Introduction
Niemann-Pick disease (NPD) is a rare inherited progressive metabolic disorder, classified into subgroups of lysosomal storage disorders that affect lipid metabolism within cells due to genetic mutations (1). There are four most commonly recognized forms of NPD on the basis of genetic cause and symptoms, namely the classic infantile form or type A (NP-A), the visceral form or type B (NP-B), the subacute or juvenile form or type C (NP-C) and the Nova Scotian variant or type D (NP-D) (2). The various types of NPD have been reported in different populations worldwide, but higher rates of incidence have been identified in certain populations, for instance, the NP-C variant in the French-Canadian population of Nova Scotia, NP-B in the Maghreb region of North Africa, and Np-C in the Spanish-American population of southern New Mexico and Colorado (2).
NP-A (MIM no. 257200) and NP-B (MIM no. 607616), also known as acid sphingomyelinase deficiencies, are caused by deficiencies of the acid sphingomyelinase (ASM) enzyme due to mutations in the sphingomyelin phosphodiesterase1 acid lysosomal gene (mapped at 11p15.4; MIM no. 607608) (3). By contrast, mutations in either the NPD type C1 (NPC1) or NPD type C1 (NPC2) gene cause NP-C, which is thus classified as NP-C1 (MIM no. 257220) or NP-C2 (MIM no. 607625), respectively (4,5); both are inherited as an autosomal recessive trait (6,7). The NPC1 gene (mapped at 18q11; MIM no. 607623) encodes NPC1 protein, which is an integral membrane protein of late endosomes involved in lipid transport; while NPC2 protein, encoded by the NPC2 gene (mapped at 14q24.3; MIM no. 601015), is a soluble non-enzymatic lysosomal cholesterol-binding protein (8). According to previous reports, more than 380 pathogenic mutations in NPC1 and more than 20 pathogenic mutations in NPC2 have been implicated in the development of NP-C (9). Approximately 95% of NP-C patients have mutations in the NPC1 gene; the remainder, presenting with a rarer form of the disease, exhibit mutations in the NPC2 gene (9). Thus, NP-C1 is more prevalent than NP-C2 in the general population. The total incidence of NP-C is estimated to be approximately 1 in 150,000 individuals (10).
Generally, patients with NP-C are unable to metabolize cholesterol and other lipids within cells. Consequently, excessive amounts of cholesterol accumulate within the liver and spleen, while other lipids accumulate in the brain (11). Additionally, NP-C causes a secondary reduction in ASM activity (11). NP-C is clinically heterogeneous, and there is a broad spectrum of phenotypes with respect to the age of onset and progression of the disease (Fig. 1) (5). Patients with NP-C usually develop a variety of progressive disabling neurological symptoms including ataxia, vertical supranuclear gaze palsy, gelastic cataplexy, spasticity, dystonia, severe liver disease, hepatosplenomegaly, interstitial lung disease, sleep disturbances, problems with speech and swallowing that worsen over time, impedance with feeding and progressive decline of intellectual function (12). Furthermore, cardiac involvement with cardiomegaly, thickened left ventricular wall and substantial endocardial fibroelastosis has been reported in NP-C patients (13). The detection of NP-C is complicated and initial diagnosis is usually based on learning disability, mild retardation, clumsiness and delayed development of fine motor skills. NP-C is typically fatal and the majority of fatalities occur before the age 20. However, in extremely rare cases, some patients reach age 40 (14). In most cases, neurological symptoms appear between the ages of 4 and 10 years, although the timing of onset may range from perinatal years to adulthood (15). Previous research has focused on low-density lipoprotein cholesterol processing in fibroblasts as the basis for the laboratory diagnosis of NP-C (16). The apparent delay in cholesterol egress clearance from the lysosomal compartment results in intracellular accumulation of cholesterol, which maybe visualized by fluorescence microscopy following filipin staining (17,18). NP-C is classically a neurovisceral disease; however, the involvement of visceral organs (the liver, spleen and sometimes lung) and neurological or psychiatric manifestations arise at different time points, and follow independent courses (5). Liver involvement with varying severity is frequently observed in the first months of life in diagnosed patients, where it constitutes the main feature of the disease (14). Regarding therapy, treatment through bone marrow and/or liver transplantation has been attempted without any effect on the neurological outcome (19,20). Miglustat, with the trade name Zavesca, is the first approved drug for treatment of progressive neurological complications in NP-C disease (21). Additionally, induced pluripotent stem cell (iPSC) lines derived from somatic cells of NP-C patients have been established and maybe used as tools to study the pathogenesis of NP-C and evaluate drug efficacy (22). Soga et al (23) produced NP-C iPSCs, and based on their findings, 2-hydroxypropyl-γ-cyclodextrin was suggested as a potential novel drug candidate for the future treatment of NP-C. However, beyond the clinical difficulties associated with NP-C, it has been demonstrated that fibroblasts derived from NP-C1 patients were resistant to Ebola virus due to mutation in the NPC1 protein, which is required for viral escape from the intracellular vesicular compartment (24).
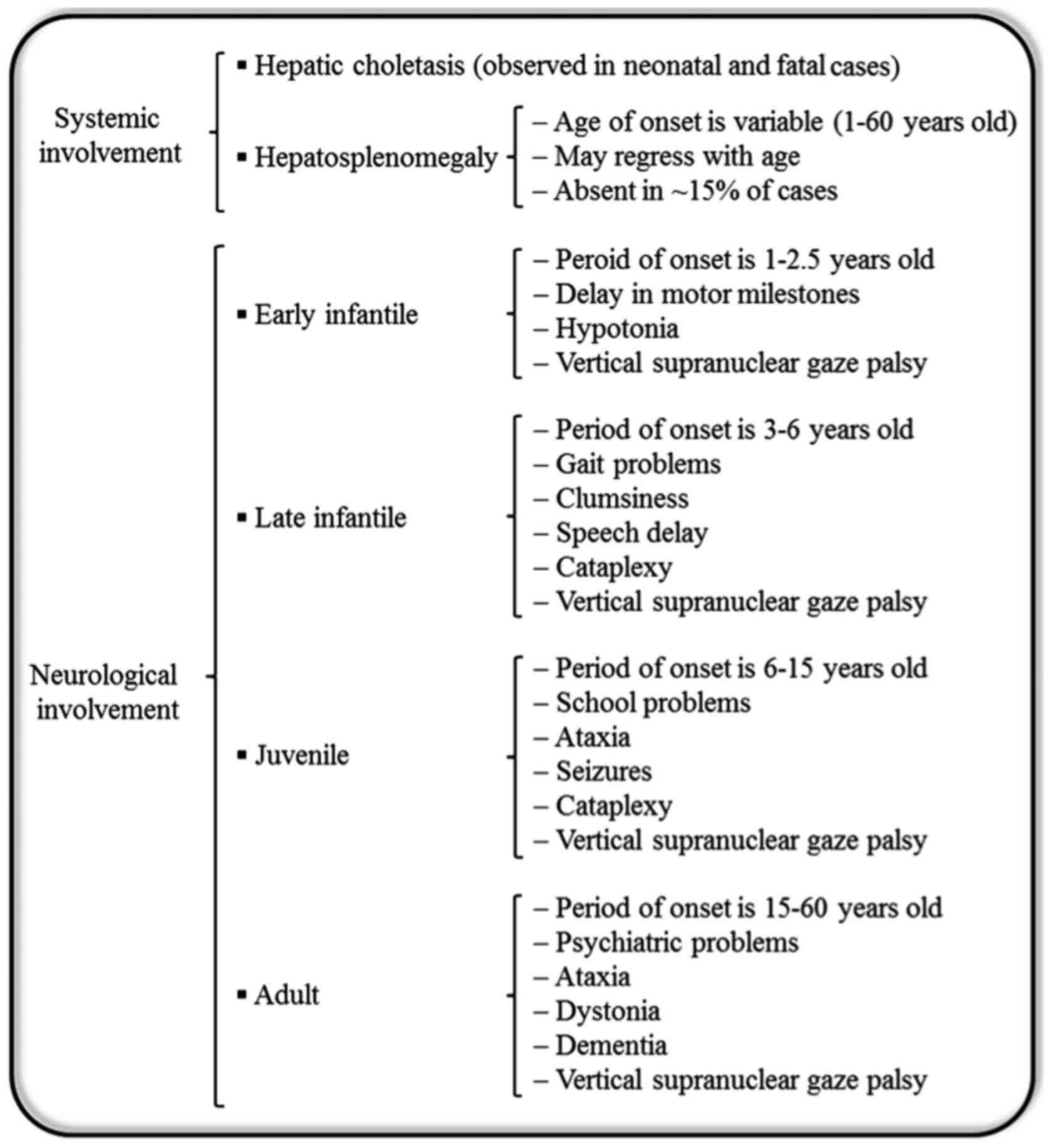 |
Figure 1.
Niemann-Pick disease type C. The diagram represents subtypes and onset ages of visceral and neurological manifestations.
|
Over the last decade, it has become apparent that cholesterol also accumulates in the mitochondria of NP-C1-deficient neurons, which may affect mitochondrial function (25,26). A possible element associated with oxidative damage in NP-C tissues may be mitochondrial dysfunction (27). Indeed, mitochondrial dysfunction and the concomitant oxidative stress appears to be a key element in many neurodegenerative diseases and pathologies associated with liver and cardiac damage (27,28). In previous studies, oxidative stress has been observed in the liver and brain of NP-C mouse models (25,29). Furthermore, Smith et al (30) reported an elevation in the level of oxidative stress markers in the serum of NP-C patients. Previous results also suggest that the mitochondrial dysfunction in NP-C plays an important role in its pathogenesis (27,28). It is well established that the mitochondrial DNA (mtDNA) contains 37 individual genes, of which 13 encode the essential subunits of the four mitochondrial respiratory chain complexes (31). The mtDNA also contains certain genes that serve primordial roles in adenosine triphosphate (ATP) synthesis, including adenosine triphosphatase (ATPase)6/8 genes. The ATPase6/8 genes encode two key subunits of the integral membrane domain F0 (subunit ‘a’ of F0 and ‘A6L’ of F0, respectively) in the mitochondrial respiratory chain complex V, which is located on the inner mitochondrial membrane. The mitochondrial complex V coordinates the final step in oxidative phosphorylation and uses the proton gradient, created together with the other four enzymatic complexes across the inner mitochondrial membrane, for ATP production (32).
Based on similarities between symptoms of NP-C and mitochondrial disorders, as well as the reported mitochondrial dysfunction and ATP deficiency in NP-C patients (25,33), the present study hypothesized that NP-C patients may carry mutations in their mitochondrial genome, particularly in the ATPase6/8 region. Therefore, the present study was undertaken to investigate the correlation between mutations in the mitochondrial ATPase6/8 genes and NP-C incidence in an Iranian population of patients with NP-C.
Materials and methods
Patient samples
Peripheral blood samples (10 ml) were collected from 150 healthy donors (75 male and 75 female) as a control group and 22 Iranian NP-C1 patients (16 male and 6 female) with no familiar relation, who were diagnosed based on the criteria of the Special Medical Center (SMC; Tehran, Iran), as described in our previous study (34). The blood samples were collected between March 2009 and March 2012 in Neurology Departments at the SMC. As all subjects in the present study were minor (between 4 months and 12 years old), their legal guardians were informed on the aims of present study, and signed a written consent form prior to blood collection agreeing to subject participation in the genetic analysis study and publication of the related results as anonymized data. This consent form was approved according to a protocol on human and patient rights by the SMC governing Ethics Committee. The study protocol was approved by the SMC board.
DNA sequencing
Total DNA was extracted from the whole blood samples via the salting out method using a Diatom DNA extraction kit (Gen Fanavaran Co., Tehran, Iran), according to the manufacturer's instructions. To analyze the variations in mitochondrial ATPase6/8 genes, polymerase chain reaction (PCR) sequencing was performed as described previously with some modifications (35). The size of the amplicon was 1,078 bp and the sequences of the oligonucleotides used for amplification of the ATPase6/8 region are listed in Table I. Sequencing of the PCR products was performed in the forward and reverse directions for confirmation of observed variations using an ABI Prism 3100 automated sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) by Kowsar Company (Tehran, Iran).
 |
Table I.
Sequences of primers used for polymerase chain reaction sequencing of the mitochondrial ATPase6/8 region.
|
Table I.
Sequences of primers used for polymerase chain reaction sequencing of the mitochondrial ATPase6/8 region.
| Primer |
Nucleotide position |
Sequence, 5′-3′ |
Ampliconsize, bp |
| Forward |
8161–8180 |
CTACGGTCAATGCTCTGAAA |
1,078 |
| Reverse |
9219–9239 |
TACTATATGATAGGCATGTGA |
|
Bioinformatics analysis
The sequences were analyzed with the FinchTV software (version 1.5.0; Geospiza, Inc., Seattle, WA, USA) and compared with human reference sequences in the GenBank database (GenBank ID: J01415.2; https://www.ncbi.nlm.nih.gov/genbank/). Finally, observed variations were compared between affected and non-affected groups. Additionally, the Mitomap database (https://www.mitomap.org/MITOMAP) was used to identify sequence variations of the mitochondrial genome in the affected and non-affected groups. The observed non-synonymous single nucleotide polymorphisms (SNPs) were analyzed with the I-Mutant 2.0 (developed by Capriotti et al (36); available online: http://folding.uib.es/i-mutant/i-mutant2.0.html) and the PolyPhen-2 software (developed by Adzhubei et al (37); available online: http://genetics.bwh.harvard.edu/pph2/) to predict the possible impacts of amino acid substitutions upon mutations on stability and function of the subjected protein based on comparative physical and evolutionary considerations, respectively. This prediction analysis at the molecular level aimed to identify SNPs affecting actual phenotypes.
Statistical analysis
For statistical analysis, the Fisher's exact test was performed using SPSS software (version 22.0.0.0; IBM Corp., Armonk, NY, USA) to assess associations between observed mutations in NP-C patients and the healthy control group. Data are presented as the mean prevalence rate (%) of each observed mutation in the affected and non-affected groups. These data are accompanied with the number of individuals who have carried the point mutation per total examined individuals in each affected and non-affected group. The frequency of each observed mutation in the affected group was compared to the frequency of same mutation in the non-affected group and a value of P<0.05 was considered to indicate statistical significance.
Results
Patient characteristics
All cases in the affected group (16 male and 6 female) presented with NP-C1 (Table II).
|
Table II.
Demographic characteristics of subjects in the affected and non-affected groups.
|
Table II.
Demographic characteristics of subjects in the affected and non-affected groups.
| Group |
Sex |
No. of individuals |
Disease |
Age range |
| Affected |
F |
6 |
NP-C1 |
5–12 years |
| |
Ma |
16 |
NP-C1 |
4 months-12 years |
| Non-affected |
F |
75 |
Healthy |
5–12 years |
| |
M |
75 |
Healthy |
5–12 years |
Gene mutations
Overall, 10 different point mutations in the ATPase6/8 genes and one point mutation in the non-coding region of mtDNA (MT-C7) were observed in the patient group, which are summarized in Table III. Among all patients, 72.7% (16/22) carried at least one point mutation in the mitochondrial genome. One of the point mutations, the A8860G mutation in ATPase6, was commonly observed in the examined individuals. In the investigated population, 72.7% (16/22) of patients as well as 74.7% (112/150) of the healthy subjects carried the SNP A8860G. In addition, the G8292A mutation, which was identified in the non-coding region of mtDNA, was observed in 9.1% (2/22) of the patient group, while none of the healthy subjects carried this mutation; thus it was deemed to be significantly increased in patients (P<0.01). Only one of the observed SNPs occurred in the ATPase8 gene, namely T8473C. This mutation leads to a synonymous amino acid change in the ATPase8 protein, and was observed in 4.6% (1/22) of the patient group while being absent from the control group (P>0.05 vs. control group). The remaining eight point mutations were observed in the ATPase6 gene, of which only one was a synonymous mutation. This mutation, C8574T, was observed in 4.6% (1/22) of patients. The other observed ATPase6 SNPs were missense mutations, which overall were detected in 9 individuals of the patient group and 18 individuals of the control group; details of their prevalence in the investigated populations are summarized in Table III. Among these missense mutations, only the C8794T variation had significantly increased frequency compared with controls (P<0.01), with a detection rate of 9.1% (2/22) in the examined NP-C1 patients.
|
Table III.
Frequency of mitochondrial ATPase6/8 SNPs in NP-C patients.
|
Table III.
Frequency of mitochondrial ATPase6/8 SNPs in NP-C patients.
| |
|
|
|
|
|
|
Prevalence, % (n/total) |
|
|
| |
|
|
|
|
|
|
|
|
|
| Gene |
Symbol |
Gene location |
Variation location |
Allele |
Type of variation |
Mutation status |
Controls |
Patients |
P-value |
Disease association (ref.) |
| MT-NC7 |
NC7 |
MT:8270–8294 |
MT:8292 |
G/A |
SNP |
Hm |
0.0 (0/150) |
9.1 (2/22) |
0.007a |
Myopathy (61), sudden infant death syndrome (62), maternally inherited diabetes and deafness (63) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8574 |
C/T |
SNP |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Diabetes (64) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8594 |
T/C |
SNP |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Friedreich's ataxia (65) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8629 |
A/G |
SNV |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Not reported |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8684 |
C/T |
SNP |
Hm |
6.0 (9/150) |
9.1 (2/22) |
1.000 |
Multiple sclerosis (66), Huntington's disease (67), autism (35), osteosarcoma (68), thyroid tumor (69) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8696 |
T/C |
SNP |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Pancreatic cancer cell line CFPAC (70) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8697 |
G/A |
SNP |
Hm |
6.0 (9/150) |
4.6 (1/22) |
0.190 |
LHON (47), thyroid tumor (69) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8794 |
C/T |
SNP |
Hm |
0.0 (0/150) |
9.1 (2/22) |
0.007a |
Coronary atherosclerosis risk (56) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8860 |
G/A |
SNP |
Hm |
74.7 (112/150) |
72.7 (16/22) |
0.000 |
Colorectal cancer (50), ovarian cancer (51), breast cancer (52), human glioma cells, glioblastoma multiforme cell line (GBM) (71), osteosarcoma (69), LHON (45) |
| MT-ATP6 |
ATPase6 |
MT:8527–9207 |
MT:8975 |
T/C |
SNP |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Deafness (72) |
| MT-ATP8 |
ATPase8 |
MT:8366–8572 |
MT:8473 |
T/C |
SNP |
Hm |
0.0 (0/150) |
4.6 (1/22) |
0.080 |
Atypical psychosis (58) |
Bioinformatics analysis
The results of analysis with I-Mutant 2.0 software indicated that all observed SNPs excluding the synonymous mutations (C8574T and T8473C) and mutation in the non-coding region of mtDNA (G8292A) may decrease the stability of the protein. In addition, PolyPhen-2 software predicted all above mentioned SNPs to decrease protein stability, possibly leading to protein dysfunction, with the exception of SNPs C8684T, C8794T and A8860G, which were predicted to be benign and to not affect protein function. The prediction results of I-Mutant 2.0 and PolyPhen-2 are summarized in Table IV.
|
Table IV.
Prediction of possible impact of amino acid substitution on protein structure stability and function for point mutations of ATPase6/8 genes.
|
Table IV.
Prediction of possible impact of amino acid substitution on protein structure stability and function for point mutations of ATPase6/8 genes.
| Location |
SNP |
Amino acid substitution |
Consequence |
I-Mutant (reliability index) |
polyphen-2 HumDiv (score) |
| MT-NC7 |
G8292A |
– |
Non-coding |
– |
– |
| MT-ATP6 |
C8574T |
G16G |
Synonymous |
– |
– |
| |
T8594C |
I23T |
Missense |
Decrease stability (8) |
Possibly damaging (0.620) |
| |
A8629G |
K35E |
Missense |
Decrease stability (5) |
Possibly damaging (0.590) |
| |
C8684T |
T53I |
Missense |
Decrease stability (0) |
Benign (0.010) |
| |
T8696C |
M57T |
Missense |
Decrease stability (0) |
Possibly damaging (0.950) |
| |
G8697A |
M57I |
Missense |
Decrease stability (4) |
Possibly damaging (0.890) |
| |
C8794T |
H90Y |
Missense |
Decrease stability (1) |
Benign (0.000) |
| |
A8860G |
T112A |
Missense |
Decrease stability (7) |
Benign (0.000) |
| |
T8975C |
L150P |
Missense |
Decrease stability (5) |
Possibly damaging (0.791) |
| MT-ATP8 |
T8473C |
P36P |
Synonymous |
– |
– |
Discussion
NP-C is a rare fatal disorder with a lack of effective treatments available. The disease is considered as a neurovisceral lipid storage disorder; however, the underlying pathogenic mechanism that links the accumulation of intracellular cholesterol/lipid with cell death in the central nervous system and liver is currently unknown. According to previous studies, oxidative stress leading to oxidative damage is observed in different tissues of NP-C patients (27,28,30), which suggests its involvement in the pathogenesis of NP-C. Furthermore, previous data suggests that mitochondrial dysfunction is associated with oxidative damage in NP-C patients and serves an important role in NP-C pathogenesis (27). Indeed, it has been reported that mitochondrial dysfunction and the associated oxidative stress appear to be key aspects in many neurodegenerative diseases and disorders associated with liver and cardiac damage (27,38,39). The role of mitochondrial dysfunction in the pathogenesis of NP-C remains unknown, but studies have identified mitochondrial dysfunction and ATP deficiency in NP-C patients (25,33). In addition, ATP deficiency in parallel with higher cholesterol content has been observed in mitochondria in NP-C mouse models, in which ATP synthesis and mitochondrial function were restored following treatment with the cholesterol chelator cyclodextrin (25). Alterations in ATP production, mitochondrial morphology and input of glutathione to the mitochondrial matrix have been reported in NP-C mouse models, and were correlated with enhanced mitochondrial cholesterol content as well as reduced ATP synthesis, enhanced reactive oxygen species generation, and increased energy demand in NP-C cells (27). These findings support the hypothesis that mitochondrial dysfunction may contribute to oxidative damage in NP-C. In the present study, the potential mutations in mitochondrial ATPase6/8 genes, which are key elements in cellular energy production, were analyzed in NP-C patients. Different mutations in the ATPase6/8 genes, which encode subunits of complex V, have been reported in various mitochondrial diseases including neuropathy, ataxia, and retinitis pigmentosa syndrome (40), Leber's hereditary optic neuropathy (LHON) (41), maternally inherited Leigh syndrome (39) and hypertrophic cardiomyopathy (42).
In the present study, 10 SNPs in mitochondrial ATPase6/8 genes and one variation in the non-coding region of mtDNA were identified in unrelated Iranian NP-C patients. Overall, 73% of examined NP-C patients carried at least one point mutation in their mtDNA. This finding suggests that mitochondria and defects in the respiratory chain may be involved in the pathogenesis of NP-C. An observed mutation was the A8860G variation in the mitochondrial ATPase6 gene, which was observed in 73% of the NP-C1 patients, and also 75% of unrelated healthy individuals. According to previous reports, this SNP is a common variation and has been identified in different populations (43). The A8860G SNP has been reported in different diseases including Alzheimer's (44), hypertrophic cardiomyopathy (45), LHON (46,47) and breast cancer (48), and also in the mitochondrial haplogroup R0H2a2a (49). The present findings also support previous data (45) that identified the A8860G SNP in a high proportion of affected and non-affected individuals. Analysis with I-Mutant 2.0 software predicted that A8860G decreases stability of ATPase6 protein. However, based on PolyPhen-2 prediction, it is a benign point mutation and has no impact on protein function. This may be due to the fact, that A8860G is located in a poorly conserved protein region. Thus it is frequently observed in various healthy and patient populations (43,49).
Amongst all observed SNPs, significant differences were observed between NP-C1 patients and healthy subjects regarding the frequencies of the SNPs G8292A and C8794T, which are located in the non-coding region of mtDNA (NC7) and in the ATPase6 gene, respectively. This suggested that the G8292A and C8794T mtDNA SNPs in patients confer genetic susceptibility to NP-C1. The G8292A SNP has been also reported in different cancers including colorectal adenomatous polyps (50), epithelial ovarian cancer (51) and sporadic breast cancer (52), and in the mitochondrial haplogroup R0a1a3 (48). Additionally, it was identified by Cerný et al (53) in LHON patients with haplogroup R0a1a1a in Yemen. These patients also carried the minor pathological mutation A3395G. No conservation rate is reported for the G8292A mutation in the Mitomap database (query no. HM185203.1). The other significant SNP, C8794T, is a homoplasmic missense mutation in the ATPase6 gene, predicted to lead to stability reduction of the protein, but also to be a benign mutation that does not change the function of the ATPase6 enzyme. However, the observed association of C8794T SNP with the NP-C phenotype indicates a potential pathophysiological effect of C8794T, which requires further investigation. The C8794T SNP has been demonstrated to be involved in different diseases including breast cancer (47), early onset cataracts and focal dystonia (54) and also to be a specific variant of mitochondrial haplogroup A in an East Asian population (55). In addition, Sawabe et al (56) reported that Japanese elderly subjects belonging to haplogroup A had a genetic risk of coronary atherosclerosis. Another report by Yu et al (57) identified the C8794T mutation in LHON Asian patients belonging to haplogroup A5, who also carried the major pathological mutation G3460A. According to the Mitomap database, the conservation rate of C8794T mutation is 73.33% (query no. GQ999962.1).
The A8629G point mutation, which was identified in the mitochondrial ATPase6 gene and only in the affected group (1/22), was reported, to the best of our knowledge, for the first time in the present study. The A8629 variant leads to a missense mutation, which was predicted to decrease stability of protein structure without affecting protein function. Although this point mutation was determined to be not significantly associated with NP-C disease, it can be regarded as a novel mitochondrial ATPase6 single nucleotide variant.
The point mutation T8473C was identified in the mitochondrial ATPase8 gene and was observed in 5% (1/22) of the affected group. This SNP causes a synonymous mutation in ATPase8 protein and was predicted to cause no functional disturbance. It has been previously detected in patients with a typical psychosis (58) and colorectal cancer (59). As among the 11 different point mutations in NP-C patients reported in the present study, only one occurred in the mitochondrial ATPase8 gene. It may be assumed that the ATPase6 gene has a higher susceptibility than the ATPase8 gene to the occurrence of mutations. Similarly, it has been reported in breast cancer patients that the ATPase6 gene had greater susceptibility to mutation than ATPase8 (48). mtDNA point mutations are typically maternally inherited, but can also occur sporadically (60). The reported mtDNA point mutations in the present study were observed only in the examined unrelated NP-C patients, whereas their relatives (especially their mothers) were not analyzed for the presence of these mutations. Therefore, the current study is unable to discriminate whether the reported mutations were sporadic or familial and maternally inherited.
In conclusion, the present study identified that 73% of the Iranian NP-C patients were carrying one or more mitochondrial mutation. Furthermore, the present study, to the best of our knowledge, is the first to report mitochondrial ATPase6/8 SNPs in NP-C patients. The current findings propose a perspective in the etiology of NP-C by indicating an association between the mitochondrial SNPs G8292A and C8794T and the incidence of NP-C disease. The detection of these SNPs may be clinically important for improving our understanding about the genotypic spectrum of NP-C, which may aid in determining the diagnosis, prognosis and treatment of NP-C patients in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants awarded to MH from the National Institute of Genetic Engineering and Biotechnology, Tehran, Iran (grant no. 521-622) and to HS from the Excellence Cluster Cardio-Pulmonary System, Germany (grant no. EXC-147), with the latter supported by the German Research Foundation (Bonn, Germany).
Availability of data and materials
The datasets used and/or analyzed during the current study included in this published article.
Authors' contributions
ShM performed the experimental procedures, data analysis and drafted the manuscript. FS participated in data interpretation and writing the manuscript. LA, SHT, PK, MRA, HS and SaM participated in coordination of the study and data analysis. MH conceived the study and aided with the study design. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The blood samples were provided by the Special Medical Center, funded by the Iranian Health Ministry.
Consent for publication
All subjects provided written informed consent to participate in the study and permitting the publication of relevant data on the terms of data anonymization.
Competing interests
All authors declare no potential conflicts of interest with respect to the research, authorship and/or publication of this study.
Glossary
Abbreviations
Abbreviations:
|
NP-C
|
Niemann-Pick disease type C
|
|
ATP
|
adenosine triphosphate
|
|
ATPase
|
adenosine triphosphatase
|
|
PCR
|
polymerase chain reaction
|
|
SNP
|
single nucleotide polymorphism
|
|
mtDNA
|
mitochondrial DNA
|
|
ASM
|
acid sphingomyelinase
|
|
SMC
|
Special Medical Center of Tehran, Iran
|
|
LHON
|
Leber's hereditary optic neuropathy
|
|
iPSC
|
induced pluripotent stem cell
|
References
|
1
|
Torres S, Balboa E, Zanlungo S, Enrich C, Garcia-Ruiz C and Fernandez-Checa JC: Lysosomal and mitochondrial liaisons in Niemann-Pick disease. Front Physiol. 8:9822017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wasserstein MP and Schuchman EH: Acid sphingomyelinase deficiencyGeneReviews®. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al: University of Washington; Seattle, WA: 1993
|
|
3
|
Schuchman EH and Wasserstein MP: Types A and B Niemann-Pick disease. Best Pract Res Clin Endocrinol Metab. 29:237–247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT and Wijburg F: NP-C Guidelines Working Group: Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol Genet Metab. 106:330–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vanier MT: Niemann-Pick disease type C. Orphanet J Rare Dis. 5:162010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patterson MC: A riddle wrapped in a mystery: Understanding Niemann-Pick disease, type C. Neurologist. 9:301–310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vanier MT and Millat G: Niemann-Pick disease type C. Clin Genet. 64:269–281. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Platt FM, Boland B and van der Spoel AC: The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 199:723–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU and Lobel P: Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci USA. 101:5886–5891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mengel E, Klünemann HH, Lourenço CM, Hendriksz CJ, Sedel F, Walterfang M and Kolb SA: Niemann-Pick disease type C symptomatology: An expert-based clinical description. Orphanet J Rare Dis. 8:1662013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patterson MC: Niemann-Pick disease overview: Types A, B and C. https://nnpdf.org/overview/February 9–2017
|
|
12
|
Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topçu M and Vanier MT: NP-C Guidelines Working Group: Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 98:152–165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guertl B, Noehammer C and Hoefler G: Metabolic cardiomyopathies. Int J Exp Pathol. 81:349–372. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Organization for Rare Disorders (NORD): Niemann Pick Disease Type CNORD's Rare Disease Database. NORD; Danbury CT: 2017, https://rarediseases.org/rare-diseases/niemann-pick-disease-type-c/
|
|
15
|
Yang CC, Su YN, Chiou PC, Fietz MJ, Yu CL, Hwu WL and Lee MJ: Six novel NPC1 mutations in Chinese patients with Niemann-Pick disease type C. J Neurol Neurosurg Psychiatry. 76:592–595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pentchev PG, Comly ME, Kruth HS, Patel S, Proestel M and Weintroub H: The cholesterol storage disorder of the mutant BALB/c mouse. A primary genetic lesion closely linked to defective esterification of exogenously derived cholesterol and its relationship to human type C Niemann-Pick disease. J Biol Chem. 261:2772–2777. 1986.PubMed/NCBI
|
|
17
|
Park WD, O'Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS, Patterson MC and Snow K: Identification of 58 novel mutations in Niemann-Pick disease type C: Correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat. 22:313–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vanier MT and Latour P: Laboratory diagnosis of Niemann-Pick disease type C: The filipin staining test. Methods Cell Biol. 126:357–375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mendes MS, Portela FX, Reis RC, Castro JD, Garcia JH and Holanda MA: Liver transplantation in a patient with Niemann-Pick disease and pulmonary involvement. J Bras Pneumol. 38:269–271. 2012.(In English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsu YS, Hwu WL, Huang SF, Lu MY, Chen RL, Lin DT, et al: Niemann-Pick disease type C (a cellular cholesterol lipidosis) treated by bone marrow transplantation. Bone Marrow Transplant. 24:103–107. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhamija R: Lysosomal storage disorderMayo Clinic Neurology Board Review: Clinical Neurology for Initial Certification and MoC. Flemming KD and Jones LK: Oxford University Press; pp. 709–716. 2015, View Article : Google Scholar
|
|
22
|
Yu D, Swaroop M, Wang M, Baxa U, Yang R, Yan Y, Coksaygan T, DeTolla L, Marugan JJ, Austin CP, et al: Niemann-Pick disease type c: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J Biomol Screen. 19:1164–1173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soga M, Ishitsuka Y, Hamasaki M, Yoneda K, Furuya H, Matsuo M, Ihn H, Fusaki N, Nakamura K, Nakagata N, et al: HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells. 33:1075–1088. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al: Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477:340–343. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K and Michikawa M: Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem. 280:11731–11739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fernández A, Llacuna L, Fernández-Checa JC and Colell A: Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J Neurosci. 29:6394–6405. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vázquez MC, del Pozo T, Robledo FA, Carrasco G, Pavez L, Olivares F, González M and Zanlungo S: Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS One. 6:e287772011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vázquez MC, Balboa E, Alvarez AR and Zanlungo S: Oxidative stress: A pathogenic mechanism for Niemann-Pick type C disease. Oxid Med Cell Longev. 2012:2057132012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC and García-Ruiz C: Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 4:185–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith D, Wallom KL, Williams IM, Jeyakumar M and Platt FM: Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol Dis. 36:242–251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schon EA, DiMauro S and Hirano M: Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat Rev Genet. 13:878–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cortés-Hernández P, Vázquez-Memije ME and García JJ: ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J Biol Chem. 282:1051–1058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Charman M, Kennedy BE, Osborne N and Karten B: MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick type C1 protein. J Lipid Res. 51:1023–1034. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Karimzadeh P, Tonekaboni SH, Ashrafi MR, Shafeghati Y, Rezayi A, Salehpour S, Ghofrani M, Taghdiri MM, Rahmanifar A, Zaman T, et al: Effects of miglustat on stabilization of neurological disorder in niemann-pick disease type C: Iranian pediatric case series. J Child Neurol. 28:1599–1606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Piryaei F, Houshmand M, Aryani O, Dadgar S and Soheili ZS: Investigation of the mitochondrial ATPase 6/8 and tRNA(Lys) genes mutations in autism. Cell J. 14:98–101. 2012.PubMed/NCBI
|
|
36
|
Capriotti E, Fariselli P and Casadio R: I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33:(Web Server issue). W306–310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Reddy PH: Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer's disease. CNS Spectr. 14 Suppl 7:8–13; discussion 16–18. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsutsui H, Kinugawa S and Matsushima S: Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 81:449–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Houstek J, Pícková A, Vojtísková A, Mrácek T, Pecina P and Jesina P: Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 1757:1400–1405. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
López-Gallardo E, Emperador S, Solano A, Llobet L, Martín-Navarro A, López-Pérez MJ, Briones P, Pineda M, Artuch R, Barraquer E, et al: Expanding the clinical phenotypes of MT-ATP6 mutations. Hum Mol Genet. 23:6191–6200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jonckheere AI, Hogeveen M, Nijtmans LG, van den Brand MA, Janssen AJ, Diepstra JH, van den Brandt FC, van den Heuvel LP, Hol FA, Hofste TG, et al: A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet. 45:129–133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jinam TA, Hong LC, Phipps ME, Stoneking M, Ameen M, Edo J and Saitou N: HUGO Pan-Asian SNP Consortium: Evolutionary history of continental southeast Asians: ‘early train’ hypothesis based on genetic analysis of mitochondrial and autosomal DNA data. Mol Biol Evol. 29:3513–3527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mostafaie N, Rossmanith W, Hombauer H, Dechat T, Raffelsberger T, Bauer K, Worofka B, Kittl E, Hofmann J, et al: Mitochondrial genotype and risk for Alzheimer's disease: cross-sectional data from the Vienna-Transdanube-Aging ‘VITA’ study. J Neural Transm (Vienna). 111:1155–1165. 2004.PubMed/NCBI
|
|
45
|
Houshmand M, Montazeri M, Kuchekian N, Noohi F, Nozar G and Zamani A: Is 8860 variation a rare polymorphism or associated as a secondary effect in HCM disease? Arch Med Sci. 7:242–246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fauser S, Luberichs J, Besch D and Leo-Kottler B: Sequence analysis of the complete mitochondrial genome in patients with Leber's hereditary optic neuropathy lacking the three most common pathogenic DNA mutations. Biochem Biophys Res Commun. 295:342–347. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Puomila A, Hämäläinen P, Kivioja S, Savontaus ML, Koivumäki S, Huoponen K and Nikoskelainen E: Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 15:1079–1089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ghaffarpour M, Mahdian R, Fereidooni F, Kamalidehghan B, Moazami N and Houshmand M: The mitochondrial ATPase6 gene is more susceptible to mutation than the ATPase8 gene in breast cancer patients. Cancer Cell Int. 14:212014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
van Oven M and Kayser M: Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 30:E386–E394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mehrabi S, Akwe JA, Adams G Jr, Grizzle W, Yao X and Aikhionbare FO: Analysis of mtDNA sequence variants in colorectal adenomatous polyps. Diagn Pathol. 5:662010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aikhionbare FO, Mehrabi S, Kumaresan K, Zavareh M, Olatinwo M, Odunsi K and Partridge E: Mitochondrial DNA sequence variants in epithelial ovarian tumor subtypes and stages. J Carcinog. 6:12007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Czarnecka AM, Klemba A, Krawczyk T, Zdrozny M, Arnold RS, Bartnik E and Petros JA: Mitochondrial NADH-dehydrogenase polymorphisms as sporadic breast cancer risk factor. Oncol Rep. 23:531–535. 2010.PubMed/NCBI
|
|
53
|
Cerný V, Mulligan CJ, Fernandes V, Silva NM, Alshamali F, Non A, Harich N, Cherni L, El Gaaied AB, Al-Meeri A and Pereira L: Internal diversification of mitochondrial haplogroup R0a reveals post-last glacial maximum demographic expansions in South Arabia. Mol Biol Evol. 28:71–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Simon DK, Tarnopolsky MA, Greenamyre JT and Johns DR: A frameshift mitochondrial complex I gene mutation in a patient with dystonia and cataracts: Is the mutation pathogenic? J Med Genet. 38:58–61. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lai CH, Huang SF, Chen IH, Liao CT, Wang HM and Hsieh LL: The mitochondrial DNA Northeast Asia CZD haplogroup is associated with good disease-free survival among male oral squamous cell carcinoma patients. PLoS One. 7:e496842012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sawabe M, Tanaka M, Chida K, Arai T, Nishigaki Y, Fuku N, Mieno MN, Kuchiba A and Tanaka N: Mitochondrial haplogroups A and M7a confer a genetic risk for coronary atherosclerosis in the Japanese elderly: An autopsy study of 1,536 patients. J Atheroscler Thromb. 18:166–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu D, Jia X, Zhang AM, Guo X, Zhang YP, Zhang Q and Yao YG: Molecular characterization of six Chinese families with m.3460G>A and Leber hereditary optic neuropathy. Neurogenetics. 11:349–356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kazuno AA, Munakata K, Mori K, Tanaka M, Nanko S, Kunugi H, Umekage T, Tochigi M, Kohda K, Sasaki T, et al: Mitochondrial DNA sequence analysis of patients with ‘atypical psychosis’. Psychiatry Clin Neurosci. 59:497–503. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vogel LK, Sæbø M, Høyer H, Kopp TI, Vogel U, Godiksen S, Frenzel FB, Hamfjord J, Bowitz-Lothe IM, Johnson E, et al: Intestinal PTGS2 mRNA levels, PTGS2 gene polymorphisms, and colorectal carcinogenesis. PLoS One. 9:e1052542014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tuppen HA, Blakely EL, Turnbull DM and Taylor RW: Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 1797:113–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kleinle S, Schneider V, Moosmann P, Brandner S, Krähenbühl S and Liechti-Gallati S: A novel mitochondrial tRNA(Phe) mutation inhibiting anticodon stem formation associated with a muscle disease. Biochem Biophys Res Commun. 247:112–115. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Opdal SH, Vege A, Arnestad M, Musse MA and Rognum TO: Mitochondrial tRNA genes and flanking regions in sudden infant death syndrome. Acta Paediatr. 96:211–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tabebi M, Mkaouar-Rebai E, Mnif M, Kallabi F, Ben Mahmoud A, Ben Saad W, Charfi N, Keskes-Ammar L, Kamoun H, Abid M and Fakhfakh F: A novel mutation MT-COIII m.9267G>C and MT-COI m.5913G>A mutation in mitochondrial genes in a Tunisian family with maternally inherited diabetes and deafness (MIDD) associated with severe nephropathy. Biochem Biophys Res Commun. 459:353–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li S, Besenbacher S, Li Y, Kristiansen K, Grarup N, Albrechtsen A, Sparsø T, Korneliussen T, Hansen T, Wang J, et al: Variation and association to diabetes in 2000 full mtDNA sequences mined from an exome study in a Danish population. Eur J Hum Genet. 22:1040–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Singh I, Faruq M, Srivastava A and Mukerji M: Analysis of mitochondrial DNA variations in Friedreich's ataxia patients in Indian families: Identification of disease associated markers. Mov Disord. 27 Suppl 1:6022012.
|
|
66
|
Ahari SE, Houshmand M, Panahi MS, Kasraie S, Moin M and Bahar MA: Investigation on mitochondrial tRNA(Leu/Lys), NDI and ATPase 6/8 in Iranian multiple sclerosis patients. Cell Mol Neurobiol. 27:695–700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kasraie S, Houshmand M, Banoei MM, Ahari SE, Panahi MS, Shariati P, Bahar M and Moin M: Investigation of tRNA(Leu/Lys) and ATPase 6 genes mutations in Huntington's disease. Cell Mol Neurobiol. 28:933–938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Guo XG, Liu CT, Dai H and Guo QN: Mutations in the mitochondrial ATPase6 gene are frequent in human osteosarcoma. Exp Mol Pathol. 94:285–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Máximo V, Soares P, Lima J, Cameselle-Teijeiro J and Sobrinho-Simões M: Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: A study with emphasis on Hürthle cell tumors. Am J Pathol. 160:1857–1865. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH and Kern SE: Detection of mitochondrial DNA mutations in pancreatic cancer offers a ‘mass’-ive advantage over detection of nuclear DNA mutations. Cancer Res. 61:1299–1304. 2001.PubMed/NCBI
|
|
71
|
DeHaan C, Habibi-Nazhad B, Yan E, Salloum N, Parliament M and Allalunis-Turner J: Mutation in mitochondrial complex I ND6 subunit is associated with defective response to hypoxia in human glioma cells. Mol Cancer. 3:192004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen J, Yuan H, Lu J, Liu X, Wang G, Zhu Y, Cheng J, Wang X, Han B, Yang L, et al: Mutations at position 7445 in the precursor of mitochondrial tRNA(Ser(UCN)) gene in three maternal Chinese pedigrees with sensorineural hearing loss. Mitochondrion. 8:285–292. 2008. View Article : Google Scholar : PubMed/NCBI
|














