Introduction
Familial adenomatous polyposis (FAP; MIM 175100) is
a rare autosomal dominant disorder, which is characterized by the
development of numerous adenomatous polyps throughout the colon and
rectum (1). It is a pre-cancerous
disease, which develops into colorectal cancer (CRC) in almost all
patients without early diagnosis and colorectal surgery (2). FAP may have extracolonic
manifestations, including osteomas, dental abnormalities,
congenital hypertrophy of the retinal pigment epithelium (CHRPE)
and upper gastrointestinal polyps (3). The incidence of FAP at birth is
estimated to be 3–10 per 100,000 individuals (4).
FAP has three phenotypes: Classic FAP (CFAP),
attenuated FAP (AFAP) and MUTYH-associated polyposis (MAP)
(5), with CFAP and AFAP being
autosomal dominant disorders. It has been identified that the
adenomatous polyposis coli (APC) gene on chromosome 5q22.2
is associated with CFAP and AFAP (6). MAP is a recessive dominant disorder
caused by mutations in the MUTYH gene (7). In the present study, mutations of the
APC gene were detected in a Chinese family with CFAP by
sequencing analysis, and a nonsense mutation was identified.
Materials and methods
Patients
A 40 year-old Chinese male patient was seen at the
Department of Emergency of the Second People's Hospital of Heifei
(Hefei, China) in August 2011, due to experiencing hematochezia for
1 day. In the past year, he had frequently suffered from moderate
diarrhea with scurrying pain around the umbilicus. His medical
history was not indicative of colitis and hemorrhoids. The patient
was a non-smoker and drank alcohol socially. Colonoscopy findings
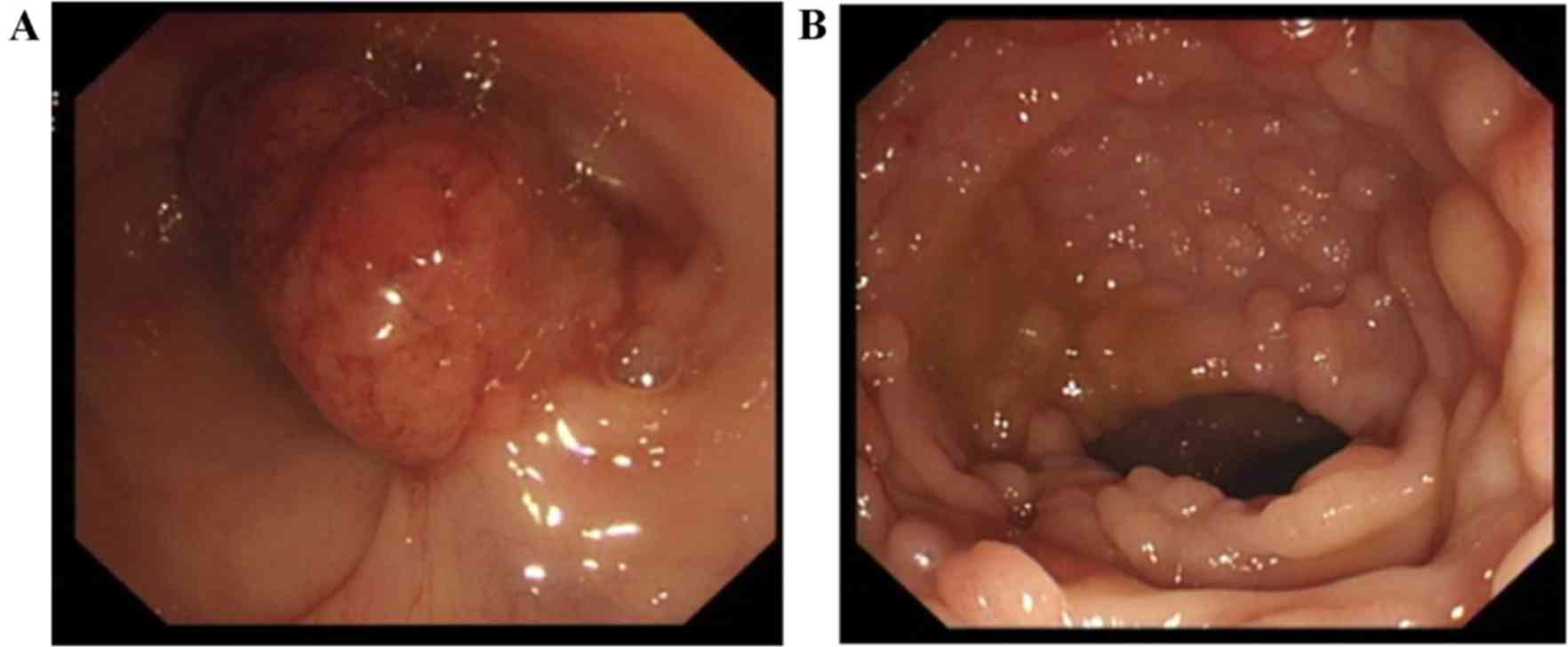
revealed a huge neoplasm with surface erosion and bleeding 35–38 cm
away from the anus (Fig. 1A), as
well as congestion, edema and diffused polyps with diameters of
0.3–0.8 cm from the ascending colon to the rectum mucosa (Fig. 1B). The primary diagnosis was FAP.
Subsequently, the patient successfully underwent laparoscopic total
colectomy and ileal anal anastomosis. Postoperative recovery was
good. Pathological findings revealed an abundance of multiple
tubular papillary adenoma with low-level intraepithelial neoplasia
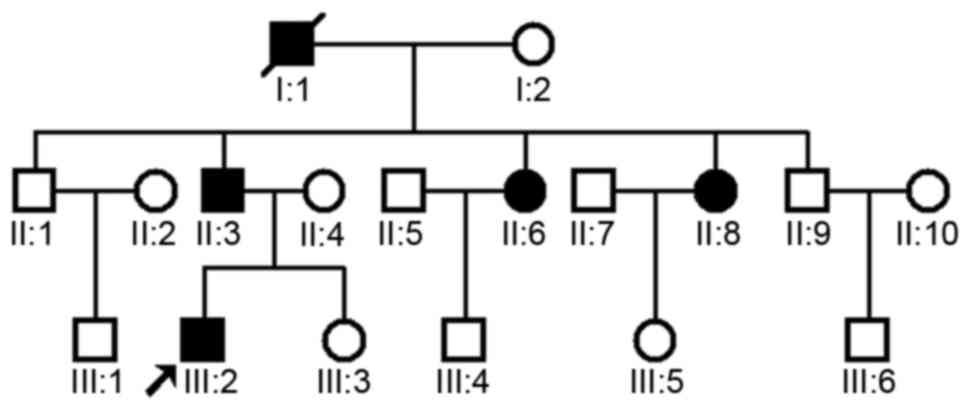
throughout the entire colon. The family comprised 18 members that
spanned three generations, including 3 male (one of which was the
proband of the present study) and 2 female individuals affected by
FAP (Fig. 2). A similar disease
course and abnormalities were found in these patients.
Mutational analysis
The protocol of the present study was approved by
the Ethics Committee of the Second People's Hospital of Heifei
(Hefei, China) and Zhongshan Hospital (Shanghai, China) and all
patients provided written informed consent to be included in the
present study. Peripheral blood samples were obtained from the four
living patients (II:3, II:6, II:8 and the proband III:2; Fig. 2). In addition, samples from 100
unrelated population-matched controls from Zhongshan Hospital were
sequenced for mutations to exclude the possibility that it is a
polymorphism in the APC gene. DNA was extracted according to
standard methods. Primers flanking all 15 coding exons and
intron-exon boundaries of the APC gene were extracted using
the web-based version of the Primer 3.0 program (http://primer3.ut.ee/). The primers used are listed in
Table I. The APC gene of this
family was analyzed by direct sequencing in reaction conditions as
previously described (8). Subsequent
to amplification, a QIAquick PCR Purification kit (Qiagen, Hilden,
Germany) was used to purify the products. The APC gene was
sequenced using an ABI PRISM® 3730 automated sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Sequence comparisons and analysis were performed using
Phred-Phrap-Consed version 12.0 software (http://www.phrap.org/phredphrapconsed.html). Mutations
were identified by comparison with the reported complementary DNA
reference sequence (GenBank accession no, NM_000038).
 | Table I.The primers sequences of APC gene. |
Table I.
The primers sequences of APC gene.
| Name | Sequence (5′-3′) | Product length
(bp) |
|---|
| APC-E02_F |
CTCTTAGATGCTGCTACTTGA | 800 |
| APC-E02_R |
GGATAGAACCAGGTACTGAC |
| APC-E03_F |
ACAGAGACTCCCCATAATCA | 587 |
| APC-E03_R |
GACTGGCAGAATAGCAACAA |
| APC-E04_F |
GTTGCTTGAAAATTCCAGTG | 642 |
| APC-E04_R |
GCTCTAAGTGTTAGCTATCAC |
| APC-E05_F |
AGCCTTTGGTGAAGTGTAAG | 640 |
| APC-E05_R |
TTGAACCCTGAGGTCCTCTA |
| APC-E06_F |
TAACCTCACTCTAACTGGAC | 676 |
| APC-E06_R |
GAAGACCACCATCTAACTCT |
| APC-E07_F |
TGATTTGACATAACCCTGAGC | 604 |
| APC-E07_R |
ACCTTCCCTGGTCTTAATGC |
| APC-E08_F |
GGATGGCATTCCTGTGAGTC | 703 |
| APC-E08_R |
GCAAACCTATTCAAGGCAAGC |
| APC-E09_F |
CTGCAGTTTAATGCTCATATGC | 377 |
| APC-E09_R |
GCAAAGTAGTCATGGCATTAGT |
| APC-E10_F |
CAGTTTGTTAGTGAGTATGC | 860 |
| APC-E10_R |
GCACATAACATTTTCCTTTG |
| APC-E11_F |
ACTTAGTCAAGGGCAGATGA | 468 |
| APC-E11_R |
GCTGATAACAGAAGTTGGTG |
| APC-E12_F |
GGAGAAACTGGCATAAAATGG | 578 |
| APC-E12_R |
TCACTACTGTGTTCCATCTG |
| APC-E13_F |
ACTTGTAGGGATCATTTCTGTG | 599 |
| APC-E13_R |
ATTGCACAACTGCCCTCTAA |
| APC-E14_F |
CAGTAACCTCAAGCTCCTGG | 828 |
| APC-E14_R |
CGAGACCAGCCTTACCAACA |
| APC-E15_F |
AAGTTCTTAATTTACCAGTG | 486 |
| APC-E15_R |
GTAGTTATCTTTTCACAGTA |
| APC-E16-1_F |
ATTGGGTCAGAATAGGAAATG | 890 |
| APC-E16-1_R |
TCTGTTGCTGGATGGTAGTT |
| APC-E16-2_F |
GTCCCAAGGCATCTCATCGT | 667 |
| APC-E16-2_R |
GCTGGGTATTGACCATAACTGC |
| APC-E16-3_F |
ATAGTGTCAGTAGTAGTGATGG | 498 |
| APC-E16-3_R |
GACACAAAGACTGGCTTACA |
| APC-E16-4_F |
ATCGAGTGGGTTCTAATCATGG | 635 |
| APC-E16-4_R |
TGGAACTTCGCTCACAGGAT |
| APC-E16-5_F |
ATCCAAGTTCTGCACAGAGT | 739 |
| APC-E16-5_R |
CTCTGAACTGCAGCATTTAC |
| APC-E16-6_F |
GCTCAAACCAAGCGAGAAGT | 750 |
| APC-E16-6_R |
TCTGCCTTCTGTAGGAATGG |
| APC-E16-7_F |
TGCTGGAGAAGGAGTTAGAG | 701 |
| APC-E16-7_R |
GGTTGGAGGTTAGTTCTGTG |
| APC-E16-8_F |
GATGATGTTGACCTTTCCAG | 574 |
| APC-E16-8_R |
CATTATCACCCTTGAGTCTTG |
| APC-E16-9_F |
ATCAGGCTATGCTCCTAAATCA | 824 |
| APC-E16-9_R |
TTTCACAGATGGCTTGGCTC |
| APC-E16-10_F |
GATTCATATTCCAGGAGTTCG | 475 |
| APC-E16-10_R |
GGCATTCTTGGATAAACCTG |
| APC-E16-11_F |
TGAGCCAACAGAACCTTACC | 777 |
| APC-E16-11_R |
AGGAAACGGTCTGAGAAGTAC |
| APC-E16-12_F |
CTCTATTTCAGGAACCAAAC | 878 |
| APC-E16-12_R |
CCTCTAACAAGAATCAAACC |
Results
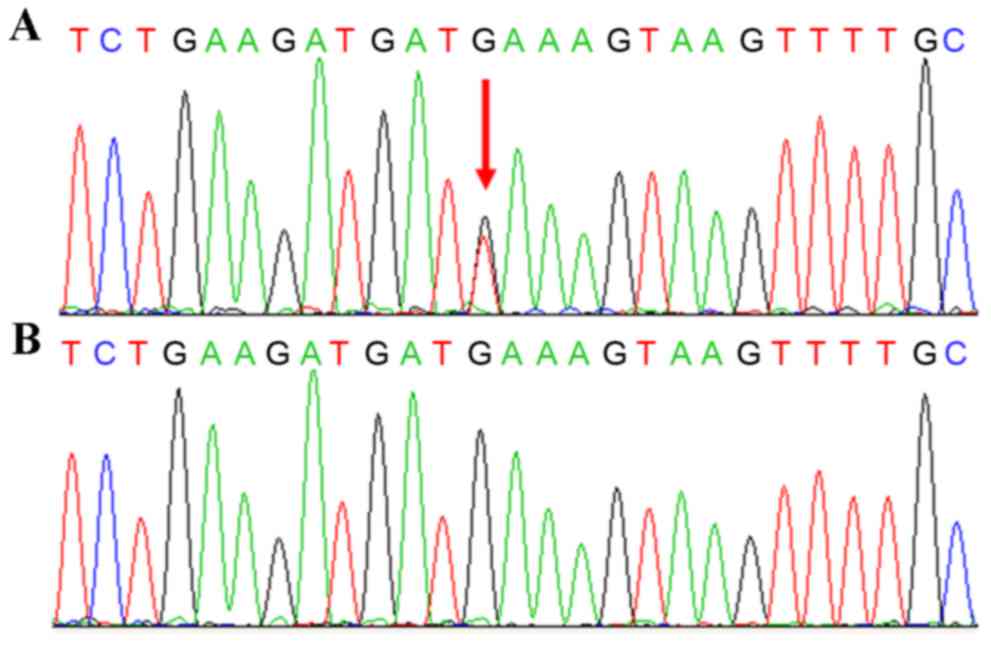
Sequencing results of the proband revealed a
nonsense mutation (c.2971G>T, p.Glu991*) located at exon 16 of
the APC gene (Fig. 3A). This
mutation was also verified in the other three patients, but
excluded in the unaffected family members and 100 unrelated
population-match controls (Fig. 3B).
This mutation forms a premature stop codon at amino acid residue
991, which results in a truncated protein short of 1,853 amino
acids. This mutation has already been reported a patient with
hereditary cancer-predisposing syndrome (https://www.ncbi.
nlm.nih.gov/clinvar/15587313/). The present study confirmed
this mutation in a Chinese family with CFAP. The result
demonstrates that this mutation may be a hotspot mutation in
diverse population.
Discussion
FAP is an autosomal dominant disease characterized
by the development of hundreds to thousands of adenomas in the
colon and rectum, and is at times accompanied with certain
extra-intestinal manifestations such as CHRPE, dental disorders and
desmoid tumors (9). APC is a
tumor suppressor gene located on the long arm of chromosome 5 in
band q21, whose mutation is responsible for CFAP and AFAP. The
length of the gene is 108,353 bp and it is divided into 15 exons
(10). The APC protein has multiple
domains that mediate oligomerization as well as binding to a
variety of intracellular proteins and has a central role in Wnt
signaling by regulating of degradation of proteins associated with
this pathway (11).
To date, according to the information available in
public databases, such as The Human Gene Mutation Database
(http://www.hgmd.cf.ac.uk/ac/index.php), >1,000
different APC mutations have been reported, among which
>100 cases were contributed by Chinese studies (12–17).
While the type of APC gene mutation varies, nonsense and
frameshift mutations are most frequently seen, and have been
predicted to produce truncated proteins, finally leading to the
development of diseases (17).
According to certain studies, most FAP patients
inherit one APC allele mutation from their parents with the
other allele being normal. Diseases would not occur until the
normal allele undergoes a new mutation (11). It has also been estimated that new
germline mutations of APC account for one third of FAP
patients who have no family history of FAP (18). Certain studies have attempted to
explore the correlation between specific APC mutations with
the clinical phenotype. Certain correlations do exist, for
instance, mutations between codons 169 and 1,578 were generally
associated with CFAP (19–21). Mutations downstream of codon 1,596
are frequently seen in AFAP (11).
Mutations between codons 1,445 and 1,578 were associated with
desmoid tumors, whereas those between codons 279 and 1,309 were
correlated with the development of duodenal polyposis (22–24).
While it appears promising to predict a patient's phenotype by the
mutation site of the APC gene, this was proven to not be
feasible in clinical practice. Considerable variability has been
found in the presentation of specific phenotypes in patients with
identical mutations (25). This
indicates that the phenotype is associated with more factors than
genetic mutations (25).
In the present study, the nonsense mutation
c.2971G>T (p.E991*) was identified in exon 15 of the APC
gene. The resulting truncated protein lacked 1,853 amino acids. The
wild-type sequence in the affected region of the APC gene is
highly evolutionarily conserved in different species, including
humans, mice, rats, frogs, zebrafish and pufferfish. Through
mutation-associated truncation, the APC protein loses its
microtubule binding domain, end binding-1 binding domain, β-catenin
degradation domain and β-catenin binding domain, which is likely to
affect the proliferation and differentiation status of cells and
eventually results in colorectal polyps and cancer (26,27). In
addition, this nonsense mutation may lead to nonsense-mediated
decay of APC transcripts. The mutation results in
haplo-insufficiency of APC, which leads to development of
diseases.
While evidence strongly links APC gene
mutations with FAP, the single factor is not sufficient to explain
the etiology of the disease. It is estimated that 10–30 percent of
patients with classical FAP do not have any detectable APC
mutation. A proportion of FAP patients have MAP, an autosomal
recessive polyposis syndrome caused by biallelic mutations in the
MUTYH gene. Therefore, it is recommended that patients who have a
recessive family history of FAP are evaluated for a MUTYH
mutation (28).
Surgery remains to be the only option to cure the
disease, although it remains debatable which surgical option is the
golden standard. However, given the substantial risk of rectal
cancer developing after colectomy and ileorectal anastomosis, most
experts recommend total proctocolectomy for typical FAP patients
with multiple rectal adenomas (29).
Diet and drugs have been shown to have a role in preventing cancer.
Caloric restriction or diet with olive oil, fruits and vegetables
significantly reduced the number of polyps in a mouse model of
multiple intestinal neoplasia with genetically manipulated APC
(30). Randomized trials have shown
that celecoxib causes regression of established adenomatous polyps
in individuals with FAP. In 2001, the US Food and Drug
Administration approved the use of celecoxib in patients with FAP
presenting with polyps (31). The
proband of the present study successfully underwent laparoscopic
total colectomy and ileal anal anastomosis and postoperative
recovery was good.
In conclusion, the present study identified a
mutation in the APC gene in a Chinese family with FAP. The
present study added novel variants to the knowledge of APC
mutations in FAP. Identification of novel mutations will be useful
to reveal the correlation between genotypes and phenotypes.
Acknowledgements
This study was supported by a grant from the
Shanghai Science and Technology Innovation Action Plan
(nano-science and technology projects; no. 12nm0501402). The
authors would like to thank all patients and control individuals
for their participation in this study.
References
|
1
|
Rhodes M and Bradburn DM: Overview of
screening and management of familial adenomatous polyposis. Gut.
33:125–131. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martellucci J, Civitelli S, Dhamo A and
Tanzini G: Familial colorectal cancer: A concept revisited.
Colorectal Dis. 11:133–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li FP, Thurber WA, Seddon J and Holmes GE:
Hepatoblastoma in families with polyposis coli. JAMA.
257:2475–2477. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gibbons DC, Sinha A, Phillips RK and Clark
SK: Colorectal cancer: No longer the issue in familial adenomatous
polyposis? Fam Cancer. 10:11–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aretz S: The differential diagnosis and
surveillance of hereditary gastrointestinal polyposis syndromes.
Dtsch Arztebl Int. 107:163–173. 2010.PubMed/NCBI
|
|
6
|
Snow AK, Tuohy TM, Sargent NR, Smith LJ,
Burt RW and Neklason DW: APC promoter 1B deletion in seven American
families with familial adenomatous polyposis. Clin Genet.
88:360–365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Inra JA, Steyerberg EW, Grover S,
McFarland A, Syngal S and Kastrinos F: Racial variation in
frequency and phenotypes of APC and MUTYH mutations in 6,169
individuals undergoing genetic testing. Genet Med. 17:815–821.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li M, Yang L, Li C, Jin C, Lai M, Zhang G,
Hu Y, Ji J and Yao Z: Mutational spectrum of the ADAR1 gene in
dyschromatosis symmetrica hereditaria. Arch Dermatol Res.
302:469–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Plawski A, Banasiewicz T, Borun P,
Kubaszewski L, Krokowicz P, Skrzypczak-Zielinska M and Lubinski J:
Familial adenomatous polyposis of the colon. Hered Cancer Clin
Pract. 11:152013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liao DX, Li B, Du XM, Yu JH, Chang H, Wu
ZQ, Hao HJ, Wang YX, Han WD, Cheng SJ and Luo CH: Two Chinese
pedigrees for adenomatous polyposis coli: New mutations at codon
1309 and predisposition to phenotypic variations. Fam Cancer.
13:361–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Half E, Bercovich D and Rozen P: Familial
adenomatous polyposis. Orphanet J Rare Dis. 4:222009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang C, Guo J, Chen H, Yao CJ, Zhuang DX,
Wang Y, Tang WJ, Ren G, Yao Y, Wu JS, et al: Gene mutation
profiling of primary glioblastoma through multiple tumor biopsy
guided by 1H-magnetic resonance spectroscopy. Int J Clin Exp
Pathol. 8:5327–5335. 2015.PubMed/NCBI
|
|
13
|
Chen QW, Zhang XM, Zhou JN, Zhou X, Ma GJ,
Zhu M, Zhang YY, Yu J, Feng JF and Chen SQ: Analysis of small
fragment deletions of the APC gene in Chinese patients with
familial adenomatous polyposis, a precancerous condition. Asian Pac
J Cancer Prev. 16:4915–4920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu T, Guo H, Zhao H, Wang L and Zhang Z:
Next-generation sequencing for molecular diagnosis of lung
adenocarcinoma specimens obtained by fine needle aspiration
cytology. Sci Rep. 5:113172015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen K, Xia G, Zhang C and Sun Y:
Correlation between smoking history and molecular pathways in
sporadic colorectal cancer: A meta-analysis. Int J Clin Exp Med.
8:3241–3257. 2015.PubMed/NCBI
|
|
16
|
Zhang Y, Lu G, Hu Q, Wang X, Li C, Mao Y
and Cui M: A de novo germline mutation of APC for inheritable colon
cancer in a Chinese family using multigene next generation
sequencing. Biochem Biophys Res Commun. 447:503–507. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song G, Yuan Y, Zheng F and Yang N: Novel
insertion mutation p. Asp610GlyfsX23 in APC gene causes familial
adenomatous polyposis in Chinese families. Gene. 516:204–208. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Queiroz Rossanese LB, De Lima Marson
FA, Ribeiro JD, Coy CS and Bertuzzo CS: APC germline mutations in
families with familial adenomatous polyposis. Oncol Rep.
30:2081–2088. 2013.PubMed/NCBI
|
|
19
|
Järvinen HJ and Peltomäki P: The complex
genotype-phenotype relationship in familial adenomatous polyposis.
Eur J Gastroenterol Hepatol. 16:5–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giardiello FM, Krush AJ, Petersen GM,
Booker SV, Kerr M, Tong LL and Hamilton SR: Phenotypic variability
of familial adenomatous polyposis in 11 unrelated families with
identical APC gene mutation. Gastroenterology. 106:1542–1547. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagase H, Miyoshi Y, Horii A, Aoki T,
Ogawa M, Utsunomiya J, Baba S, Sasazuki T and Nakamura Y:
Correlation between the location of germ-line mutations in the APC
gene and the number of colorectal polyps in familial adenomatous
polyposis patients. Cancer Res. 52:4055–4057. 1992.PubMed/NCBI
|
|
22
|
Caspari R, Olschwang S, Friedl W, Mandl M,
Boisson C, Böker T, Augustin A, Kadmon M, Möslein G, Thomas G, et
al: Familial adenomatous polyposis: Desmoids tumours and lack of
ophthalmic lesions (CHRPE) associated with APC mutations beyond
codon 1444. Hum Mol Genet. 4:337–340. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soravia C, Berk T, Madlensky L, Mitri A,
Cheng H, Gallinger S, Cohen Z and Bapat B: Genotype-phenotype
correlations in attenuated adenomatous polyposis coli. Am J Hum
Genet. 62:1290–1301. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Touriño R, Conde-Freire R,
Cabezas-Agrícola JM, Rodríguez-Aves T, López-Valladares MJ,
Otero-Cepeda JL and Capeans C: Value of the congenital hypertrophy
of the retinal pigment epithelium in the diagnosis of familial
adenomatous polyposis. Int Ophthalmol. 25:101–112. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeichner SB, Raj N, Cusnir M, Francavilla
M and Hirzel A: A de novo germline APC mutation (3927del5) in a
patient with familial adenomatous polyposis: Case report and
literature review. Clin Med Insights Oncol. 6:315–323. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grady WM and Markowitz SD: Hereditary
colon cancer genes. Methods Mol Biol. 222:59–83. 2003.PubMed/NCBI
|
|
27
|
Näthke I: APC at a glance. J Cell Sci.
117:4873–4875. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Russell AM, Zhang J, Luz J, Hutter P,
Chappuis PO, Berthod CR, Maillet P, Mueller H and Heinimann K:
Prevalence of MYH germline mutations in Swiss APC mutation-negative
polyposis patients. Int J Cancer. 118:1937–1940. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aziz O, Athanasiou T, Fazio VW, Nicholls
RJ, Darzi AW, Church J, Phillips RK and Tekkis PP: Meta-analysis of
observational studies of ileorectal versus ileal pouch-anal
anastomosis for familial adenomatous polyposis. Br J Surg.
93:407–417. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mai V, Colbert LH, Berrigan D, Perkins SN,
Pfeiffer R, Lavigne JA, Lanza E, Haines DC, Schatzkin A and
Hursting SD: Calorie restriction and diet composition modulate
spontaneous intestinal tumorigenesis in Apc (Min) mice through
different mechanisms. Cancer Res. 63:1752–1755. 2003.PubMed/NCBI
|
|
31
|
Steinbach G, Lynch PM, Phillips RK,
Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y,
Fujimura T, et al: The effect of celecoxib, a cyclooxygenase-2
inhibitor, in familial adenomatous polyposis. N Engl J Med.
342:1946–1952. 2000. View Article : Google Scholar : PubMed/NCBI
|