Introduction
Allgrove syndrome (AS; OMIM no. 231550), first
described in 1978 (1), is a rare
autosomal recessive congenital disease characterized by the triad
of adrenal insufficiency (Addison's disease), failure of the lower
esophageal sphincter to relax (achalasia) and absence of tear
secretion (alacrima). AS is also known as 3A syndrome or, with the
addition of autonomic and central nervous system impairment, 4A
syndrome (2). The syndrome usually
presents itself in the first decade of an individual's life, but
cases have been also identified in adulthood, usually due to
childhood symptoms having been overlooked. AS occurs worldwide, and
its various clinical manifestations range from mild to severe
(occasionally fatal) (1–4). The 3A syndrome is a progressive
disorder and it can take years for the full clinical effects to
develop (5). The pathogenic gene of
AS, AAAS, is located on chromosome 12q13, consists of 16 exons and
encodes the protein ‘alacrima, achalasia, adrenal insufficiency and
neurologic disorder’ (ALADIN). The gene has been found to be
ubiquitously expressed throughout the body and not just in the
adrenal glands, as previously believed (6). A variety of mutations scattered
throughout the gene have been reported, except in exon 3 (7). There does not appear to be a
correlation between the different mutations found within AAAS and
the clinical manifestations of the disease (6). In 2006, the Beijing Children's Hospital
of Capital Medical University (Beijing, China) reported the first
case of AS in the Han population from the mainland of China
(8). Two additional cases (one male
and one female patient) were admitted in 2012. In the present
study, the clinical characteristics of these 3 cases were
summarized and the literature was reviewed in order to enhance the
understanding of the pathogenic and clinical characteristics of AS
in the Chinese population and to improve diagnosis of the
disease.
Case report
Methods
The present study comprises a retrospective analysis
of 3 cases of AS diagnosed in 2006 or 2012. Written informed
consent for all tests and treatments, as well as the genetic
analysis, was provided from the parents of the patients. Whole
blood samples were obtained from the patients and their parents for
the AAAS gene analysis. Table I
provides a summary of the symptoms and results of the genetic
analysis for each case. Genomic DNA was isolated from peripheral
blood using the Takara Blood Genome DNA Extraction kit (Takara Bio,
Inc., Otsu, Japan). The entire coding region of the AAAS gene
(NM_015665) was amplified and sequenced using the Sanger method
(9). The primers and sequencing
conditions were as previously reported (10). The patients' sequences were blasted
against the GenBank Database, and the mutations in the gene of each
patient were confirmed by sequencing the same fragment of their
parents' DNA (NM_015665).
 | Table I.Clinical summary for patients 1, 2 and
3. |
Table I.
Clinical summary for patients 1, 2 and
3.
| Patient | Gender/age at
admission (years) | Initial symptoms | Alacrima | Achalasia | Adrenal
dysfunction | Neuropathy | Genetic mutation |
|---|
| 1 | F/7.25 | Vomiting
Hyperpigmentation | + | + | + | + | c.771delG Exon 8 |
| 2 | M/2.60 | Vomiting
Hyperpigmentation | + | + | + | – | c.771delG Exon 8 |
| 3 | F/4.30 | Vomiting
Hyperpigmentation | + | + | + | – | c.1366C>T Exon
15 |
Patient 1
A Chinese girl, aged 7 years and 3 months, was
diagnosed with AS on February 11, 2006. This was the first case of
AS to be diagnosed on the mainland of China, to the best of our
knowledge (6). Two years and 9
months prior to diagnosis, the patient had been admitted to a local
hospital due to generalized hyperpigmentation and emaciation, in
addition to a 3-min period of unconsciousness. The laboratory
examination revealed that the blood electrolyte and serum cortisol
levels of the patient, as well as the liver and kidney function,
were normal; however, the levels of adrenocorticotropin (ACTH) were
markedly elevated. A computed tomography scan of both adrenal
glands and a magnetic resonance imaging (MRI) scan of the head
revealed no significant anomalies. The patient was diagnosed with
Addison's disease, and the hyperpigmentation slowly dissipated with
oral hydrocortisone acetate (50 mg/m2).
Nine months prior to admission to the Beijing
Children's Hospital, the patient had a vomiting incident without
abdominal pain or regurgitation, which lasted 5–6 days. Treatment
with an increased dose of hydrocortisone was not effective, and the
patient exhibited increasing levels of weakness, fatigue,
difficulty in eating and weight loss.
The parents of the patient were healthy (father, 30
years old; mother, 32 years old). Gravida 1 (G1) and (G2) were
aborted; the patient was G3 para 1 (P1) with full-term, normal
delivery, and her intelligence was determined to be normal, as
compared with children of the same age. Consanguineous
relationships between the parents were found to exist for at least
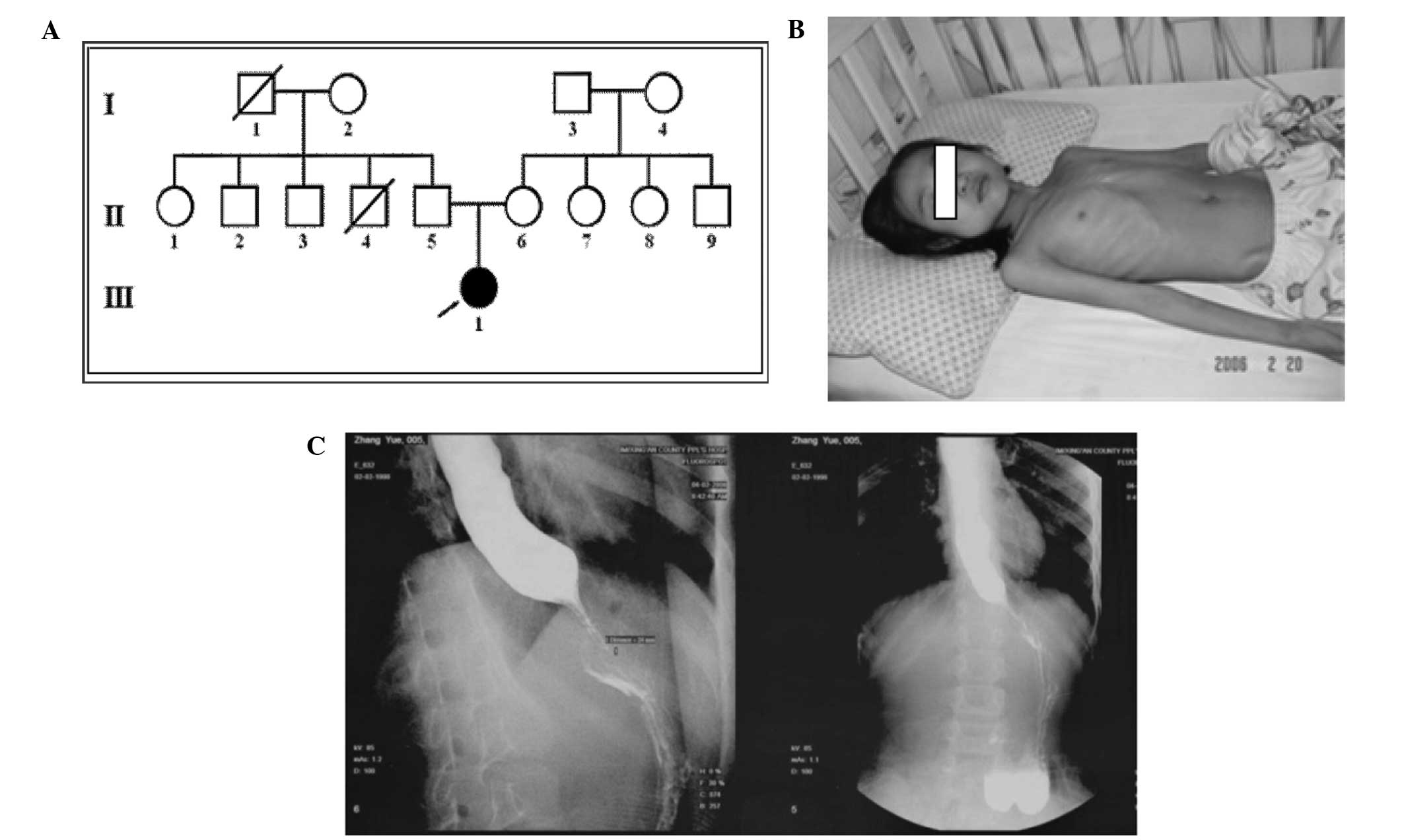
the previous three generations (Fig.
1A).
The findings of the physical examination were as
follows: Blood pressure (BP), 90/60 mmHg; height, 120 cm; weight,
15 kg. No abnormalities were observed in the heart or lungs of the
patient. The breast development stage of the patient was determined
to be Tanner stage I (11). The
mental status of the patient was evaluated as alert but weak.
Hyperpigmentation of the skin and thin subcutaneous fat were noted
(Fig. 1B). Normal muscle strength
and tone were found in all extremities, but muscle volume was
reduced and a flattening of the thenar eminence was observed. The
abdominal reflex was positive, while the knee jerk, Achilles tendon
and radial periosteal reflexes were found to be hyperreflexive. The
patient was negative for meningism and positive for the Babinski
sign on the right side. No abnormal gait was observed. The
electroencephalograms (EEGs), brain MRI scan and evoked potential
and nerve conduction studies showed no specific abnormalities.
Esophagography showed achalasia of the lower esophagus at the
cardia (Fig. 1C). Funduscopy
indicated that the optic nerve discs were pale, suggesting optic
nerve atrophy. The results of a bilateral Schirmer test were 0 mm,
which led to a diagnosis of alacrima. Further medical history
revealed that, not long after birth, the patient was unable to
produce tears when crying. The suspicion that the patient was
positive for AS was confirmed by genetic testing, which revealed a
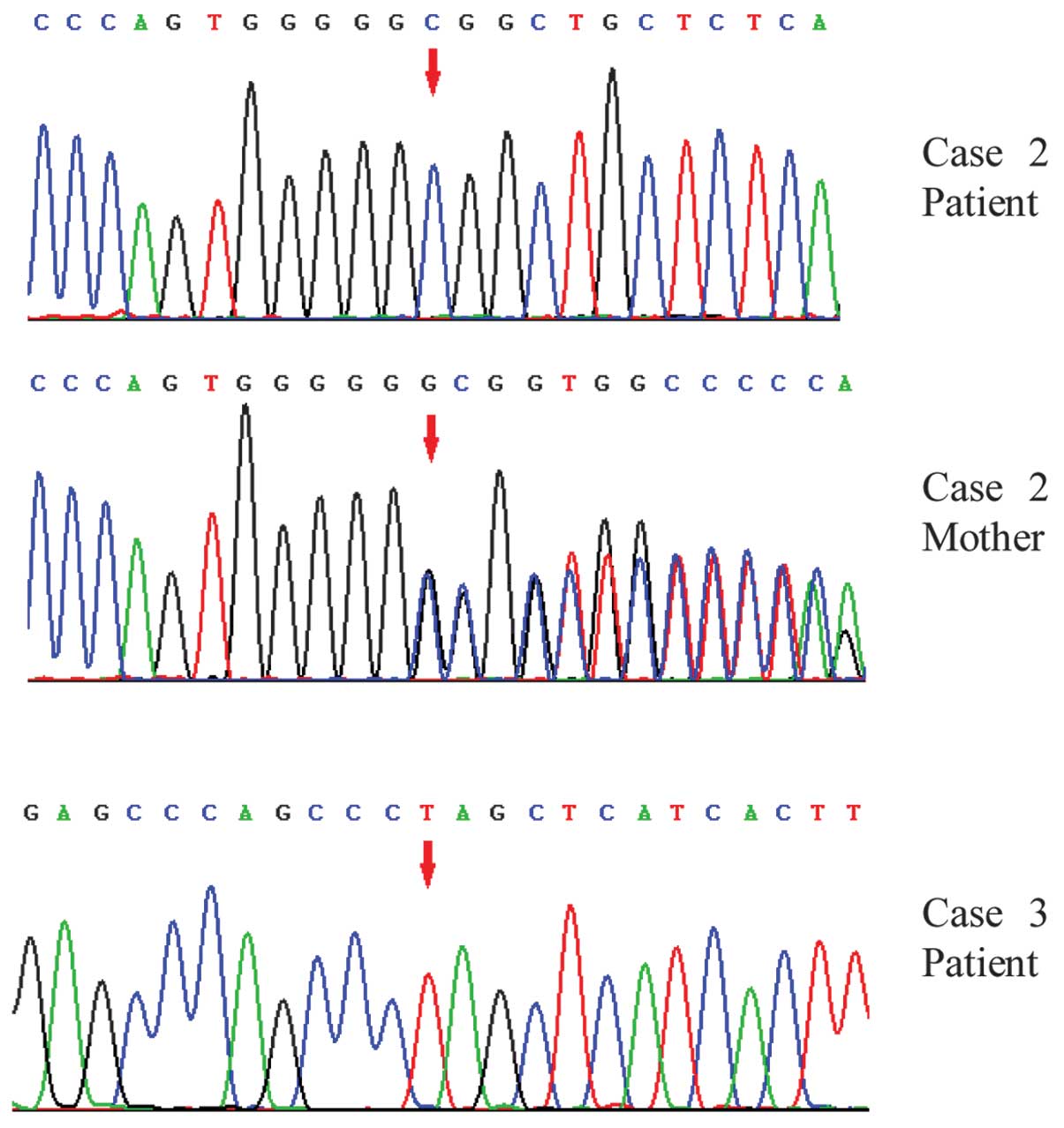
c.771delG mutation in exon 8 of the AAAS gene (6). The parents refused follow-up care for
this patient, due to ‘familial reasons’.
Patient 2
A male patient, aged 2 years and 7 months, was
admitted to the hospital on March 2, 2013 with a 1-year history of
vomiting after either overeating or rapid eating. No apparent
causes or obvious weight loss had been observed.
Six months prior to admission, the parents noted
hyperpigmentation in the lips, photophobia and frequent blinking
following birth. The patient was treated for dry-eye syndrome due
to alacrima. There was no documented family history of adrenal
gland disease. The parents did not have a consanguineous
relationship and were in good health. The patient was G1P1 and the
birth weight was recorded at 3.45 kg; the intellectual and physical
development of the child was determined to be normal, as compared
with children of the same age.
The findings of the physical examination were as
follows: BP, 86/50 mmHg; height, 92 cm; weight, 13 kg. The heart,
lungs, abdomen, nervous system and external genitalia of the
patient were found to be normal. Hyperpigmentation was observed
over most of the body. Esophagography showed achalasia of the lower
esophagus at the cardia. The patient was unable to produce tears
when crying but refused to undergo the Schirmer test; therefore, an
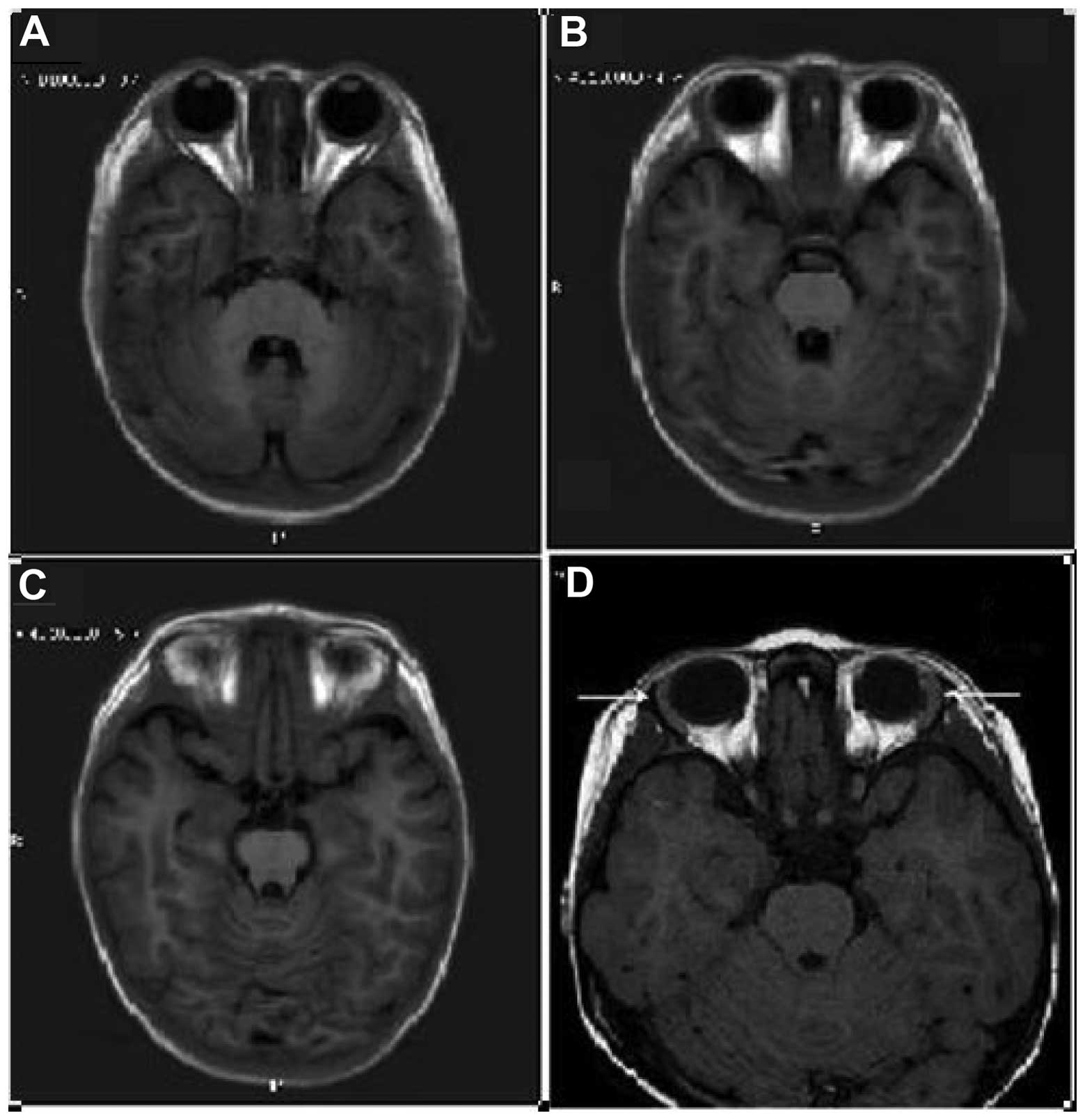
MRI scan (Fig. 2) was performed in
order to confirm lacrimal gland hypoplasia and diagnose alacrima.
The clinical diagnosis was AS. Genetic testing confirmed the
presence of a c.771delG mutation in exon 8 of the AAAS gene.
(Fig. 3). On day 3 after admission,
the patient had a hypoglycemic episode and convulsion occurred due
to an adrenal crisis. In response, the patient was treated with 100
mg/m2 hydrocortisone; the ACTH level decreased from
>1,250 to 6.8 pg/ml (normal range, 0–46 pg/ml), and the
hydrocortisone dosage was reduced to 20 mg/m2. Although
the vomiting continued, without relationship with overeating, the
patient was able to eat every day. The growth and nutritional
status of the patient was found to be normal after 18 months of
follow-up care.
Patient 3
A female patient, aged 4 years and 4 months, was
admitted to the hospital with a 3-year history of vomiting small
amounts following every meal. No obvious causes of the vomiting
were identified. Approximately 2 years prior to admission, the
patient exhibited hyperpigmentation of the lips, in addition to
fatigue, with a slow/stagnating increase in weight and height.
One month prior to admission, the patient had a
seizure and was diagnosed with secondary epilepsy, based on an
abnormal EEG. The patient was successfully treated with Topamax but
continued to vomit >10 times/day. The patient was G2P2 with
full-term normal delivery and a birth weight of 3.35 kg. The
intellectual and physical development of the patient was similar to
that of healthy children of the same age. There was no family
history of genetic diseases, and the parents were in a
non-consanguineous marriage.
The findings of the physical examination were as
follows: BP, 85/55 mmHg; height, 92.7 cm; weight, 12 kg. The
patient was mentally alert, and no significant anomalies were
observed in the heart, lungs, abdomen, nervous system or external
genitalia. A generalized hyperpigmentation was observed.
Esophagography showed achalasia of the lower esophagus at the
cardia. In the hospital, the patient was observed not to produce
tears when crying. The results of a bilateral Schirmer test were 0
mm, which led to the diagnosis of alacrima. Further medical history
confirmed that the alacrima had manifested at birth. Following the
diagnosis of AS, the patient was treated with hydrocortisone
replacement therapy (25 mg/m2), and the vomiting was
alleviated. Genetic testing confirmed a c.1366C>T mutation in
exon 15 of the AAAS gene (Fig. 3).
No additional follow-up information is available for this
patient.
Discussion
AS is a rare autosomal recessive congenital disease
characterized by adrenal insufficiency (Addison's disease), failure
of the lower esophageal sphincter to relax (achalasia) and an
absence of tear secretion (alacrima). AS does not appear to be
age-, ethnicity- or gender-specific (12), but varies widely in severity, with
some patients developing no symptomology and others suffering a
fatal outcome (13–15). Pediatric patients with AS often
present with the classic triad of symptoms, while patients with the
late-onset or adult-onset condition exhibit symptoms that involve
the nervous system (16). The
pathogenic gene for AS, AAAS, is located on chromosome 12q13 and
consists of 16 exons, which encode the protein ALADIN (17). A variety of mutations scattered
throughout the gene have been reported, with exon 3 being the
exception, as no AS-related mutations have yet been reported
therein. Of note, little correlation has been found among the
genotype, phenotype and variable clinical expression of family
members with AS (7,18).
In the present study, the early onset of the disease
was a common characteristic among all 3 cases recorded. All
patients had a history of excessive vomiting with no apparent
etiology, which is a common symptom of AS in pediatric patients.
Despite the fact that adrenal insufficiency was observed, all cases
were misdiagnosed for >1 year. The patients were found to be
unable to produce tears shortly after birth. Two of the patients
(cases 1 and 3) were confirmed to have alacrima based on the
results of the Schirmer test, while the other patient (case 2)
refused to undergo the test. An MRI scan was instead performed to
confirm lacrimal gland hypoplasia. Radiography of the upper
digestive tract indicated delayed opening of the lower esophageal
sphincter at the cardia, confirming the diagnosis of achalasia.
Examination of patient 1 revealed tendon hyperreflexia and optic
never atrophy, which indicated that this case of AS also involved a
neurological manifestation, more commonly found in adult patients.
Patients 2 and 3 did not exhibit any neurological symptoms. Vishnu
et al (19) suggested that
neurological symptoms may manifest in certain subgroups of patients
with a less severe and chronic course of the disease. Based on the
triad of adrenal insufficiency, achalasia and alacrima, all 3
patients described in the present study were diagnosed with AS.
Due to poor patient cooperation, patient 2 required
an MRI scan. The lacrimal glands are located in the orbital
lacrimal fossa, and their features on the T1- and T2-weighted MR
images are similar to those of the extraocular muscles and cerebral
gray matter density (20). Kim et
al (20) described two cases of
lacrimal gland agenesis in a single family. With regard to the
present cases, the Schirmer test (bilateral) detected <1 mm of
wetting in 5 min in patients 1 and 3, and the orbital MRI indicated
the absence of both lacrimal glands in patient 2. Sahinoglu et
al (21) performed lacrimal
gland biopsy through a left superotemporal extraperiosteal
approach. The histopathology showed no evidence of lacrimal gland
tissue and an orbital MRI scan revealed the absence of both
lacrimal glands. The fact that the study by Sahinoglu et al
indicated a correlation between the MRI and biopsy findings
provided the basis for the use of MRI to prove alacrima in the
patient who would not cooperate with the Schirmer test in the
present study.
AS is known to be caused by AAAS gene mutations that
encode abnormal ALADIN proteins. ALADIN proteins belong to the
tryptophan-aspartic acid-repeat-containing family of proteins
(14) and contribute to the nuclear
pore complex (NPC) (22). The ALADIN
protein anchors to the cytoplasmic side of the NPC, and the
mislocalization of a mutant ALADIN affects the exchange of nuclear
material (23). Among the 16 exons
of the AAAS gene, mutations have been identified in all except exon
3, which has a relatively shorter sequence (6,7). In
addition, pathogenic mutations have been reported in introns 4, 5,
11 and 14 (24). Mutations include
point, frame-shift and nonsense mutations, as well as DNA fragment
deletions. Although no specific recognition of prominent hot-spot
mutations has been found, there appears to be a relatively high
frequency of mutations at the following loci: Exon 1 (c.43C>A,
p.Q15K), exon 8 (c.787T>C, p.S263P) and intron 14 (IVS
14+1G>A) (6,7,24–27). The
mutations in exons 1 and 8 have been reported in >20 families
with no significant regional discrepancy (28–30). The
same deletion in exon 8, a region that has been associated with a
high frequency of mutations, was observed in patients 1 and 2. The
most commonly observed mutation in exon 8 (c.787T>C, p.S263P)
has been reported in cases predominantly from European countries
(16); however, the mutation in exon
8 (c.771delG) found in the present study has only been reported in
China, to date.
Among the mutations of the 3 cases reported in the
present study, two of the patients exhibited the same homozygous
point mutation and one exhibited a homozygous nonsense mutation.
The patients in cases 1 (8) and 2
came from two unrelated families and exhibited the same homozygous
point mutation, c.771delG, in exon 8, which caused a frame shift
that subsequently generated a stop codon after 32 amino acids,
resulting in a truncated protein, Arg258GlyfsX33, of 289 amino-acid
residues. In both cases, the parents of the patients were found to
carry heterozygous mutations. Case 3 had a homozygous c.1366C>T
mutation in exon 15, which has also been reported in the United
States in 2003 (31). This mutation
causes a dysfunctional ALADIN protein truncated at amino acid 456.
Residues 478–499 of the C-terminus of a normal ALADIN protein are
essential for its membrane-anchoring function (14). Including the cases of the present
study, 3 out of 4 AS cases reported in the Chinese Han population
have exhibited the c.771delG mutation, suggesting that it may be a
mutation specific to the Chinese (32).
AAAS, the pathogenic gene of AS, is widely expressed
in human tissues, and the clinical manifestations of the disease
are highly variable. There is currently no specific cure for the
disease, only treatments that relieve its symptoms. The detection
of AS through genetic analysis in pediatric patients that do not
exhibit all the symptoms is likely to provide an opportunity for
earlier diagnosis. Hormone replacement therapy to treat adrenal
insufficiency appears to be the most effective treatment in the
patients with AS. One case from the present study demonstrated
cortisone resistance and required a higher dose of hydrocortisone
therapy. Artificial tears can improve eye irritation, reduce eye
blink rate and prevent eye infections and corneal ulcers in
patients with alacrima. With regard to patients with achalasia,
surgical intervention is generally not advocated, since there are
less invasive and more effective treatment options, such as the
application of a balloon to dilate the lower esophageal sphincter.
If this treatment has no effect or fails, patients can be treated
surgically with a lower esophageal ring myotomy. No effective
treatments for the neuropathological symptoms of AS have been
found. Due to the fact that autonomic dysfunction can cause the
most severe symptoms, a neuropsychological and psychopathological
assessment of patients with this syndrome is advised. Although
genetic analysis of the AAAS gene in AS patients at a young age may
facilitate an earlier disease diagnosis and comprehensive
treatment, the problems in neurological and gastrointestinal
systems require further investigation in order to facilitate the
development of more effective novel treatments.
Acknowledgements
The authors would like to thank the parents of the
patients that participated in the present study for cooperating
with the genetic analyses.
References
|
1
|
Allgrove J, Clayden GS, Grant DB and
Macaulay JC: Familial glucocorticoid deficiency with achalasia of
the cardia and deficient tear production. Lancet. 1:1284–1286.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gazarian M, Cowell CT, Bonney M and Grigor
WG: The ‘4A’ syndrome: Adrenocortical insufficiency associated with
achalasia, alacrima, autonomic and other neurological
abnormalities. Eur J Pediatr. 154:18–23. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sandrini F, Farmakidis C, Kirschner LS, Wu
SM, TullioPelet A, Lyonnet S, Metzger DL, Bourdony CJ, Tiosano D,
Chan WY, et al: Spectrum of mutations of the AAAS gene in Allgrove
syndrome: Lack of mutations in six kindreds with isolated
resistance to corticotropin. J Clin Endocrinol Metab. 86:5433–5437.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bizzarri C, Benevento D, Terzi C, Huebner
A and Cappa M: Triple A (Allgrove) syndrome: An unusual association
with syringomyelia. Ital J Pediatr. 39:392013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazzone L, Postorino V, De Peppo L,
Vassena L, Fatta L, Armando M, Scirè G, Cappa M and Vicari S:
Longitudinal neuropsychological profile in a patient with triple A
syndrome. Case Rep Pediatr. 2013:6049212013.PubMed/NCBI
|
|
6
|
Papageorgiou L, Mimidis K, Katsani KR and
Fakis G: The genetic basis of triple A (Allgrove) syndrome in a
Greek family. Gene. 512:505–509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huebner A, Kaindl AM, Knobeloch KP,
Petzold H, Mann P and Koehler K: The triple A syndrome is due to
mutations in ALADIN, a novel member of the nuclear pore complex.
Endocr Res. 30:891–899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gong CX, Wen YR, Zhao XL, Su C, Cao BY and
Zhang X: Allgrove syndrome in the mainland of China: Clinical
report and mutation analysis. Zhonghua Er Ke Za Zhi. 45:422–425.
2007.(In Chinese). PubMed/NCBI
|
|
9
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dumić M, Barišić N, Rojnić-Putarek N,
Kušec V, Stanimirović A, Koehler K and Huebner A: Two siblings with
triple A syndrome and novel mutation presenting as hereditary
polyneuropathy. Eur J Pediatr. 170:393–396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marshall WA and Tanner JM: Variations in
pattern of pubertal changes in girls. Arch Dis Child. 44:291–303.
1969. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakamura K, Yoshida K, Yoshinaga T,
Kodaira M, Shimojima Y, Takei Y, Morita H, Kayanuma K and Ikeda S:
Adult or late-onset triple A syndrome: Case report and literature
review. J Neurol Sci. 297:85–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ismail EA, TulliotPelet A, Mohsen AM and
Al-Saleh Q: Allgrove syndrome with features of familial
dysautonomia: A novel mutation in the AAAS gene. Acta Paediatr.
95:1140–1143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cronshaw JM and Matunis MJ: The nuclear
pore complex protein ALADIN is mislocalized in triple A syndrome.
Proc Natl Acad Sci USA. 100:5823–5827. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Toromanovic A, Tahirovic H, Milenkovic T,
Koehler K, Kind B, Zdravkovic D, Hasanhodzic M and Huebner A:
Clinical and molecular genetic findings in a 6-year-old Bosnian boy
with triple A syndrome. Eur J Pediatr. 168:317–320. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dumic M, Barišic N, Kusec V, Stingl K,
Skegro M, Stanimirovic A, Koehler K and Huebner A: Long-term
clinical follow-up and molecular genetic findings in eight patients
with triple A syndrome. Eur J Pediatr. 171:1453–1459. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weber A, Wienker TF, Jung M, Easton D,
Dean HJ, Heinrichs C, Reis A and Clark AJ: Linkage of the gene for
the triple A syndrome to chromosome 12q13 near the type II keratin
gene cluster. Hum Mol Genet. 5:2061–2066. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luigetti M, Pizzuti A, Bartoletti S,
Houlden H, Pirro C, Bottillo I, Madia F, Conte A, Tonali PA and
Sabatelli M: Triple A syndrome: A novel compound heterozygous
mutation in the AAAS gene in an Italian patient without adrenal
insufficiency. J Neurol Sci. 290:150–152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vishnu VY, Modi M, Prabhakar S, Bhansali A
and Goyal MK: ‘A’ motor neuron disease. J Neurol Sci. 336:251–253.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SH, Hwang S, Kweon S, Kim TK and Oh J:
Two cases of lacrimal gland agenesis in the same
family-clinicoradiologic findings and management. Can J Ophthalmol.
40:502–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahinoglu N, Tuncer S, Alparslan N and
Peksayar G: Isolated form of congenital bilateral lacrimal gland
agenesis. Indian J Ophthalmol. 59:522–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu X, Mitchell JM, Wozniak RW, Blobel G
and Fan J: Structural evolution of the membrane-coating module of
the nuclear pore complex. Proc Natl Acad Sci USA. 109:16498–16503.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kind B, Koehler K, Lorenz M and Huebner A:
The nuclear pore complex protein ALADIN is anchored via NDC1 but
not via POM121 and GP210 in the nuclear envelope. Biochem Biophys
Res Commun. 390:205–210. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Babu K, Murthy KR, Babu N and Ramesh S:
Triple A syndrome with ophthalmic manifestations in two siblings.
Indian J Ophthalmol. 55:304–306. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Palka C, Giuliani R, Brancati F, Mohn A,
Di Muzio A, Calabrese O, Huebner A, De Grandis D, Chiarelli F,
Ferlini A and Stuppia L: Two Italian patients with novel AAAS gene
mutation expand allelic and phenotypic spectrum of triple A
(Allgrove) syndrome. Clin Genet. 77:298–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brooks BP, Kleta R, Stuart C, Tuchman M,
Jeong A, Stergiopoulos SG, Bei T, Bjornson B, Russell L, Chanoine
JP, et al: Genotypic heterogeneity and clinical phenotype in triple
A syndrome: A review of the NIH experience 2000–2005. Clin Genet.
68:215–221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brooks BP, Kleta R, Caruso RC, Stuart C,
Ludlow J and Stratakis CA: Triple-A syndrome with prominent
ophthalmic features and a novel mutation in the AAAS gene: A case
report. BMC Ophthalmol. 4:72004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Handschug K, Sperling S, Yoon SJ, Hennig
S, Clark AJ and Huebner A: Triple A syndrome is caused by mutations
in AAAS, a new WD-repeat protein gene. Hum Mol Genet. 10:283–290.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Houlden H, Smith S, De Carvalho M, Blake
J, Mathias C, Wood NW and Reilly MM: Clinical and genetic
characterization of families with triple A (Allgrove) syndrome.
Brain. 125:2681–2690. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prpic I, Huebner A, Persic M, Handschug K
and Pavletic M: Triple A syndrome: Genotype-phenotype assessment.
Clin Genet. 63:415–417. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
ReshmiSkarja S, Huebner A, Handschug K,
Finegold DN, Clark AJ and Gollin SM: Chromosomal fragility in
patients with triple A syndrome. Am J Med Genet A. 117A:30–36.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lam YY, Lo IF, Shek CC, Tong TM, Ng DK,
Tong TF, Choi MS, Lam ST and Ho CS: Triple-A syndrome - The first
Chinese patient with novel mutations in the AAAS gene. J Pediatr
Endocrinol Metab. 19:765–770. 2006. View Article : Google Scholar : PubMed/NCBI
|