr Neurol.
29:337–340. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu YR, Chen CM, Chao CY, Lyu RK and
Lee-Chen GJ: Pantothenate kinase-associated neurodegeneration in
two Taiwanese siblings: Identification of a novel PANK2 gene
mutation. Mov Disord. 24:940–941. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang YH, Tang BS, Zhao AL, Xia K, Long
ZG, Guo JF, Westaway SK and Hayflick SJ: Novel compound
heterozygous mutations in the PANK2 gene in a Chinese patient with
atypical pantothenate kinase-associated neurodegeneration. Mov
Disord. 20:819–821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma LY, Wang L, Yang YM, Lu Y, Cheng FB and
Wan XH: Novel gene mutations and clinical features in patients with
pantothenate kinase-associated neurodegeneration. Clin Genet.
87:93–95. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thomas M, Hayflick SJ and Jankovic J:
Clinical heterogeneity of neurodegeneration with brain iron
accumulation (Hallervorden-Spatz syndrome) and pantothenate
kinase-associated neurodegeneration. Mov Disord. 19:36–42. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hallervorden J and Spatz H: Eigenartige
Erkrankung im extrapyramidalen System mit besonderer Beteiligung
des Globus pallidus und der Substantia nigra. Z Ges Neurol
Psychiatr. 79:254–302. 1922. View Article : Google Scholar
|
|
13
|
Popławska-Domaszewicz K,
Florczak-Wyspiańska J and Kozubski W: Update on neurodegeneration
with brain iron accumulation. Neurol Neurochir Pol. 48:206–213.
2014.PubMed/NCBI
|
|
14
|
Hartig MB, Hörtnagel K, Garavaglia B,
Zorzi G, Kmiec T, Klopstock T, Rostasy K, Svetel M, Kostic VS,
Schuelke M, et al: Genotypic and phenotypic spectrum of PANK2
mutations in patients with neurodegeneration with brain iron
accumulation. Ann Neurol. 59:248–256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamashita S, Maeda Y, Ohmori H, Uchida Y,
Hirano T, Yonemura K, Uyama E and Uchino M: Pantothenate
kinase-associated neurodegeneration initially presenting as
postural tremor alone in a Japanese family with homozygous N245S
substitutions in the pantothenate kinase gene. J Neurol Sci.
225:129–133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee CH, Lu CS, Chuang WL, Yeh TH, Jung SM,
Huang CL and Lai SC: Phenotypes and genotypes of patients with
pantothenate kinase-associated neurodegeneration in Asian and
Caucasian populations: 2 cases and literature review.
ScientificWorldJournal. 2013:8605392013. View Article : Google Scholar : PubMed/NCBI
|