Introduction
Hemophilia A (HA; OMIM: 306700) is a disorder of the
coagulation system that is experienced worldwide (1). HA, the result of reduced factor VIII
(FVIII) activity (2), is an X-linked
recessive bleeding disorder characterized by defects in the FVIII
gene, which codes for the FVIII protein. Membrane-bound FVIII
performs a critical function in blood coagulation as the
pro-cofactor to the serine-protease, factor IXa (FIXa) in the
FVIIIa-FIXa complex assembled on the activated platelet membrane
(3). HA is rare (4), although it is the most common inherited
bleeding disorder (5), and has a high
treatment cost (6). The incidence of
HA is ~1 in 10,000 live male births (7). Typically, affected patient experience
prolonged bleeding caused by lack or reduced residual activity of
the coagulant FVIII (FVIII:C). The severity of the disease is
defined based on the quantity of the residual FVIII:C level
(8). As recessive X-linked disease,
the HA phenotype is manifested in hemizygous males whereas
heterozygous females (carrier) are usually asymptomatic, showing
normal or intermediate FVIII activity (FVIII:C) levels (9). As a carrier, there is 50% probability of
transmitting the abnormal allele to the child (10). HA is rare in females (11); however, there are various potential
genetic mechanisms leading to HA in certain females, as follows: i)
homozygous mutations as consequence of consanguineous marriage
(12); ii) heterozygous FVIII mutation
combined with skewed inactivation of X chromosome (13); and iii) compound heterozygous mutations
(11). FVIII, a plasma glycoprotein
coded by a 186-kb gene with 26 exons is located at the Xq28
position (chrX: 154,835,795–155,022,753; University of California,
Santa Cruz genome browser, GRCh38/hg38). Since the publication of
sequence FVIII gene by Gitschier et al (14) at Genetech in 1984, numerous mutations
causing HA have been identified. The most common is the inversion
of intron 22, which occurs in 40–50% of patients with severe HA
(15), whereas the inversion of intron
1 is present in just 1–5% of patients (16).
Currently, it is proposed that the diagnosis of HA
should be extended to genetic testing, to establish the causative
mutation (17,18). Determination of the mutation
responsible for HA is important for various reasons, including: i)
Enabling a preliminary assessment of the risk of FVIII inhibitor
development; and ii) identifying a definitive diagnosis of HA
carrier status (17).
Carrier diagnosis is critical for preventing the
birth of children affected by coagulation disorders. There are two
genetic methods for the analysis of genes involved in HA, direct
and indirect. Direct sequence analysis of the FVIII gene is a gold
standard for genetic diagnosis of HA (19). However, in ~2–5% of patients with
severe HA a causative mutation is occasionally not identified in
FVIII gene (20). Direct sequencing of
the FVIII gene for carrier detection in developing countries is
limited by the large size and mutational heterogeneity of the gene.
This limitation is overcome by indirect analysis of polymorphisms
linked to the FVIII gene, rendering carrier detection economically
practicable (19).
Linkage analysis requires other affected family
members with informative polymorphic markers to track the affected
allele (21). Short tandem repeats
(STRs), restriction fragment length polymorphism (RFLP), and
variable number tandem repeats (VNTR) are used as polymorphic
markers. Linkage analysis is a common indirect method for detection
of female carriers in families with HA (22). In the present study, a combination of
intragenic and extragenic STR markers was used in linkage analysis
for carrier diagnosis in a Chinese HA family.
Materials and methods
Subjects
The present study was conducted in two generations
of a HA family; the mother was a carrier, the son was the proband,
the father was healthy and the daughter was the suspected carrier.
Written informed consent was obtained from each member of the
family. Venous blood samples (~10 ml) were drawn from each member
and collected in EDTA vacutainers at Zhongnan Hospital, Wuhan
University (Wuhan, China). Genomic DNA was extracted from white
blood cells according to the conventional phenol-chloroform method
using an ultracentrifuge (Hema Medical Instrument Co., Ltd.,
Guangdong, China).
Polymorphic markers and primers
The STR markers selected to assess their efficacy as
genetic diagnosis markers in the present study were STR22, an
intragenic marker, and six extragenic STR polymorphic markers,
DXS1073, DXS15, DXS8091, DXS1227, DXS991 and DXS993. These markers
were selected based on the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/), Genome Database
(The University of California, Santa Cruz genome browser) and
Ensembl (http://www.ensembl.org/index.html). The DXS15 and
STR22 primers were synthesized and labeled with fluorescent dye,
5′6-FAM (Sangong Company Shanghai, China) and are presented in
Table I. The DXS1073, DXS8091,
DXS1227, DXS991 and DXS993 primers were from an ABI Linkage Mapping
Set v2.5 MD10 (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
 | Table I.Primer sequences of STR22 and DXS15
polymorphic markers with fluorescent dyes. |
Table I.
Primer sequences of STR22 and DXS15
polymorphic markers with fluorescent dyes.
| Primer names | Forward primers | Reverse primers |
|---|
| STR22 |
5′-GTACTGGGAATGCACAGCCTA-3′ | 5′-(FAM)
CCAGACATGTCAAGGTGTCAA-3′ |
| DXS15 | 5′-(5′6-FAM)
AGCACATGGTATAATGAACCTCCACG-3′ |
5′-CAGTGTGAGTAGCATGCTAGCATTTG-3′ |
Polymerase chain reaction (PCR)
amplification and capillary electrophoresis
PCR was conducted in a total volume of 25 µl
containing 1 µl genomic DNA (40–50 ng/µl), 1 µl primer pair mix (10
µM each primer), 0.5 µl dNTPs (10 mmol/l), 2.5 µl MgCl2
(25 mM) and 10X PCR Buffer, (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) 1 µl Taq polymerase (1 U/µl), and 15.5 µl
ddH2O. After 5 min of denaturation at 95°C, 35 cycles of
the following were performed: Denaturation at 94°C for 30 sec;
annealing at 55°C for DXS1073, DXS8091, DXS1227, DXS991, and
DXS993, at 60°C for STR22 or at 58°C for DXS15, for 30 sec;
extension at 72°C for 30 sec; and final extension at 72°C for 5
min. Each of the PCR products was diluted 10-fold using
ddH2O. Subsequently, GeneScan™ 500 LIZ® Size
Standards (Thermo Fisher Scientific, Inc.) and formamide (Thermo
Fisher Scientific, Inc.) were combined at a ratio of 1:3, and 3 µl
diluted PCR products was mixed with 9 µl GeneScan™ 500
LIZ® Size Standards/formamide. Following denaturation at
95°C for 5 min and rapid cooling on ice, the mixture was
electrophoresed and separated by capillary electrophoresis on an
ABI 3130 Genetic Analyzer (Thermo Fisher Scientific, Inc.) and
results were analyzed using GeneMapper ID software v3.2 (Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to a previous
study (23).
Results
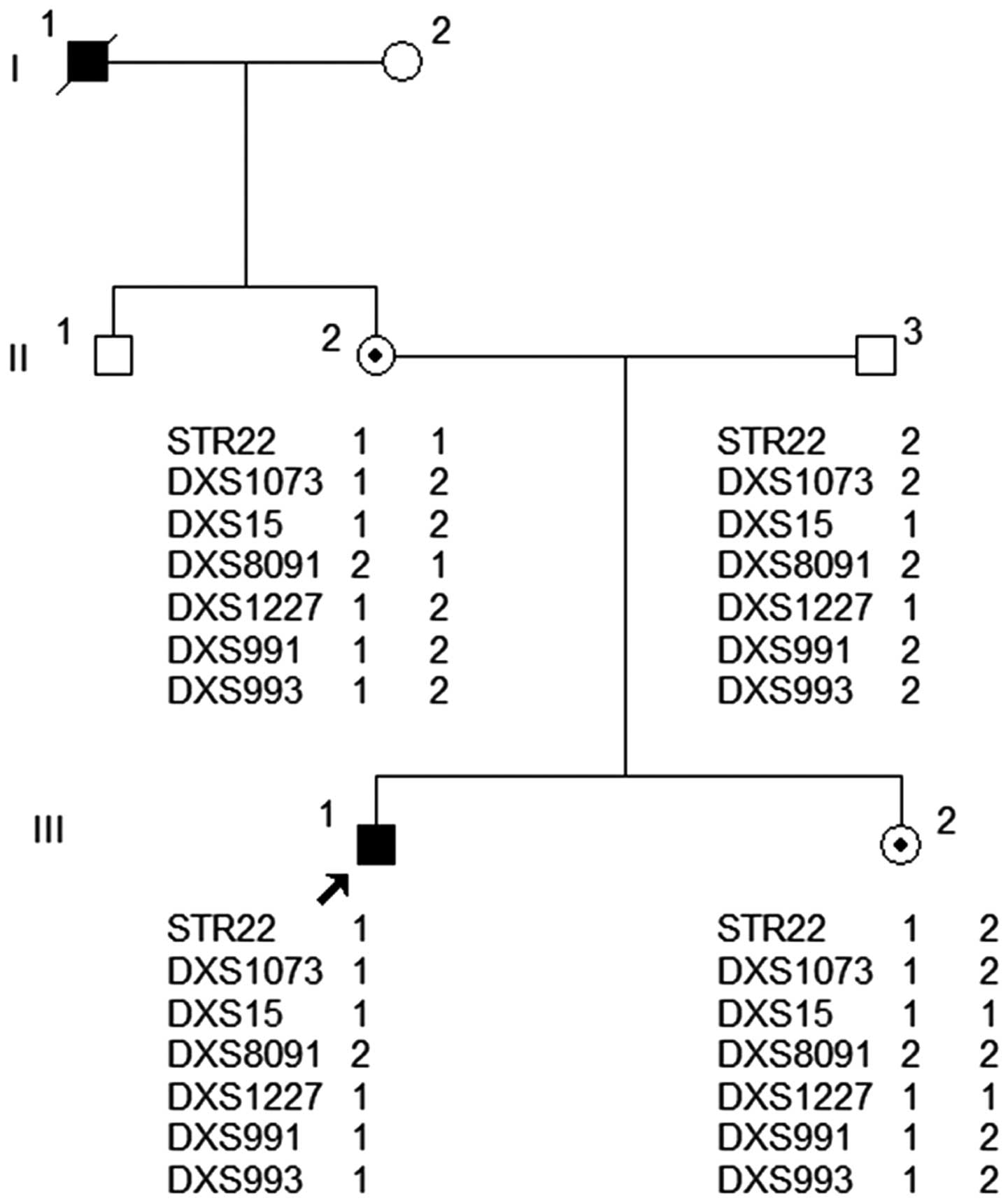
Six extragenic markers and one intragenic marker
were employed for carrier diagnosis of the daughter who was thought
to be an HA carrier. Electrophoretogram results obtained from
capillary electrophoresis are presented in Fig. 1 and demonstrate that all of the STR
markers employed in the present study were informative for linkage
analysis of pathogenic and healthy haplotypes among the HA family
members (Table II). For the seven STR
markers (Fig. 2), the haplotype in the
son (III-1), the proband, and that of the mother (II-2), who was a
carrier, are identical. The sister (III-2) of the proband carried
the same haplotype as the proband, which came from her mother, an
obligate carrier. STR22, an intragenic marker, and DXS15 and
DXS1073 are closer to the FVIII gene with low chances of
recombination, indicating that these three markers possess more
diagnostic characteristic features, which increased the accuracy of
the result. Thus, the use of these markers (STR22, DXS15 and
DXS1073) assisted with identifying that the proband's sister may be
a carrier of HA.
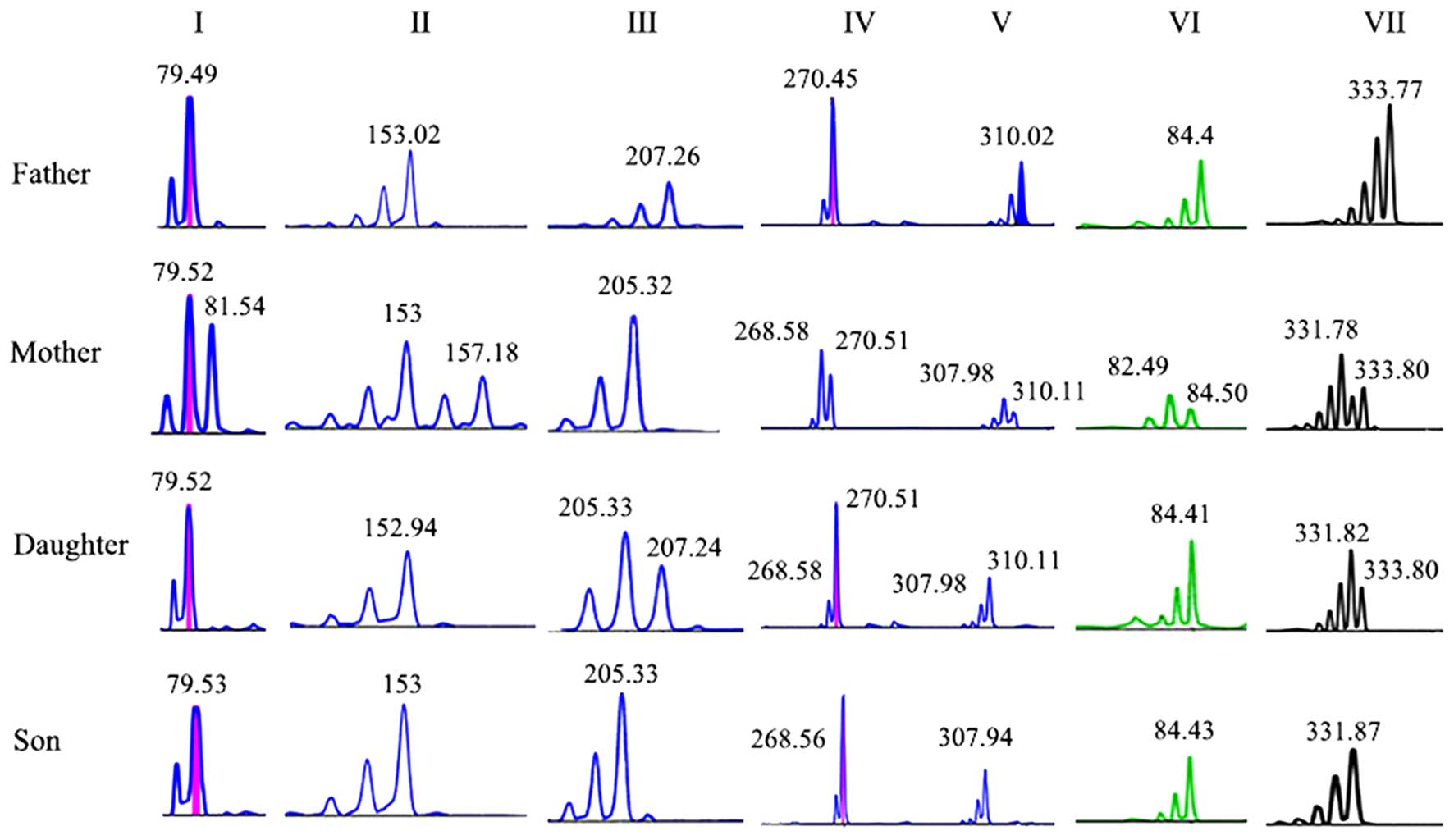 | Figure 1.Electrophoretogram of seven short
tandem repeat markers in the family with hemophilia A. I, DXS1227;
II, DXS15; III, STR22; IV, DXS993; V, DXS1073; VI, DXS8091; VII,
DXS991. Father, healthy; mother, carrier; daughter, suspected
carrier; son, proband. |
| Table II.STR linkage result from the HA
family. |
Table II.
STR linkage result from the HA
family.
| STR marker | Father | Mother | Daughter | Son | Loci |
|---|
| STR22 | 207.26 | 205.32/205.32 | 205.33/207.24 | 205.33 |
|
| DXS1073 | 310.02 | 307.98/310.11 | 307.98/310.11 | 307.94 | X:
154,835,788–155,026,940 |
| DXS15 | 153.02 |
153/157.18 | 152.94/152.94 | 153 | X:
153,366,650–153,366,809 |
| DXS8091 | 84.4 | 84.50/82.49 | 84.41/84.41 |
84.43 | X:
148,521,335–148,521,437 |
| DXS1227 |
79.49 | 79.52/81.54 | 79.52/79.52 |
79.53 | X:
141,714,259–141,714,432 |
| DXS991 | 333.77 | 331.78/333.80 | 331.82/333.80 | 331.87 | X:
55,492,619–55,492,898 |
| DXS993 | 207.45 | 268.58/270.51 | 268.58/270.51 | 268.56 | X:
41,288,430–41,288,735 |
Discussion
HA is the most common bleeding disorder with
X-linked recessive inheritance caused by a wide variety of
mutations in the FVIII gene located in chromosome Xq28. FVIII is
the only gene known to be associated with HA (7). Given the hemizygous nature of X-linked
disorders, males are primarily affected, while females are commomly
heterozygous for the gene mutation and are typically referred as
carriers (24). The female carriers
are usually asymptomatic with bleeding events occurring in only
~10% of cases (25). There is a 50%
chance that a carrier mother will transmit the defective X-linked
gene to the male or female child. All female offspring born to a
hemophilic father are obligate carriers. To identify the females at
risk of being a carrier, it is important to understand the
heriditary of the disorder. Although sporadic cases result from
de novo mutations (20),
carrier detection and prenatal diagnosis is critical for reducing
the number of births of children with hemophilia in developing
countries, where patients with this particular coagulation disorder
rarely survive beyond childhood (25).
Molecular analysis techniques, including the direct
and indirect analysis of the FVIII gene sequence have increased the
detection rate of HA carriers (22).
Genetic counseling, carrier testing, and prenatal diagnosis of
hemophilia have become an integrated aspect of the comprehensive
care for hemophilia during the past three decades.
Direct sequence analysis of the FVIII gene is a gold
standard for genetic diagnosis of HA (19). However, due to heterogenous nature of
mutations, the large size of inversions and the complexity of FVIII
gene, direct recognition of the disease-associated mutation is
complicated. Linkage analysis is an auxillary strategy to direct
mutation analysis for genetic counseling of HA (26). Indirect linkage analysis using highly
informative polymorphic markers is the method of choice for carrier
detection of HA in developing countries, as direct DNA or mRNA
sequence analysis is significantly more costly and difficult when
compared with indirect gene tracking (19). Polymorphic markers are slight DNA
sequence variations usually present in the non-coding regions of a
gene in the population. The sequences are stable and inherited
according to Mendel's laws. However linkage analysis has certain
limitations, for example this technique requires entire families
and cannot be conducted on isolated individuals. Furthermore, it
presumes that the stated relationships are correct. The frequency
of recombination is a major concern. Recombination events between
FVIII and the extragenic site occur in ≤5% of meiosis (leads to
misdiagnosis), although these have not been observed between
hemophilic and intragenic sites (27).
Thus, the probability of meiotic recombination should be considered
while using linked markers for HA diagnosis.
Following observation of normal FVIII (67.7%) and
FIX (90.9%) activity in the proband's sister, the aim of the
present study was to use STR markers to perform linkage analysis to
establish whether the sister of the proband was a carrier of HA.
For this investigation a combination of one intragenic STR marker
and six extragenic markers was used. The intragenic marker, STR22
(a dinucleotide repeats sequence in intron 22) and DXS1073, DXS15,
DXS8091, DXS1227, DXS991 and DXS993 extragenic markers are linked
to the FVIII gene. Following analysis of the haplotypes obtained
from an electrophoretogram, the STR haplotypes were able to
effectively isolate the healthy and pathogenic haplotypes among the
family members, particularly STR22, an intragenic marker, and DXS15
and DXS1073, which are closer to the FVIII gene; thus providing
sufficient evidence to establish the carrier status.
As a consequence of genetic variation among
different populations, a high degree of genetic difference has been
identified between various ethnic groups, such as Chinese, Asian
Indian, Turkish, Russian, Japanese and Caucasian. Therefore, there
is a requirement to establish the specific markers linked to the
FVIII gene in each population (22).
The heterozygosity rate (HR) of each marker in the control
population requires investigation to identify suitable markers for
genetic diagnosis in the given study population in order to perform
efficient tracking of the chromosome carrying the mutant gene.
The efficacies of various polymorphic markers linked
to the FVIII gene have been studied in different ethnic groups and
found to differ significantly. In the study by Li et al
(28). HRs of 88 and 62% were observed
for DXS15 and DXS1073 markers, respectively in a Chinese
population. DXS15 with HR 77.4% was observed in a Hungarian
population (29). In the study done by
Liang et al (30) a HR of 43.6%
for STR22 was observed in a Chinese population. A higher HR is
indicative of a higher efficiency of DNA diagnosis in HA (31).
Additional polymorphic markers were used in numerous
studies; in a study of a Chinese population by Sun et al
(32) HRs of 26.3 and 15.8% for
BclI (polymorphic site in intron 18) and HindIII
(polymorphic site in intron 19), respectively were observed;
whereas, in a Turkish population, HRs were 47 and 35% for
BclI and HinIII, respectively (22). It is possible that these two
polymorphism markers are less informative in a Chinese population.
Similarly, Sun et al (32),
used STR markers in intron 1 and observed an HR of 27.7% and the
marker in intron 24 was not helpful with diagnosing any of the
families in that study. Highly informative extragenous VNTR St14
(DXS52) (heterozygosity ≤90%) burdened with a high risk of
recombination (3–5%) and is used in exceptional cases only where
other VNTR markers were not enough for molecular diagnosis
(25). The risk of recombination of
the external markers limits the use of RFLP in tracking the
defective allele. Additional informative intragenic markers, such
as STR increase the accuracy of analysis (33). Furthermore, Dai et al (34) observed the HR of 21.57% for G6PD and
35.29% for DXS1108 in Chinese population, which was consistent with
previous report (35).
Thus, accurate carrier detection of HA and effective
early prenatal diagnosis represent the most effective forms of
disease control. Linkage analysis, with a combination of intragenic
and extragenic markers, is considered to be optimal for avoiding
misdiagnosis due to recombination and hence results in an accurate
diagnosis. In conclusion, multiflourescent PCR employing STR22,
DXS15 and DXS1073 polymorphic markers, during linkage analysis,
which was conducted in the present study was identified to be
convenient and efficient, and may be performed in clinical
laboratories for carrier detection in Chinese HA families. Despite
certain limitations in the linkage analysis method for indirect
carrier detection, it is a widely used approach and provides an
alternative strategy when direct mutation is not feasible for
genetic counseling of HA.
References
|
1
|
Mansouritorghabeh H: Clinical and
laboratory approaches to hemophilia a. Iran J Med Sci. 40:194–205.
2015.PubMed/NCBI
|
|
2
|
Paroskie A, Oso O, Almassi B, DeBaun MR
and Sidonio RF Jr: Both hemophilia health care providers and
hemophilia a carriers report that carriers have excessive bleeding.
J Pediatr Hematol Oncol. 36:e224–e230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dalm D, Galaz-Montoya JG, Miller JL,
Grushin K, Villalobos A, Koyfman AY, Schmid MF and Stoilova-McPhie
S: Dimeric organization of blood coagulation factor VIII bound to
lipid nanotubes. Sci Rep. 5:112122015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karaman K, Akbayram S, Garipardıç M and
Öner AF: Diagnostic evaluation of our patients with hemophilia A:
17-year experience. Turk Pediatri Ars. 50:96–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shastry SP, Kaul R, Baroudi K and Umar D:
Hemophilia A: Dental considerations and management. J Int Soc Prev
Community Dent. 4(Suppl 3): S147–S152. 2014.PubMed/NCBI
|
|
6
|
Zhou ZY, Koerper MA, Johnson KA, Riske B,
Baker JR, Ullman M, Curtis RG, Poon JL, Lou M and Nichol MB: Burden
of illness: Direct and indirect costs among persons with hemophilia
A in the United States. J Med Econ. 18:457–465. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lheriteau E, Davidoff AM and Nathwani AC:
Haemophilia gene therapy: Progress and challenges. Blood Rev.
29:321–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pezeshkpoor B, Pavlova A, Oldenburg J and
El-Maarri O: F8 genetic analysis strategies when standard
approaches fail. Hamostaseologie. 34:167–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radic CP, Rossetti LC, Abelleyro MM,
Tetzlaff T, Candela M, Neme D, Sciuccati G, Bonduel M,
Medina-Acosta E, Larripa IB, et al: Phenotype-genotype correlations
in hemophilia A carriers are consistent with the binary role of the
phase between F8 and X-chromosome inactivation. J Thromb Haemost.
13:530–539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roualdes O, Nougier C, Fretigny M,
Talagrand E, Durand B, Negrier C and Vinciguerra C: Usefulness of
an in vitro cellular expression model for haemophilia A carrier
diagnosis: Illustration with five novel mutations in the F8 gene in
women with isolated factor VIII:C deficiency. Haemophilia.
21:e202–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qiao SK, Ren HY, Ren JH and Guo XN:
Compound heterozygous hemophilia A in a female patient and the
identification of a novel missense mutation, p.Met1093Ile. Mol Med
Rep. 9:466–470. 2014.PubMed/NCBI
|
|
12
|
Gurjar V and Gurjar M: Consanguineous
marital union resulting in a progeny of whistling-face syndrome and
hemophilia: A case report. J Int Oral Health. 7:78–80.
2015.PubMed/NCBI
|
|
13
|
Zheng J, Ma W, Xie B, Zhu M, Zhang C, Li
J, Wang Y, Wang M and Jin Y: Severe female hemophilia A patient
caused by a nonsense mutation (p.Gln1686X) of F8 gene combined with
skewed X-chromosome inactivation. Blood Coagul Fibrinolysis.
26:977–978. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gitschier J, Wood WI, Goralka TM, Wion KL,
Chen EY, Eaton DH, Vehar GA, Capon DJ and Lawn RM: Characterization
of the human factor VIII gene. Nature. 312:326–330. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar P, Husain N, Soni P, Faridi NJ and
Goel SK: New protocol for detection of intron 22 inversion mutation
from cases with hemophilia A. Clin Appl Thromb Hemost. 21:255–259.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Odnoczko E, Stefańska-Windyga E, Baran B,
Górska-Kosicka M, Sowińska I, Bykowska K and Windyga J: Detection
of inversion mutations (INV22 and INV1) in F8 gene using IS-PCR
method in Polish patients with severe hemophilia A. Acta Haematol
Pol. 46:372–377. 2015. View Article : Google Scholar
|
|
17
|
Edyta Odnoczko JW: Genetic testing in
hemophilia A diagnostic. Hematologia. 5:193–202. 2014.
|
|
18
|
Sawecka J, Skulimowska J, Windyga J and
Koscielak J: Inversion of intron 1 of factor VIII gene in patients
with severe hemophilia A. Acta Haematologica Polonica. 37:61–65.
2006.
|
|
19
|
Bugvi SM, Imran M, Mahmood S, Hafeez R,
Fatima W and Sohail S: Screening of intron 1 inversion and three
intragenic factor VIII gene polymorphisms in Pakistani hemophilia A
families. Blood Coagul Fibrinolysis. 23:132–137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jayandharan GR and Srivastava A and
Srivastava A: Role of molecular genetics in hemophilia: From
diagnosis to therapy. Semin Thromb Hemost. 38:64–78. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kessler L, Adams R, Mighion L, Walther S
and Ganguly A: Prenatal diagnosis in haemophilia A: Experience of
the genetic diagnostic laboratory. Haemophilia. 20:e384–391. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moharrami T, Derakhshan SM, Pourfeizi AA
and Khaniani MS: Detection of hemophilia a carriers in Azeri
Turkish population of Iran: usefulness of HindIII and BclI markers.
Clin Appl Thromb Hemost. 21:755–759. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rong Y, Gao J, Jiang X and Zheng F:
Multiplex PCR for 17 Y-chromosome Specific Short Tandem Repeats
(STR) to enhance the reliability of fetal sex determination in
maternal plasma. Int J Mol Sci. 13:5972–5981. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sidonio RF, Mili FD, Li T, Miller CH,
Hooper WC, DeBaun MR and Soucie M: Hemophilia Treatment Centers
Network: Females with FVIII and FIX deficiency have reduced joint
range of motion. Am J Hematol. 89:831–836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saxena R and Ranjan R: Prenatal Diagnosis
of Hemophilia A and B. J Mol Biol & Mol Imaging. 1:1–6.
2014.
|
|
26
|
Ding QL, Lu YL, Dai J, Xi XD, Wang XF and
Wang HL: Characterisation and validation of a novel panel of the
six short tandem repeats for genetic counselling in Chinese
haemophilia A pedigrees. Haemophilia. 18:621–625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klopp N, Oldenburg J, Uen C, Schneppenheim
R and Graw J: 11 hemophilia A patients without mutations in the
factor VIII encoding gene. Thromb Haemost. 88:357–360.
2002.PubMed/NCBI
|
|
28
|
Li SY, Ma XY, Zhang HM, Wang XM and Li Q:
A multifluorescent STR-PCR for prenatal gene diagnosis of
hemophilia A carriers. J Pract Med. 27:20502011.
|
|
29
|
Bors A, Andrikovics H, Illés Z, Jáger R,
Kardos M, Marosi A, Nemes L and Tordai A: Carrier and prenatal
diagnostic strategy and newly identified mutations in Hungarian
haemophilia A and B families. Blood Coagul Fibrinolysis.
26:161–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang Y, Zhao Y, Yan M, Fan XP, Xiao B and
Liu JZ: Prenatal diagnosis of haemophilia A in China. Prenat Diagn.
29:664–667. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Carvalho FM, de Vargas Wolfgramm E,
Paneto GG, de Paula Careta F, Spagnol Perrone AM, de Paula F and
Louro ID: Analysis of factor VIII polymorphic markers as a means
for carrier detection in Brazilian families with haemophilia A.
Haemophilia. 13:409–412. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun P, Ma L, Diao G, Li CQ and Lin FZ:
Application of indirect linkage analysis and direct genotyping to
hemophilia A carrier detection in Sichuan, China. Genet Mol Res.
14:8229–8235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rabbani B, Rezaeian A, Khanahmad H,
Bagheri R, Kamali E and Zeinali S: Analysing two dinucleotide
repeats of FVIII gene in Iranian population. Haemophilia.
13:740–744. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dai J, Lu Y, Ding Q, Wang H, Xi X and Wang
X: The status of carrier and prenatal diagnosis of haemophilia in
China. Haemophilia. 18:235–240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fang Y, Wang XF, Dai J and Wang HL: A
rapid multifluorescent polymerase chain reaction for genetic
counselling in Chinese haemophilia A families. Haemophilia.
12:62–67. 2006. View Article : Google Scholar : PubMed/NCBI
|