Introduction
Lung cancer is one of the leading causes of
cancer-associated mortalities worldwide, with ~1 million new cases
annually in terms of incidence and mortality (1–4). Lung
cancer accounted for 13% (1.6 million) of the total cases and 18%
(1.8 million) of mortalities in 2008 (1). Non-small-cell lung cancer (NSCLC)
accounts for ~80% of all lung cancer cases (5). The management of patients with NSCLC is
based on systemic chemotherapy; the standard treatment for patients
with advanced NSCLC includes platinum-based combination
chemotherapy, which has demonstrated modest but significant
improvements in survival rates over best supportive care (6). However, the survival rates remain low
(7). In addition, although
chemotherapy may prolong the survival of patients with advanced
disease, clinically significant adverse side effects, including
excessive toxicity, reduce its effectiveness (8). Therefore, novel agents are urgently
required for the treatment of this disease.
The epithelial-to-mesenchymal transition (EMT) has
an important role in numerous physiological and pathological
processes in the human body, including regulating the transcription
of genes involved in embryonic development (6), the inflammatory response (8), tissue regeneration (9), organ fibrosis (10,11), and
tumor invasion and metastasis. During EMT, epithelial cells loosen
and cell-cell adhesion is lost (12). Furthermore, EMT is characterized by
the downregulation of E-cadherin and cytokeratins, and the
upregulation of mesenchymal proteins, including vimentin,
fibronectin and N-cadherin (13,14). A
previous study demonstrated that activation of the epidermal growth
factor (EGF) receptor (EGFR) by EGF promoted EMT in hepatocellular
carcinoma cell lines by altering the expression levels and
molecular structures of EMT-associated markers, including the
E-cadherin/β-catenin complex (14).
AXL is a member of the receptor tyrosine kinase
family (9). Previous studies
demonstrated that downregulation of AXL expression in solid tumors
by RNA interference (RNAi) was able to decrease cell invasion and
proliferation, and increase the chemosensitivity of cells via the
activation of AKT and mitogen-activated protein kinase (MAPK)
pathways (10,11,13). In
addition, AXL was identified as an oncogene in human leukemia cells
(14–16), and the expression levels of AXL were
shown to be increased in >50% of NSCLC cell lines (17). Similarly, a previous study reported
that the expression levels of AXL were upregulated in 48.3% of lung
adenocarcinoma tissues, in which they were correlated with lymph
node metastasis and the stage of the disease (16). These reports highlight the clinical
importance of AXL. Targeting of AXL with RNAi or specific
monoclonal antibodies has previously been shown to inhibit the
proliferation of NSCLC cells and tumor cells in a mouse xenograft
model (18). A previous study on
NSCLC reported that increased activation of AXL was associated with
acquired resistance to EGFR-targeted therapy (19).
The present study aimed to investigate the role of
AXL in NSCLC, in particular the molecular mechanism underlying its
involvement in EMT. Reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) and western blot analysis demonstrated
that AXL was upregulated in NSCLC cells. Subsequently, inhibition
of AXL by RNAi was conducted in order to investigate the
effects of downregulation of AXL on cell viability and the
expression levels of EMT-associated genes.
Materials and methods
Agents
Doxorubicin, paclitaxel, vincristine, cisplatin and
3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT)
were from Sigma-Aldrich, Inc. (Shanghai, China). Dulbecco's
modified Eagle's medium (DMEM) and RPMI-1640 medium were from Gibco
(Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cell lines
The human H1299, A549 and PC9 NSCLC cell lines, and
normal lung cells (as the control), were purchased from the Cancer
Research Institute of China Medical University (Shenyang, China).
The cells (1×105 cells/ml) were cultured in DMEM
supplemented with 10% (m/v) fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin (Sangong Biotech, Shanghai, China) at 37°C in a 5%
CO2 atmosphere with stable humidity.
MTT assay
Cell viability was assessed using the MTT assay, as
described in a previous study (20).
Briefly, the cells were plated at a density of 3,000 cells/well
into 96-well plates. At the end of treatment, the supernatant was
removed using a pipette and 20 µl MTT and 270 ml fresh DMEM were
added to the supernatant. Following incubation for 4 h at 37°C, 120
µl dimethyl sulfoxide was placed in each well to dissolve the
tetrazolium crystals, after which the absorbance at 570 nm was
recorded using a Multi-Well Plate Reader (Tecan Group, Ltd.,
Männedorf, Switzerland). Each experiment was performed four times.
The results are presented as the percentage growth inhibition with
respect to the untreated cells.
RNAi procedure
Double-strained small interfering RNA (siRNA) probes
targeting the human AXL gene (GenBank accession no.
NM-021913) and the Twist gene (GenBank accession no.
NM_000474.3) were synthesized using the Silencer siRNA Construction
kit (Ambion; Thermo Fisher Scientific, Inc.), as described in a
previous study (21). Briefly,
single-stranded gene-specific sense and antisense RNA oligomers
were synthesized by in vitro transcription using T7 RNA
polymerase (Sangong Biotech). In order to promote annealing of the
siRNA, sense and antisense single-stranded RNA oligomers were mixed
and incubated at 37°C overnight, after which the siRNA was
ethanol-precipitated and resuspended in nuclease-free water. The
integrity of the siRNA was assessed by gel electrophoresis (Sangong
Biotech), and it was quantified of by measuring the absorbance at
260 nm. Subsequently, the cells were transfected with four pooled
siRNA duplexes (20 nM; Invitrogen; Thermo Fisher Scientific, Inc.)
using the TransIT-TKO® Transfection Reagent (Mirus Bio,
LLC, Madison, WI, USA), in order to target the endogenous
AXL and Twist genes. The cells were transfected twice
at 3-day intervals, and were then collected at 72 h following the
second transfection. Specific silencing of the target genes was
confirmed using qPCR and western blot analysis. A mock transfection
with the transfection reagent alone served as the control.
Flow cytometry (FCM) analysis
Log phase PC9 cells were collected at a final
concentration of 2×105 cells/ml, washed with
phosphate-buffered saline (PBS) and fixed with 70% ethanol. The
cells were centrifuged at 8,000 × g for 5 min at 4°C to
remove ethanol, washed with PBS, and stained with propidium iodide
(PI; Sangong Biotech) in the dark for 30 min prior to FCM analysis.
The FACSCalibur™ platform (BD Biosciences, Franklin Lakes, NJ, USA)
was used to detect the cell cycle distribution. The cells were
sampled using the CellQuest Pro software, version 3.0 (BD
Biosciences), and the proportion of cells in the various stages of
the cell cycle were quantified using ModFit LT 3.0 (Verity Software
House, Inc., Topsham, ME, USA) (22). Each experiment was performed four
times.
Evaluation of apoptosis
PC9 cells (1×106) were collected by
centrifugation at 8,000 × g for 5 min at 4°C. The cell
pellets were lysed using DNA lysis buffer (10 mM Tris, pH 7.5, 400
mM EDTA and 1% Triton X-100), followed by centrifugation at 6,000 ×
g for 8 min at 4°C. The supernatant was incubated overnight
with proteinase K (0.1 mg/ml; Sangong Biotech), then with RNase
(0.2 mg/ml; Sangong Biotech) for 2 h at 37°C. DNA was extracted
using phenol:chloroform (1:1), separated by 2% agarose gel
electrophoresis (Sangong Biotech), and visualized by ultraviolet
illumination after staining with 10% ethidium bromide and washing
twice with water. Quantitative assessment of apoptotic cells was
conducted using the terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) method (BD ApoAlert DNA Fragmentation Assay
kit; cat. no. 630107; BD Biosciences), which examines DNA-strand
breaks during apoptosis.
RT-qPCR
RNA extraction from the cells was carried out using
TRIzol reagent (Life Technologies; Thermo Fisher Scientific, Inc.)
according to the manufacturer's recommendations. Genomic DNA was
eliminated and RT conducted using the PrimeScript RT Reagent kit
with gDNA Eraser, and qPCR was conducted using the SYBR Green PCR
kit (both Takara Biotechnology Co., Ltd., Dalian, China) according
to the manufacturer's recommendations. The process was conducted
using a 7500 Real-Time PCR System (Applied Biosystems; Thermo
Fisher Scientific, Inc. The primers used in the PCR were as
follows: AXL forward, 5′-TCGCCAGTGGCATGGAGTATCTG-3′ and
reverse, 5′-GGCGATACGTCCCTGGCGGTA-3′; Twist forward,
5′-CATCCACACCGTCCCCTCCC-3′ and reverse,
5′-GACTGGCGAGCTGGACACGTC-3′; and β-actin forward,
5′-TGAAGTACCCCATCGAGCAC-3′ and reverse, 5′-CTTGGGGTTCAGGGGGGCCT-3′
(GenBank accession no. BC004251.1). β-actin was used as an
internal reference.
The reaction mix consisted of 4 µl cDNA, 0.5 µl
forward and reverse primer mix (20 µM of each), 1 µl 50X ROX
Reference Dye II and 25 µl 2X SYBR Green PCR mix in a final volume
of 50 µl. All reactions were setup in triplicate and every sample
was replicated in parallel three times to ensure statistical
relevance. A negative control without cDNA template, and RT control
without RT transcription were included. The following thermal
conditions were used for all PCR reactions: 30 sec at 95°C,
followed by 40 cycles of 30 sec at 95°C and 34 sec at 60°C. Primer
specificity was confirmed by RT-PCR amplification prior to qPCR,
which generated single amplicons of the expected size for each
primer set. Furthermore, the amplicons were sequenced by a
commercial sequencing service (BGI, Shenzhen, China) in order to
validate their specific amplification, and the specificity of qPCR
was confirmed by the presence of dissociation curves with single
peaks and the sequencing of its products with unique bands of the
expected size. Amplicon dissociation curves were obtained after
cycle 40 using default settings suggested by the instrument. Data
were analyzed using the SDS software (version 2.0.6; Applied
Biosystems; Thermo Fisher Scientific, Inc.). All quantifications
were normalized against the quantity of β-actin using the
2−ΔΔCq method (23).
Western blot analysis
Cell lysates were prepared in
radioimmunoprecipitation assay buffer [50 mM Tris-HCl buffer, pH
7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1%
sodium dodecyl sulfate (SDS)], supplemented with 1X Halt protease
inhibitor and 1X Halt phosphatase inhibitor cocktails (Pierce
Biotechnology, Inc., Rockford, IL, USA). The Bio-Rad protein assay
(cat. no. 500-0006; Bio-Rad Laboratories, Inc., Hercules, CA, USA)
was used to determine protein concentrations. Proteins (100 ng)
were separated by 10–12% SDS-polyacrylamide gel electrophoresis and
transferred to polyvinylidene difluoride (PVDF) membranes (GE
Healthcare Life Sciences, Chalfont, UK). The membranes were blocked
for 1 h at room temperature using bovine serum albumin (BSA;
Sangong Biotech). Subsequently, the membranes were incubated with
specific primary antibodies (1:100 dilution) targeting Axl (cat.
no. 4566), Twist (cat. no. 46702) and β-actin (cat. no. 4790; all
from Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C
overnight. After washing the membrane three times with TBST (5 min
per wash), it was incubated with the recommended dilution (1:100)
of horseradish peroxidase-conjugated secondary antibodies (cat. no.
7071; Cell Signaling Technology, Inc.) at room temperature for 1 h,
and then washed as described above. The protein bands were
visualized using the Immobilon Western Chemiluminescent HRP
Substrate (EMD Millipore, Billerica, MA, USA). The
chemiluminescence of proteins transferred to the PVDF membranes was
detected using the ECL Plus Western Blot analysis Reagent (GE
Healthcare Life Sciences). Relative protein expression levels were
semi-quantified by densitometry using ImageJ software (version
2.1.4.7; National Institutes of Health, Bethesda, MA, USA).
Statistical analysis
Data were analyzed using SPSS statistical software,
version 16.0 (SPSS, Inc., Chicago, IL, USA). The results are
expressed as the mean ± standard deviation. The mean was compared
using the Student's t-test or by analysis of variance. The
statistical significance of the studies was determined using the
parametric unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
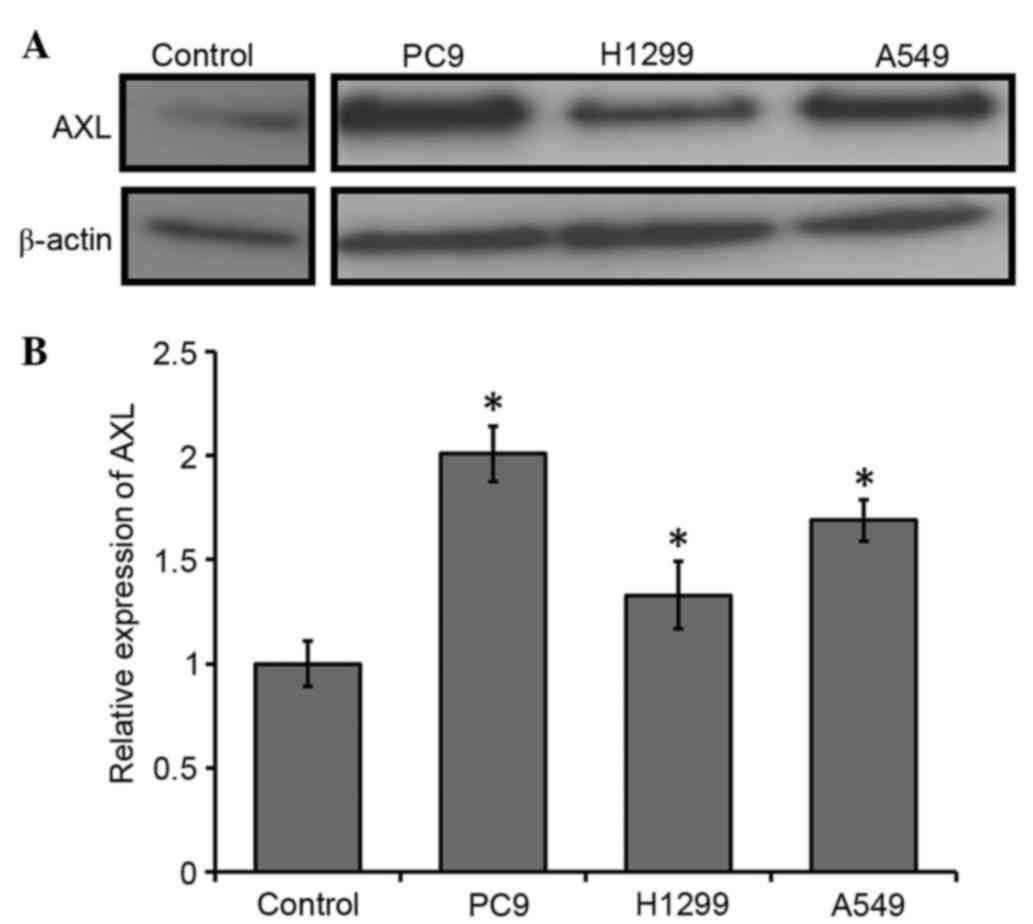
AXL is upregulated in NSCLC cells
To evaluate the expression of AXL in the NSCLC cell
lines, RT-qPCR and western blot analysis were conducted. AXL
protein expression levels were markedly upregulated in the NSCLC
cell lines compared with the control (Fig. 1A). Consistent with this, the mRNA
expression levels of AXL were significantly upregulated in
the NSCLC cells, as compared with the control (P<0.05; Fig. 1B). PC9 exhibited the highest level of
AXL transcripts (~2-fold upregulation), as compared with the
control; the AXL transcripts were ~1.7-fold upregulated in
the A549 cells and 1.3-fold upregulated in the H1299 cells, as
compared with the control (Fig. 1B).
These results suggest that AXL is upregulated in NSCLC cells.
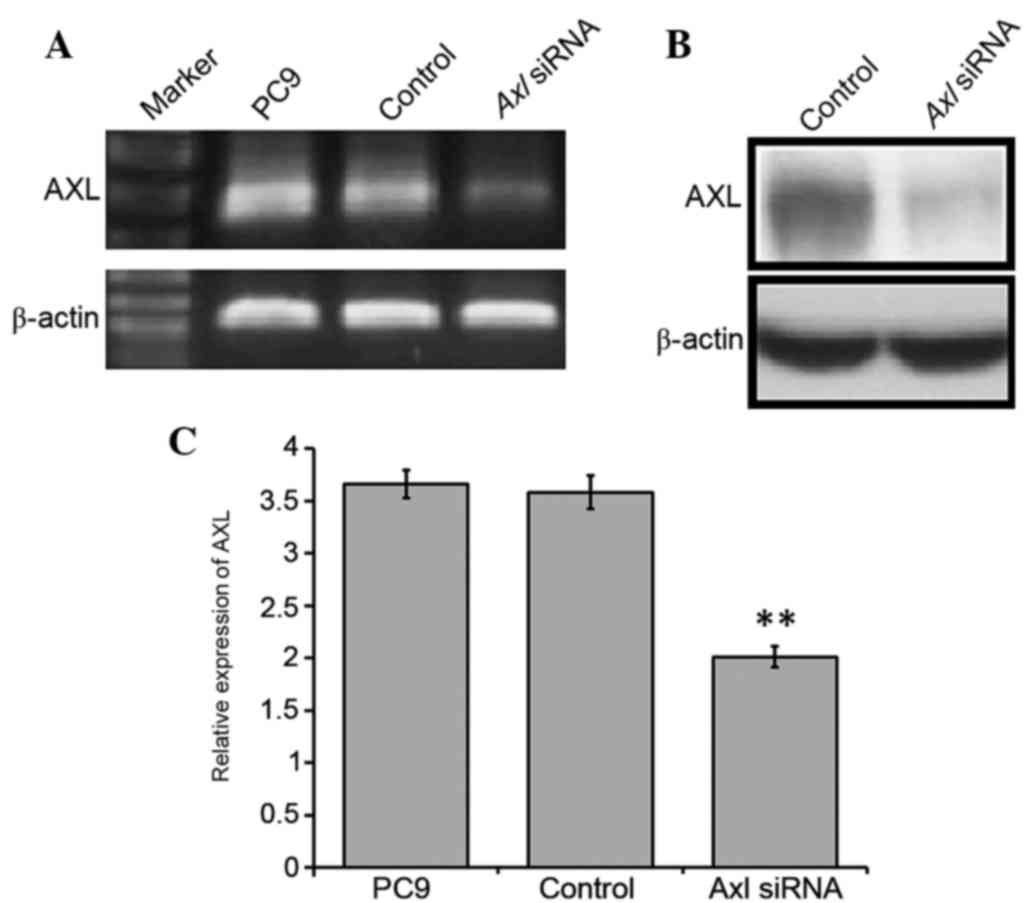
AXL promotes NSCLC cell survival
To characterize the role of AXL in NSCLC cells, a
gene-silencing experiment using double-stranded siRNA to silence
AXL in the PC9 cells was conducted. PC9 cells were selected
for further experimentation, as the greatest upregulation of
AXL expression levels was observed in this cell line. PC9
cells were transfected with the siRNA and, after 3–7 days, RNA was
extracted from control and transfected cell lines. A marked
reduction was detected in the mRNA and protein expression levels of
AXL in the siRNA-transfected cells, as compared with the control
cells (Fig. 2A and B), and this was
shown to be significant using RT-qPCR (P<0.01; Fig. 2C). In particular, ~1.83-fold
downregulation of AXL was detected in the transfected cells,
as compared with the control (Fig.
2). These results suggested that knockdown of AXL expression in
the PC9 cells was successful.
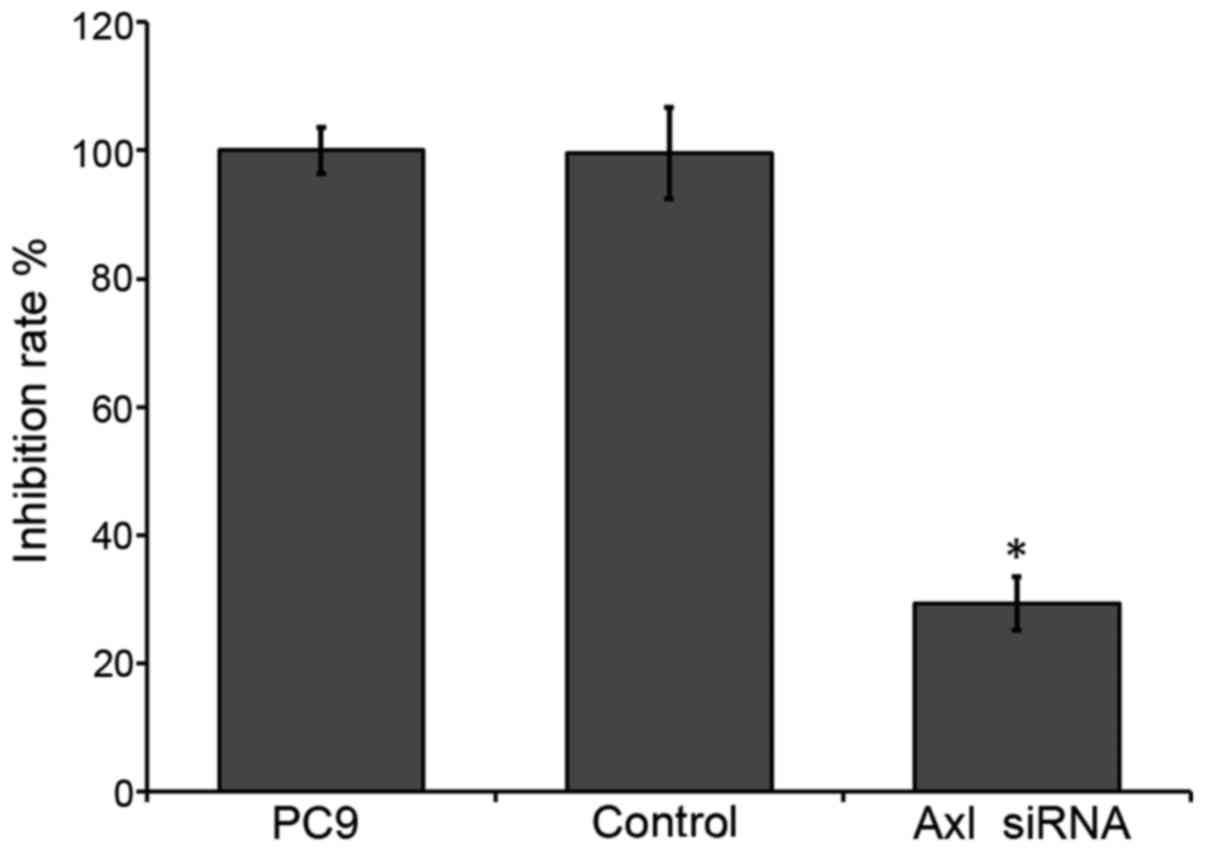
The viability of the PC9 cells was analyzed using
the MTT assay. Downregulation of endogenous AXL in PC9 cells
significantly reduced cell survival (~29.3%), as compared with the
control (P<0.05; Fig. 3). These
results suggest that inhibition of AXL by RNAi may reduce the
survival of NSCLC cells.
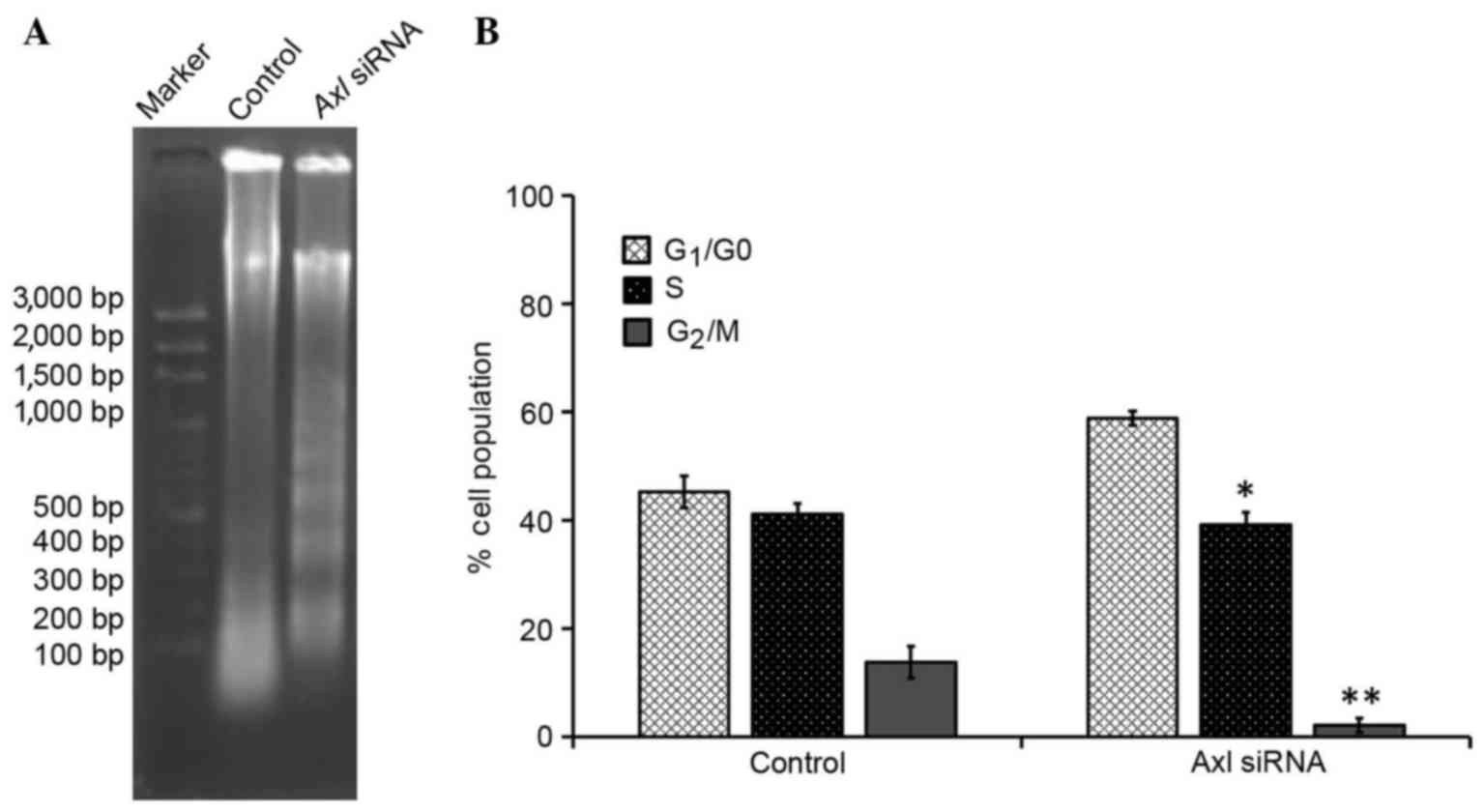
AXL is involved in NSCLC cell apoptosis. To further
investigate the involvement of AXL in the inhibition of cell
proliferation, cell-cycle distribution was evaluated using FCM
analysis. As compared with the control group, the number of cells
in the G1/G0 phase was significantly
increased (P<0.05), and the number of cells in the S and
G2/M phases was significantly decreased (P<0.05), in
the PC9 cells treated with AXL siRNA; the majority of cells were in
the G1 phase (Table I).
Furthermore, the proportion of diploid cells slightly increased,
with a reduction in the number of aneuploid cells, and the cell
debris was markedly increased in the PC9 cells treated with AXL
siRNA, as compared with the control group.
 | Table I.Differences in cell cycle induced by
downregulated AXL. |
Table I.
Differences in cell cycle induced by
downregulated AXL.
| Group | Cell debris
(%) | Apoptosis (%) | Diploid (%) | Aneuploid (%) |
|---|
| Control | 1.88±0.2167 | 0.0267±0.0252 | 94.7867±4.5027 | 5.2133±4.5027 |
| Axl siRNA | 46.9533±5.0537 | 0.06934±0.0723 | 99.9101±0.1558 | 0.0901±0.1558 |
The effect of downregulating AXL expression on the
induction of apoptosis in PC9 cells was investigated using a DNA
fragmentation assay. Agarose gel electrophoresis demonstrated that
knockdown of AXL resulted in the formation of DNA fragments
in PC9 cells (Fig. 4A). In addition,
a quantitative evaluation was conducted using TUNEL to detect
DNA-strand breaks. As compared to with the control cells, 30.7% of
PC9 G2 cells treated with AXL siRNA were
apoptotic cells (Fig. 4B). These
results suggest that interfering with AXL expression may inhibit
cell-cycle progression in PC9 G2 cells, thereby
resulting in a marked increase in the percentage of cells in the
G1 phase.
Downregulation of AXL inhibits
EMT-inducing Twist
The acquisition of mesenchymal cell characteristics
by epithelial cells, in particular the ability to migrate as single
cells and invade the extracellular matrix, is the functional
hallmark of EMT (24). EMT-inducing
genes that have essential roles in EMT, including Twist and
Snail, are termed master EMT genes (25). These genes function as
transcriptional repressors of the cell-cell adhesion glycoprotein,
E-cadherin whose functional loss is one of the hallmarks of EMT
(26). Therefore, the status of
Twist expression in PC9 cells was investigated in the
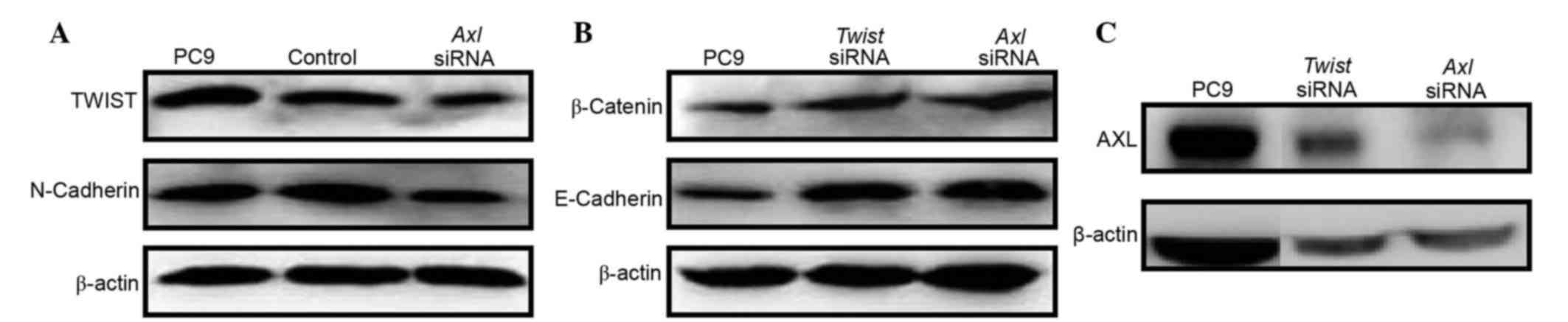
present study. PC9 cells that were stably expressing Twist
exhibited an EMT phenotype characterized by upregulation of the
mesenchymal marker N-cadherin (Fig.
5A), and downregulation of the epithelial markers E-cadherin
and β-cadherin (Fig. 5B). Notably,
Axl was markedly upregulated in the Twist-expressing PC9
cells (Fig. 5C). Conversely, in the
PC9 cells treated with AXL siRNA, downregulation of Twist and
N-cadherin (Fig. 5A), and
upregulation of E-cadherin and β-cadherin (Fig. 5B) were observed. However, knockdown
of Twist did not affect the expression levels of AXL
(Fig. 5C), despite the fact that
E-cadherin and β-cadherin were upregulated (Fig. 5B). These results suggest that
downregulation of AXL may abrogate EMT program induction by
inhibiting the expression of Twist in NSCLC cells.
Discussion
This study investigated the role of AXL in NSCLC
cells, in particular the molecular mechanisms underlying the
involvement of AXL in the EMT. AXL was shown to be upregulated in
all NSCLC cell lines used in the present study, which is consistent
with a previous study (27),
suggesting that AXL may be used as a biomarker of NSCLC.
Furthermore, in the present study, downregulation of AXL by RNAi
was shown to inhibit PC9 cell survival, which may have been due to
accelerated cell apoptosis or cell proliferation inhibition at the
G1 stage. A previous study reported that human NSCLC
with activating mutations in the EGFR did not respond to treatment
with EGFR-targeted tyrosine kinase inhibitors (TKIs), due to the
acquisition of resistance (27).
Furthermore, activation of the AXL kinase was associated with
resistance to EGFR-targeted therapy in patients with lung cancer
(27), suggesting that increased AXL
expression levels may contribute to drug resistance in lung cancer.
Therefore, downregulation of AXL expression may be considered an
effective tool for lung cancer therapy (28). Alterations in AXL-associated
signaling pathways have been observed in ~20% of patients with
acquired resistance to EGFR-TKI (29), although it remains to be determined
whether these patients would benefit from AXL inhibition. In
EGFR-TKI resistance, AXL may act as a bypass to activate downstream
signals associated with cell survival and growth (30). Therefore, combined treatment with
EGFR and AXL inhibitors may effectively abrogate the growth of
tumor cells. A similar phenomenon was observed in hepatocyte growth
factor receptor-mediated resistance (31).
EMT is an embryonic developmental process involving
changes in cell morphology and expression of EMT-associated genes
(25). EMT also occurs during the
progression of several types of human cancer, in which it confers
motility and invasiveness on cancer cells, leading them to acquire
the ability to metastasize to distant sites (32). A previous study reported that lung
cancer cell lines may be classified into mesenchymal and epithelial
phenotypes based on the expression status of E-cadherin and
Vimentin, which are markers of epithelial and mesenchymal
phenotypes, respectively (33). A
genetic study of early development reported the existence of a
number of EMT-inducing genes encoding transcription factors that
are capable of inducing EMT when ectopically expressed in
epithelial cells (34). Therefore,
the present study investigated the expression levels of several
master EMT genes, as well as AXL, in order to elucidate the
potential role of AXL in EMT. The master EMT gene Twist was
upregulated in the PC9 NSCLC cells, which was accompanied by
upregulation of AXL. Conversely, inhibition of AXL by
RNAi resulted in the downregulation of Twist in PC9 cells;
thus suggesting that downregulation of AXL inhibits EMT-inducing
Twist. Therefore, AXL may regulate EMT via the EMT-associated
genes. The results of the present study may have clinical
significance in the design of therapeutic strategies targeting AXL
and EMT signaling pathways.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pujol JL, Barlesi F and Daurès JP: Should
chemotherapy combinations for advanced non-small cell lung cancer
be platinum-based? A meta-analysis of phase III randomized trials.
Lung Cancer. 51:335–345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Le Chevalier T, Scagliotti G, Natale R,
Danson S, Rosell R, Stahel R, Thomas P, Rudd RM, Vansteenkiste J,
Thatcher N, et al: Efficacy of gemcitabine plus platinum
chemotherapy compared with other platinum containing regimens in
advanced non-small-cell lung cancer: A meta-analysis of survival
outcomes. Lung Cancer. 47:69–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ettinger DS, Akerley W, Borghaei H, Chang
AC, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Ganti AK,
Govindan R, et al NCCN (National Comprehensive Cancer Network), :
Non-small cell lung cancer. J Natl Compr Canc Netw. 10:1236–1271.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chevalier TL, Brisgand D, Douillard JY,
Pujol JL, Alberola V, Monnier A, Riviere A, Lianes P, Chomy P and
Cigolari S: Randomized study of vinorelbine and cisplatin versus
vindesine and cisplatin versus vinorelbine alone in advanced
non-small-cell lung cancer: Results of a European multicenter trial
including 612 patients. J Clin Oncol. 12:360–367. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Non-small Cell Lung Cancer Collaborative
Group, . Chemotherapy in non-small cell lung cancer: A
meta-analysis using updated data on individual patients from 52
randomised clinical trials. BMJ. 311:899–909. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cappuzzo F, Hirsch FR, Rossi E, Bartolini
S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini
I, et al: Epidermal growth factor receptor gene and protein and
gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer
Inst. 97:643–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiller JH, Harrington D, Belani CP,
Langer C, Sandler A, Krook J, Zhu J and Johnson DH; Eastern
Cooperative Oncology Group, : Comparison of four chemotherapy
regimens for advanced non-small-cell lung cancer. N Engl J Med.
346:92–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robinson DR, Wu YM and Lin SF: The protein
tyrosine kinase family of the human genome. Oncogene. 19:5548–5557.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Valverde P, Obin MS and Taylor A: Role of
Gas6/Axl signaling in lens epithelial cell proliferation and
survival. Exp Eye Res. 78:27–37. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goruppi S, Ruaro E and Schneider C: Gas6,
the ligand of Axl tyrosine kinase receptor, has mitogenic and
survival activities for serum starved NIH3T3 fibroblasts. Oncogene.
12:471–480. 1996.PubMed/NCBI
|
|
12
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, Del Barrio MG, Portillo F and Nieto MA: The
transcription factor Snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goruppi S, Ruaro E, Varnum B and Schneider
C: Requirement of phosphatidylinositol 3-kinase-dependent pathway
and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3
fibroblasts. Mol Cell Biol. 17:4442–4453. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Donnell K, Harkes IC, Dougherty L and
Wicks IP: Expression of receptor tyrosine kinase Axl and its ligand
Gas6 in rheumatoid arthritis: Evidence for a novel endothelial cell
survival pathway. Am J Pathol. 154:1171–1180. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Janssen JW, Schulz AS, Steenvoorden AC,
Schmidberger M, Strehl S, Ambros PF and Bartram CR: A novel
putative tyrosine kinase receptor with oncogenic potential.
Oncogene. 6:2113–2120. 1991.PubMed/NCBI
|
|
16
|
O'Bryan JP, Frye RA, Cogswell PC, Neubauer
A, Kitch B, Prokop C, Espinosa R, Le Beau MM, Earp HS and Liu ET:
axl, a transforming gene isolated from primary human myeloid
leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell
Biol. 11:5016–5031. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shieh YS, Lai CY, Kao YR, Shiah SG, Chu
YW, Lee HS and Wu CW: Expression of axl in lung adenocarcinoma and
correlation with tumor progression. Neoplasia. 7:1058–1064. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ye X, Li Y, Stawicki S, Couto S,
Eastham-Anderson J, Kallop D, Weimer R, Wu Y and Pei L: An anti-Axl
monoclonal antibody attenuates xenograft tumor growth and enhances
the effect of multiple anticancer therapies. Oncogene.
29:5254–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meyer AS, Miller MA, Gertler FB and
Lauffenburger DA: The receptor AXL diversifies EGFR signaling and
limits the response to EGFR-targeted inhibitors in triple-negative
breast cancer cells. Sci Signal. 6:ra662013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Janmaat ML, Kruyt FA, Rodriguez JA and
Giaccone G: Response to epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells: Limited antiproliferative
effects and absence of apoptosis associated with persistent
activity of extracellular signal-regulated kinase or Akt kinase
pathways. Clin Cancer Res. 9:2316–2326. 2003.PubMed/NCBI
|
|
21
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Danielsen T, Hvidsten M, Stokke T, Solberg
K and Rofstad EK: Hypoxia induces p53 accumulation in the S-phase
and accumulation of hypophosphorylated retinoblastoma protein in
all cell cycle phases of human melanoma cells. Br J Cancer.
78:1547–1558. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-tie quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu MM, Mao GX, Liu J, Li JC, Huang H, Liu
YF and Liu JH: Low expression of the FoxO4 gene may contribute to
the phenomenon of EMT in non-small cell lung cancer. Asian Pac J
Cancer Prev. 15:4013–4018. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vandewalle C, Van Roy F and Berx G: The
role of the ZEB family of transcription factors in development and
disease. Cell Mol Life Sci. 66:773–787. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cerchia L, Esposito CL, Camorani S, Rienzo
A, Stasio L, Insabato L, Affuso A and de Franciscis V: Targeting
Axl with an high-affinity inhibitory aptamer. Mol Ther.
20:2291–2303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hata A, Katakami N, Yoshioka H, Takeshita
J, Tanaka K, Nanjo S, Fujita S, Kaji R, Imai Y, Monden K, et al:
Rebiopsy of non-small cell lung cancer patients with acquired
resistance to epidermal growth factor receptor-tyrosine kinase
inhibitor: Comparison between T790M mutation-positive and
mutation-negative populations. Cancer. 119:4325–4332. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niederst MJ and Engelman JA: Bypass
mechanisms of resistance to receptor tyrosine kinase inhibition in
lung cancer. Sci Signal. 6:10.1126/scisignal.20046522013.
View Article : Google Scholar
|
|
31
|
Qi J, McTigue MA, Rogers A, Lifshits E,
Christensen JG, Jänne PA and Engelman JA: Multiple mutations and
bypass mechanisms can contribute to development of acquired
resistance to MET inhibitors. Cancer Res. 71:1081–1091. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takeyama Y, Sato M, Horio M, Hase T,
Yoshida K, Yokoyama T, Nakashima H, Hashimoto N, Sekido Y, Gazdar
AF, et al: Knockdown of ZEB1, a master epithelial-to-mesenchymal
transition (EMT) gene, suppresses anchorage-independent cell growth
of lung cancer cells. Cancer Lett. 296:216–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mani SA, Guo W, Liao M-J, Eaton EN,
Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et
al: The epithelial-mesenchymal transition generates cells with
properties of stem cells. Cells. 133:704–715. 2008. View Article : Google Scholar
|