Introduction
Ischemic heart disease is one of the major causes of
mortality in humans worldwide (1).
The ischemic myocardium will finally lead to myocardial infarction.
However, timely reperfusion exacerbates the heart failure in
patients, though the infarct size has not enlarged (2). Post-ischemic reperfusion contributes
greatly to the cardiomyocyte death through the
mitochondria-mediated pathway (3).
The mechanisms of myocardial ischemia and reperfusion injury
include overproduction of reactive oxygen species (ROS), overload
of intracellular calcium, collapse of mitochondrial membrane
potential (MMP) and prolonged opening of the mitochondrial
permeability transition pore (mPTP) among other processes (4). Natural compounds, such as berberine
(5), tanshinone IIA (6) and lycopene (7), serve a pivotal role in the development
of effective therapeutics for myocardial ischemia and reperfusion
injury.
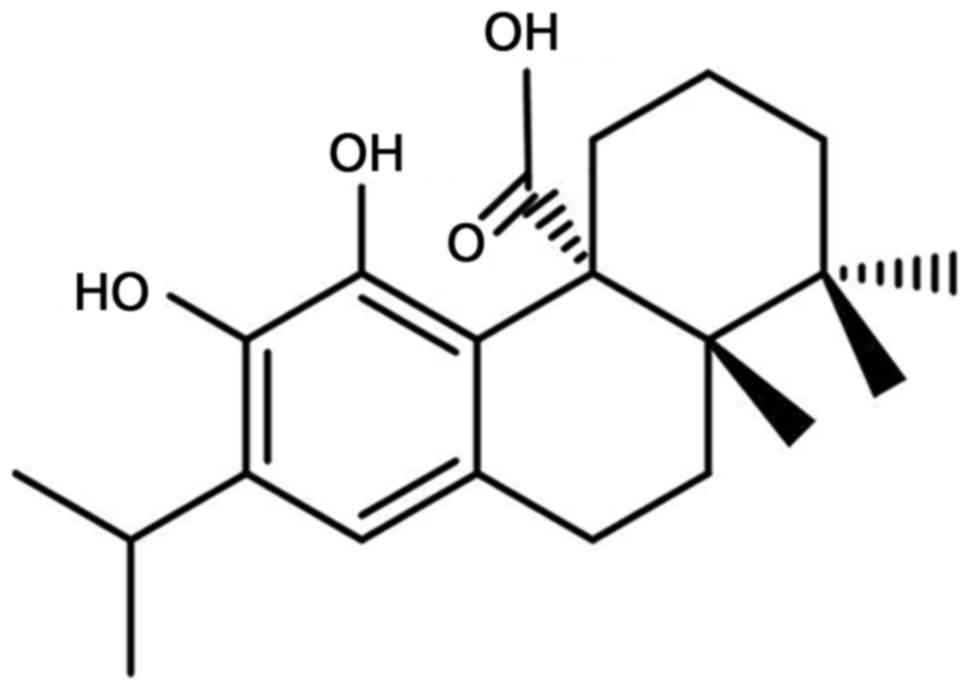
Carnosic acid is a natural diterpenoid (Fig. 1) that has been identified as the
major bioactive phytochemical in numerous medicinal plants,
including Rosmarinus officinalis (8), Salvia fruticosa (9) and Ocimum sanctum (10). Previous pharmacological studies have
demonstrated that carnosic acid affords various biological
activities, such as neuroprotection (11), prevention of advanced glycation
end-product formation (12),
attenuation of Alzheimer's disease (13), anticancer activity (14), anti-inflammation (15) and renoprotection (16). Furthermore, carnosic acid presented a
cardioprotective effect in an isoproterenol-induced myocardial
stress mouse model via preventing oxidative stress and apoptosis
(17).
In an attempt to identify novel therapeutic
approaches for myocardial ischemia and reperfusion injury, the
present study assessed the myocardial protection exerted by
carnosic acid and the associated underlying mechanisms using H9c2
cardiomyocytes subjected to hypoxia/reoxygenation.
Materials and methods
Chemicals and reagents
Carnosic acid was purchased from J&K Scientific
Ltd. (Beijing, China). Dulbecco's modified Eagle's medium (DMEM)
and fetal bovine serum were supplied by Thermo Fisher Scientific,
Inc. (Waltham, MA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). The Fluo-3 acetoxymethyl (AM),
ROS assay kit, lactate dehydrogenase (LDH) activity assay kit, BCA
protein concentration assay kit, MMP assay kit with JC-1, and
Caspase-3 Activity assay kit, as well as the cleaved caspase-3 (cat
no. AF0081), B-cell lymphoma 2 (Bcl-2; cat no. AF0060),
Bcl-2-associated X protein (Bax; cat no. AF0054) and β-actin (cat
no. AF003) antibodies, were obtained from Beyotime Institute of
Biotechnology (Nantong, China). Calcein-AM was obtained from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan).
Cell culture and model
establishment
Rat H9c2 cardiomyocytes were obtained from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China) and
cultured in DMEM containing 10% fetal bovine serum, 1%
penicillin/streptomycin under humid conditions with 5%
CO2 and 95% air at 37°C. Next, the cells in logarithmic
phase were incubated in 96-well plates at a density of
1×105 cells/ml. The cells were divided into the control
group, hypoxia/reoxygenation model group (H/R group), and three
experimental groups pretreated with 0.1, 1 and 10 µM carnosic acid
in DMSO for 4 h prior to hypoxia/reoxygenation. To establish the
hypoxia/reoxygenation model, H9c2 cells in the H/R and experimental
groups were incubated in an atmosphere with 95% N2 and
5% CO2 at 37°C for 4 h, and then exposed to 95% air and
5% CO2 at 37°C for a further 4 h. The control group was
cultured under normoxic conditions.
Cell viability assay
To detect the protective effects of carnosic acid on
H9c2 cardiomyocytes exposed to hypoxia/reoxygenation, an MTT assay
was performed. Subsequent to the aforementioned treatments, the
cells were incubated with 0.2 ml MTT (0.5 mg/ml) for 4 h at 37°C.
Next, 200 µl DMSO was added into each well in order to dissolve the
formazan crystals. The optical density (OD) was recorded on a
BioTek ELx800 microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA) at 490 nm. The results are expressed as the
relative percentage of the control group.
Extracellular LDH activity
To further confirm the protective effects of
carnosic acid, the LDH activity in the culture medium was measured
by an LDH assay kit, according to the manufacturer's protocol.
Briefly, following treatment, the culture medium was centrifuged at
400 × g and room temperature for 5 min, and then 20 µl supernatant
was collected and mixed with 20 µl 2,4-dinitrophenylhydrazine.
After incubation at 37°C for 15 min, 250 µl NaOH (0.4 M) was added
into the mixture and incubated for a further 15 min at 37°C. The
mixture was maintained at room temperature for 5 min, and
subsequently the OD was recorded on a microplate reader at 450 nm.
The activity of LDH was derived from the OD values and expressed as
U/l.
Intracellular calcium level
To monitor the intracellular calcium in H9c2
cardiomyocytes, the fluorescence dye Fluo-3 AM was employed,
following the manufacturer's protocol. Briefly, the pretreated H9c2
cardiomyocytes were loaded with 5 µM Fluo-3 AM at 37°C for 30 min
in the dark, and then washed with PBS for three times to remove any
excessive dye. The fluorescence intensity of Fluo-3 chelated with
calcium was recorded on a PerkinElmer EnVision fluorescence
microplate reader (PerkinElmer, Llantrisant, UK) at excitation and
emission wavelengths of 488 and 525 nm, respectively.
Measurement of ROS production
The production of ROS was detected by a fluorescence
method using a ROS assay kit. Subsequent to treatment, the medium
was replaced and the cells were rinsed with PBS. Next, 10 µM
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) in DMEM was
added and incubated at 37°C for 30 min. The cells were then washed
again with PBS to remove the excessive dye, and the fluorescence
intensity was recorded on a fluorescence microplate reader at 485
nm (excitation wavelength) and 520 nm (emission wavelength).
MMP assay
The MMP was also detected using a MMP assay kit with
JC-1. JC-1 accumulates in the mitochondrial matrix of normal cells
and forms aggregates, which emit red fluorescence under excitation.
When the MMP collapses, JC-1 exists as a monomer and thus no red
fluorescence is observed under excitation (18). Following the treatment, the H9c2
cardiomyocytes were washed with PBS and then loaded with JC-1 at
37°C for 20 min. Subsequent to washing with JC-1 buffer solution,
the fluorescence intensity was read at an excitation wavelength of
488 nm and an emission wavelength of 530 nm. The MMP is expressed
as a percentage compared with the fluorescence intensity of the
control group.
mPTP opening
The mPTP opening was directly evaluated through
monitoring the release of mitochondrial calcein. Briefly,
cardiomyocytes were incubated with 2 µM calcein-AM and 1 mM
CoCl2 at room temperature for 30 min. The free
calcein-AM and CoCl2 were washed with PBS to be removed,
and the cells were incubated with 1 mM CoCl2 for a
further 20 min at 37°C to specifically quench the fluorescence of
free calcein in the cytosol. Subsequently, the fluorescence
intensity of mitochondrial calcein in the cardiomyocytes was
measured on a fluorescence microplate reader at 490 nm for
excitation and 515 nm for emission. The loss of calcein
fluorescence in cardiomyocytes indicated the opening of mPTP. The
results are expressed as the fluorescence intensity percentage of
the control group.
Caspase-3 activity
Caspase-3 activity was assessed by a colorimetric
assay kit following the manufacturer's instructions. H9c2
cardiomyocytes were pretreated as described earlier and washed with
PBS, followed by lysis using a Caspase-3 Assay kit (cat no. C1116;
Beyotime Institute of Biotechnology) and centrifugation at 16,000 ×
g for 10 min at 4°C. The supernatant was then collected and
incubated with substrate (Ac-DEVD-pNA) at 37°C for 2 h. The OD
values were measured on a microplate reader at 405 nm, and the
activity of caspase-3 is expressed as the relative percentage of
the OD value of the control group.
Western blot analysis
H9c2 cardiomyocytes were treated as described
earlier and then subjected to western blot analysis to detect the
expression levels of caspase-3, Bcl-2 and Bax. Briefly, the cells
were lysed with lysis buffer solution containing 20 mM Tris-HCl (pH
7.4), 150 mM NaCl, 1% Triton and 1 mM phenylmethane sulfonyl
fluoride on ice for 30 min. Next, the cell lysate was centrifuged
at 12,000 × g for 15 min at 4°C, and the supernatant was collected
as the total protein for the subsequent analysis of cleaved
caspase-3, Bcl-2, and Bax. Following quantification by a BCA assay
kit, the samples were separated by 15% SDS-PAGE and transferred to
polyvinylidene difluoride membranes. Subsequent to blocking with
defatted milk at room temperature for 1 h, the membranes were
incubated overnight at 4°C with primary antibodies against cleaved
caspase-3, Bcl-2, Bax and β-actin (diluted at 1:1,000). The
membranes were then incubated with the respective secondary
antibody conjugated to horseradish peroxidase (diluted at 1:3,000;
cat no. A0192; Beyotime Institute of Biotechnology) at room
temperature for 1 h, and visualized by an enzyme-link
chemiluminescence substrate (cat no. P0203; Beyotime Institute of
Biotechnology) on a Bio-Rad ChemiDoc XRS+ imaging system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). β-actin was used as the
internal control.
Statistical analysis
The results are expressed as the mean ± standard
deviation, and were analyzed by GraphPad Prism version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA). Comparisons were
implemented by Student's t-test for single components or one-way
analysis of variance followed by Dunnett's test for multiple
components. P<0.05 was considered to be an indicator of
statistically significant differences. All data are the result of
at least six independent experiments.
Results
Carnosic acid improves the viability
of H9c2 cardiomyocytes induced by hypoxia/reoxygenation
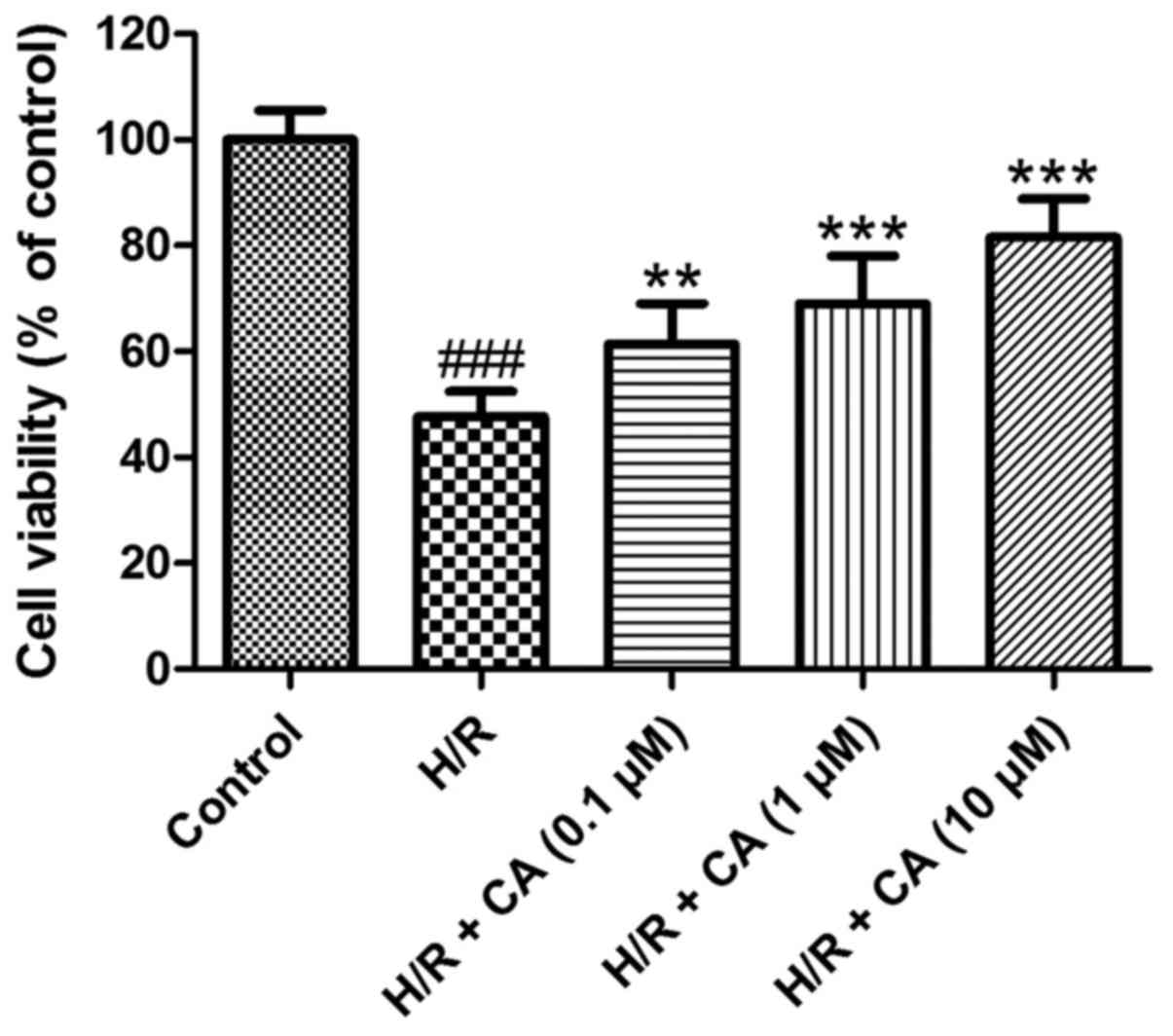
As shown in Fig. 2,
the viability of H9c2 cardiomyocytes was decreased when exposed to
hypoxia/reoxygenation (P<0.001). In the presence of different
carnosic acid concentrations, the poor survival of H9c2
cardiomyocytes was significantly ameliorated compared with the H/R
group, in a dose-dependent manner. In particular, the viability in
the group treated with 10 µM carnosic acid reached 81.55±7.31% of
the control group value (P<0.001). These results indicated that
carnosic acid exhibited protective effects on H9c2 cardiomyocytes
injured by hypoxia/reoxygenation.
Carnosic acid reduces LDH release in
H9c2 cardiomyocytes induced by hypoxia/reoxygenation
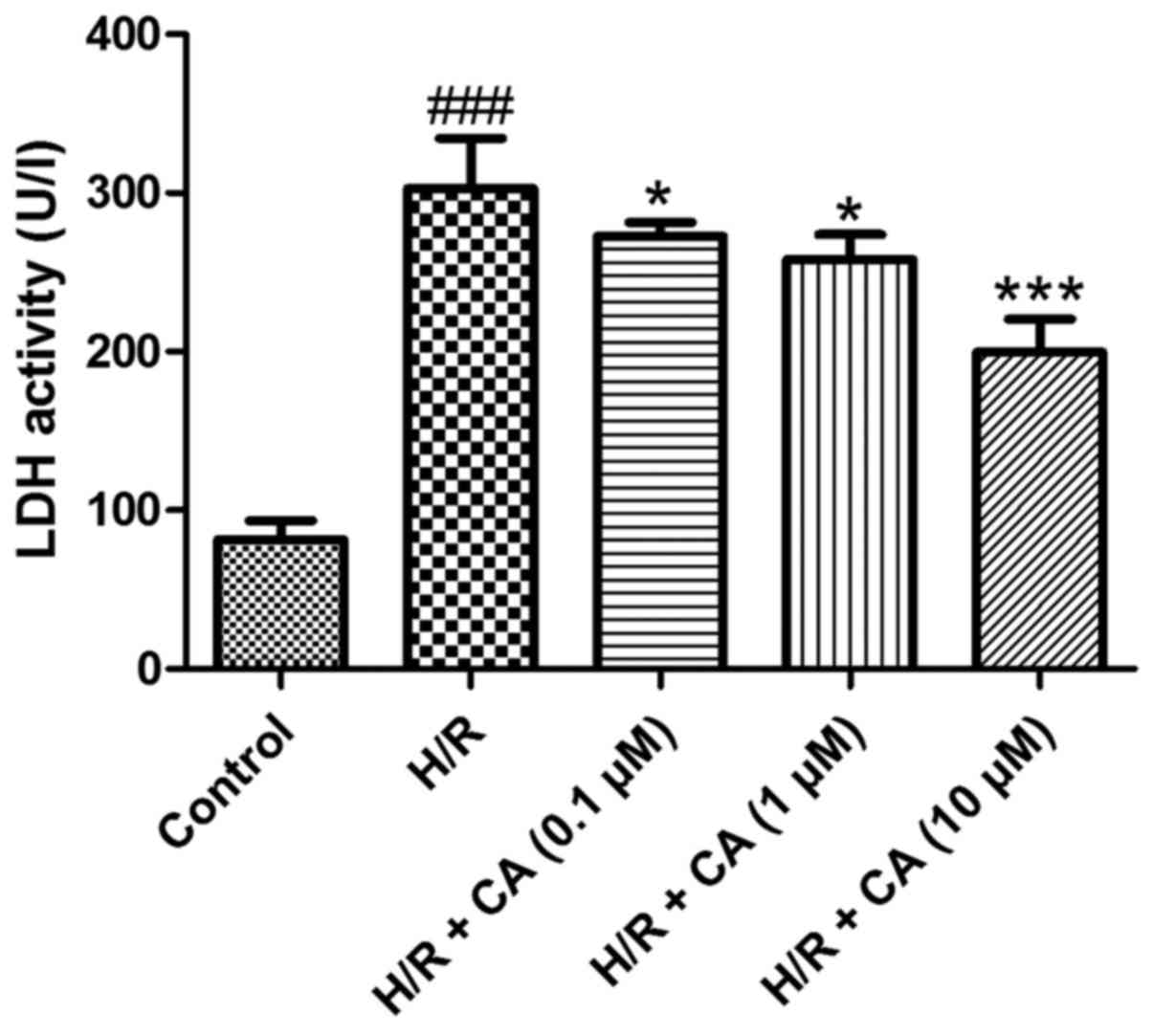
Under hypoxia/reoxygenation, the activity of LDH in
the culture medium of H9c2 cardiomyocytes increased to
approximately three times greater than the control group activity,
which demonstrated that leakage of LDH from the cytosol occurred in
the H/R group and cell survival decreased (P<0.001). When
treated with carnosic acid, the release of LDH in H9c2
cardiomyocytes was significantly decreased in a dose-dependent
manner (P<0.05; Fig. 3). These
results indicated the carnosic acid affected the LDH release in
H9c2 cardiomyocytes induced by hypoxia/reoxygenation.
Carnosic acid attenuates the overload
of intracellular calcium in H9c2 cardiomyocytes induced by
hypoxia/reoxygenation
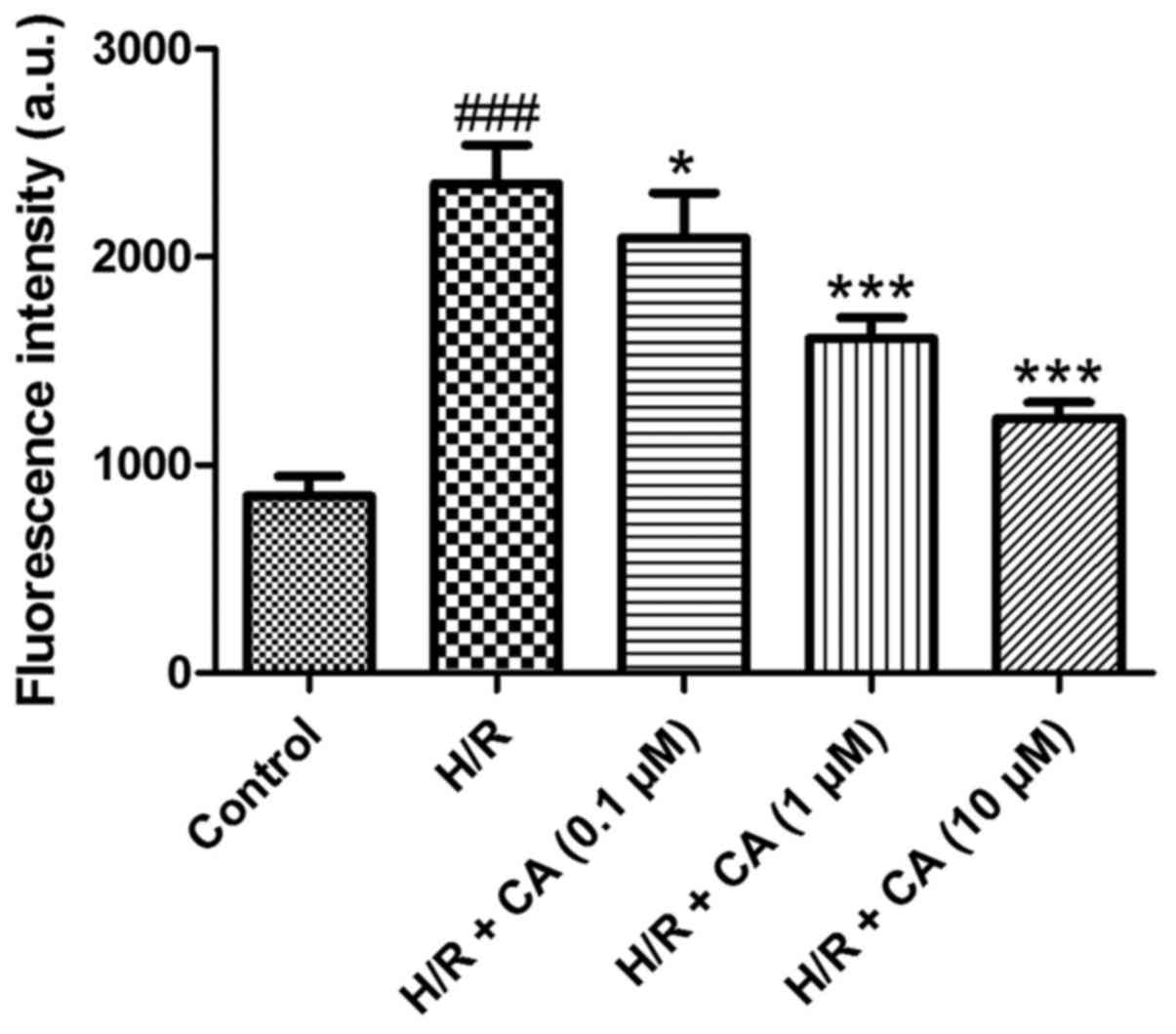
To determine the intracellular calcium level, the
fluorescence probe Fluo-3 AM was used. Following treatment of
hypoxia/reoxygenation, the fluorescence intensity of the
intracellular calcium in the H/R group was significantly elevated
(2,352.85±185.48) when compared with the control group
(850.00±96.86; P<0.001; Fig. 4).
However, compared with the H/R group, carnosic acid treatment
dose-dependently attenuated the overload of intracellular calcium
as observed by the reduced fluorescence intensity (2,090.10±218.08,
1,609.07±98.73 and 1,224.07±76.41 at 0.1, 1 and 10 µM,
respectively; P<0.05; Fig. 4).
These results suggested that carnosic acid was able to reduce the
overload of intracellular calcium in H9c2 cardiomyocytes induced by
hypoxia/reoxygenation.
Carnosic acid alleviates the
overproduction of ROS in H9c2 cardiomyocytes induced by
hypoxia/reoxygenation
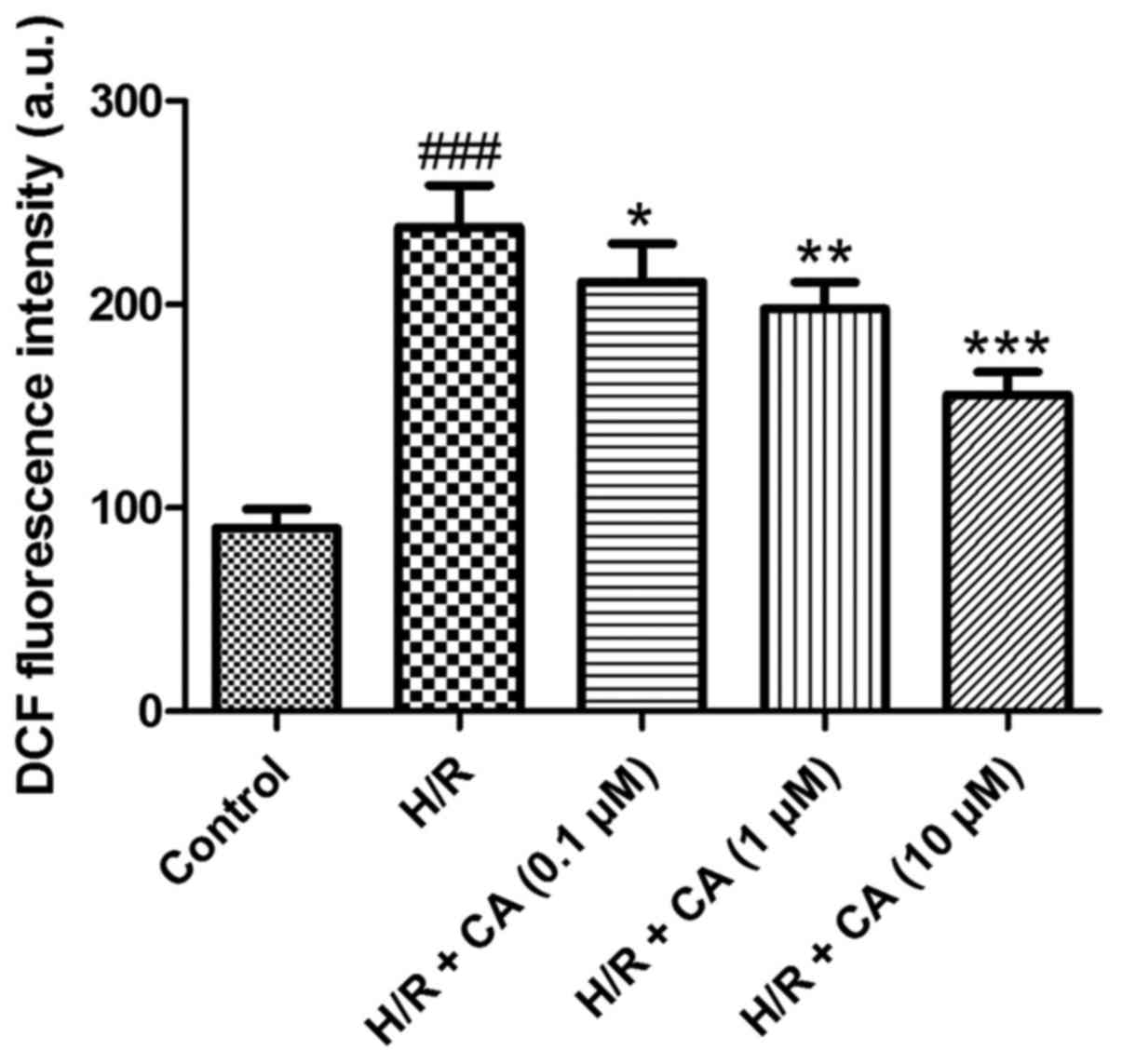
The production of intracellular ROS was measured
with the fluorescence probe DCFH-DA according to the manufacturer's
protocol. DCFH-DA passes through the cellular membrane and is
hydrolyzed to DCFH by intracellular esterases. DCFH is then unable
to cross the membrane and is thus reserved in the cytosol. With the
excessive production of ROS, DCFH is quantitatively oxidized into
the fluorescent dichlorofluorescein (DCF) (19). The results of the present experiments
revealed that the fluorescence intensity in the H/R group
(238.12±20.51) was markedly higher in comparison with that in the
control group (90.00±9.43). Upon treatment with different
concentrations of carnosic acid, the fluorescence intensity of DCF
was reduced in a dose-dependent manner (Fig. 5), implying that carnosic acid was
able to alleviate the overproduction of intracellular ROS in H9c2
cardiomyocytes induced by hypoxia/reoxygenation.
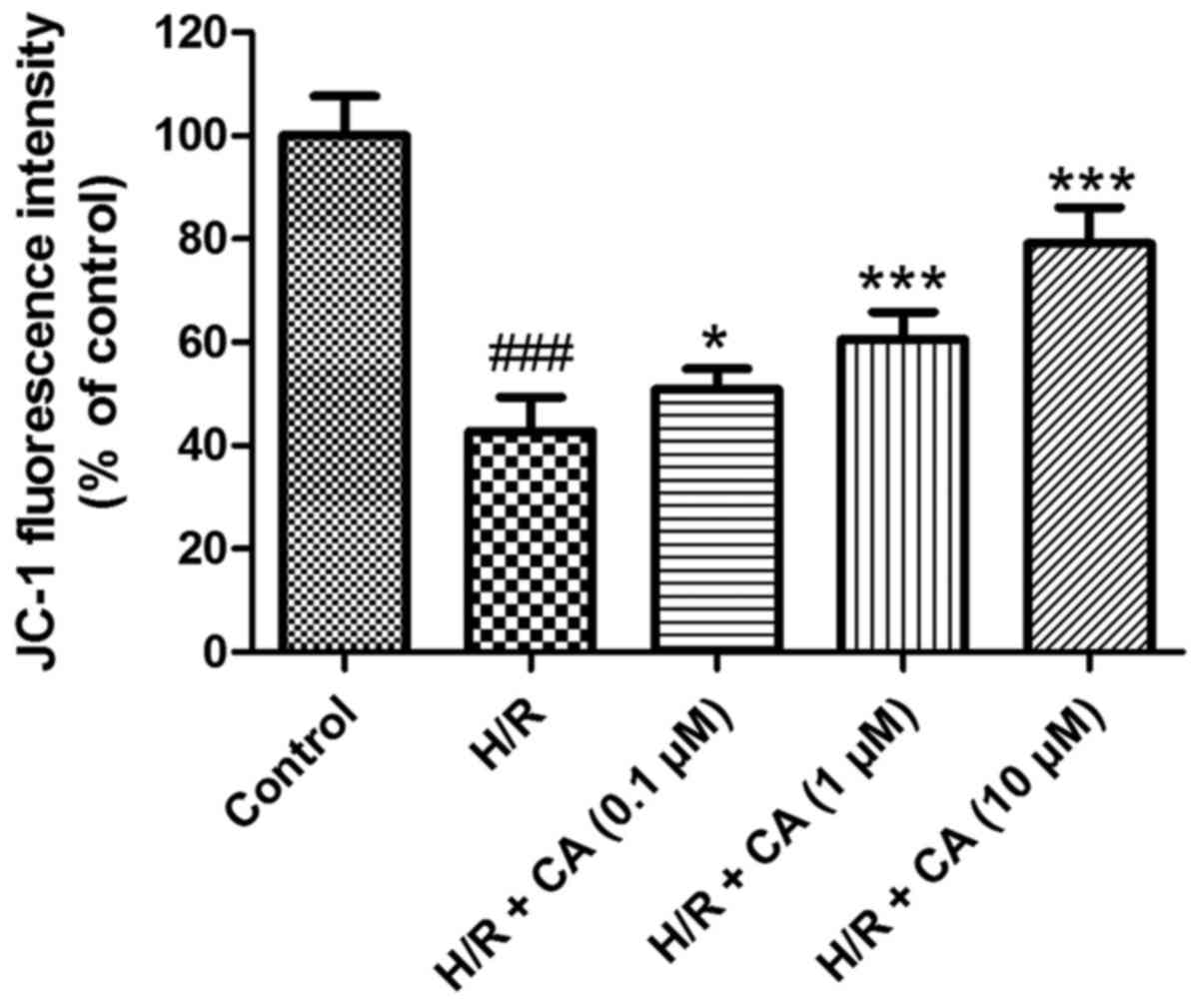
Carnosic acid ameliorates the collapse
of MMP in H9c2 cardiomyocytes induced by hypoxia/reoxygenation
The MMP in H9c2 cardiomyocytes was evaluated to
investigate the effects of carnosic acid on the mitochondrial
function of H9c2 cardiomyocytes induced by hypoxia/reoxygenation.
In contrast to the control group, the fluorescence intensity in the
H/R group (42.78±6.54%) was evidently diminished (P<0.001),
which implicated the collapse of MMP in the H/R group after
induction of hypoxia/reoxygenation. However, in the presence of
different carnosic acid concentrations, the fluorescence intensity
was significantly increased to different extents (Fig. 6). In particular, the experimental
group with 10 µM carnosic acid treatment exhibited ~80% of the
fluorescence intensity of the control group. These results revealed
that the collapse of MMP in hypoxia/reoxygenation-treated H9c2
cardiomyocytes was relieved by carnosic acid.
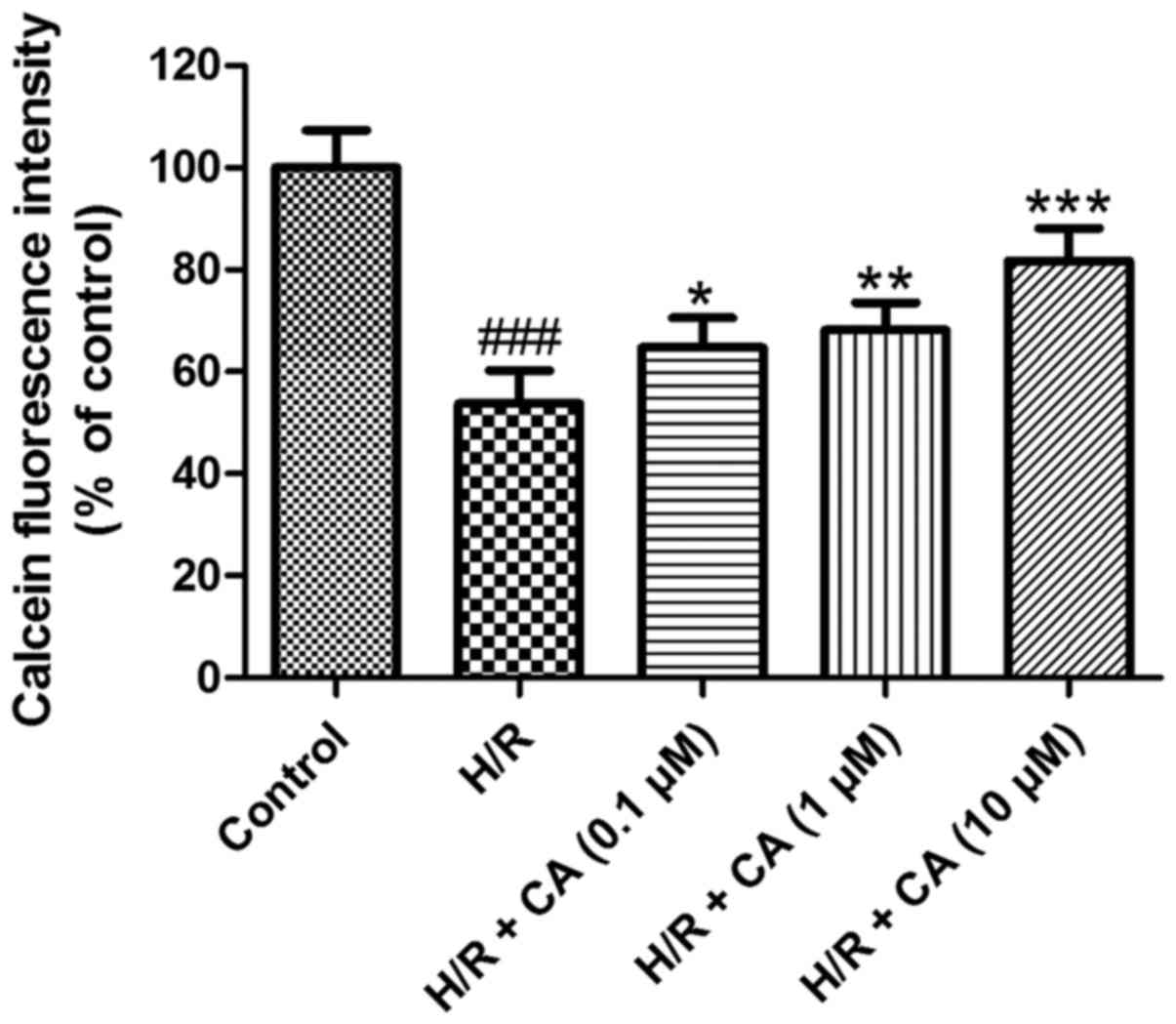
Carnosic acid relieves the opening of
mPTP in H9c2 cardiomyocytes induced by hypoxia/reoxygenation
The mPTP opening was assessed based on the
fluorescence intensity of mitochondrial free calcein. As shown in
Fig. 7, following induction of
hypoxia/reoxygenation, the fluorescence intensity of mitochondrial
calcein in the H/R group was close to half that of the control
group (53.71±6.49%; P<0.001), which revealed the opening of the
mPTP. Upon exposure with increasing carnosic acid doses, the
opening of the mPTP was ameliorated in a dose-dependent manner, as
indicated by the reduced fluorescence intensity of mitochondrial
free calcein. Treatment of 10 µM carnosic acid resulted in a
fluorescence intensity at 81.74±6.40% of the control group value
(P<0.001). These results implied that carnosic acid inhibited
the mPTP opening that was induced by hypoxia/reoxygenation.
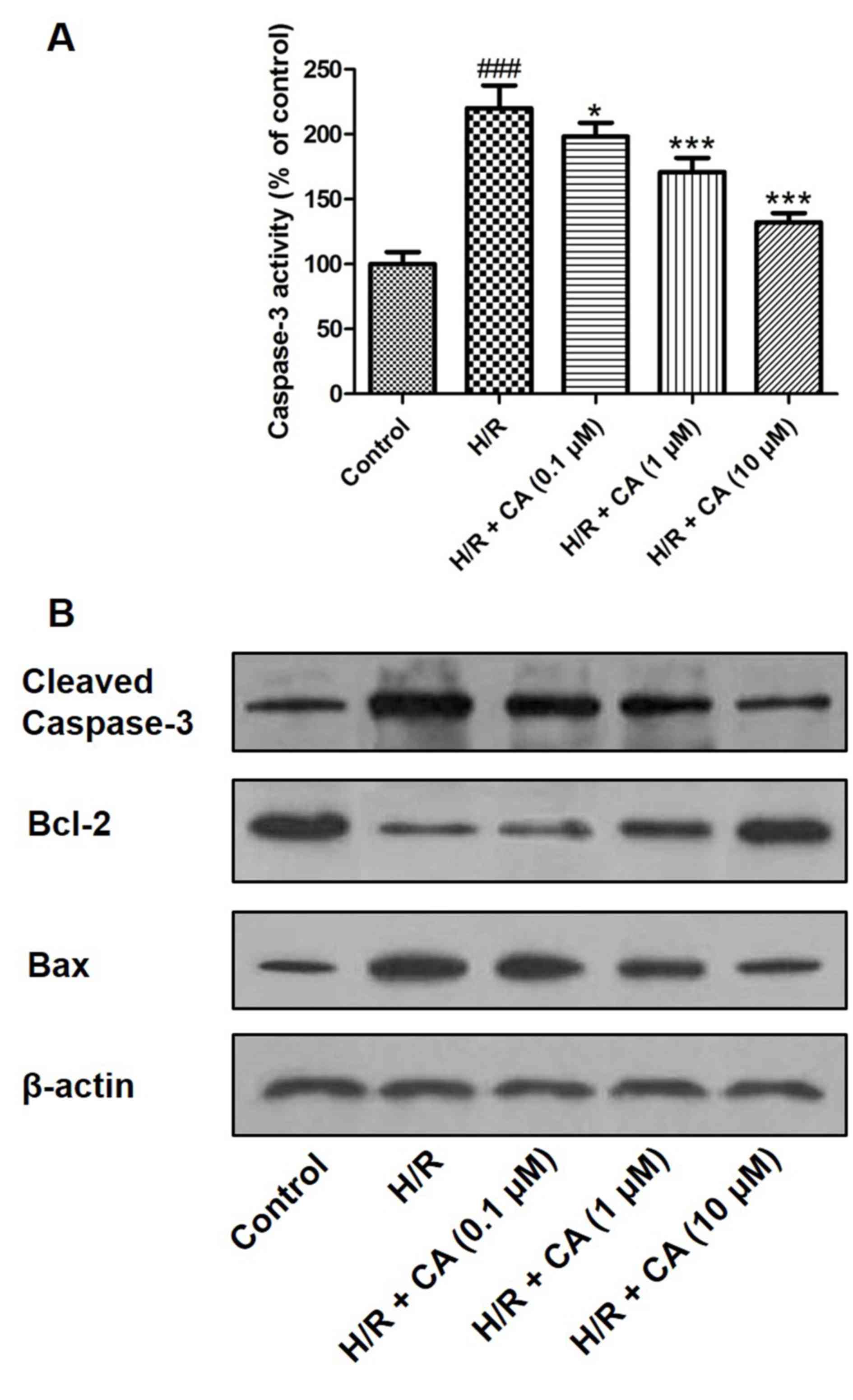
Carnosic acid diminishes caspase-3
activity and affects the expression levels of caspase-3, Bcl-2 and
Bax
Caspase-3 is an executioner enzyme in apoptosis and
is activated through hydrolysis. The results of the colorimetric
assay revealed that the activity of caspase-3 was markedly enhanced
in H9c2 cardiomyocytes following treatment of
hypoxia/reoxygenation. However, when pretreatment with carnosic
acid was performed, the activity of caspase-3 was significantly
inhibited (Fig. 8A). Western blot
analysis also demonstrated that hypoxia/reoxygenation upregulated
the expression of caspase-3, while carnosic acid treatment
suppressed this expression in a dose-dependent manner (Fig. 8B). In addition, western blot analysis
indicated the expression changes of Bcl-2 and Bax, two proteins
associated with apoptosis. Hypoxia/reoxygenation downregulated the
expression of Bcl-2 and upregulated Bax expression. On the
contrary, different concentrations of carnosic acid resulted in the
upregulation of Bcl-2 and downregulation of Bax (Fig. 8B).
Discussion
Myocardial ischemia and reperfusion injury is the
major cause of cardiomyocyte apoptosis in myocardial infarction
(20). Timely reperfusion disrupts
the redox homeostasis and accumulates excessive ROS (3). As the major site of ROS production, the
function of mitochondria, including MMP and mPTP, is then severely
affected; thus, myocardial cells undergo apoptosis via the
mitochondria-mediated pathway (21,22). In
addition, during ischemia and reperfusion, the cytosolic calcium
overloads and interacts with oxidative stress, which accentuates
the dysfunction of the mitochondria via the collapse of MMP and
mPTP opening (2,3).
As a member of the cysteinyl aspartate specific
protease family, caspase-3 serves a crucial role in apoptosis.
Cleavage at specific sites will activate caspase-3 and promote cell
apoptosis (23,24). Bcl-2 and Bax are members of the Bcl-2
protein family that are involved in mitochondria-mediated
apoptosis. Bcl-2 inhibits apoptosis and suppresses the activation
of caspase-3, while Bax enhances the cell apoptosis (25).
In the discovery of novel therapeutics for
myocardial ischemia and reperfusion injury, various natural
phytochemicals present promising protection via various pathways.
Berberine displayed the protective effects through activating janus
kinase 2/signal transducer and activator of transcription 3
signaling and attenuating endoplasmic reticulum stress (5). Furthermore, tanshinone IIA can
attenuate the injury by activating the phosphoinositide
3-kinase/Akt/mammalian target of rapamycin signaling pathway
(6). Previous findings have
suggested that gypenoside protects the cardiomyocytes against the
injury via the mitogen-activated protein kinase-mediated nuclear
factor-κB pathway (26). In
addition, as the convergent pathway of final ischemia and
reperfusion injury, mitochondria-mediated apoptosis has been
demonstrated to be involved in protective effects of most
phytochemicals, including clematichinenoside (27) and ilexsaponin A (28).
In the present study, carnosic acid was observed to
improve the cell viability and leakage of LDH in H9c2
cardiomyocytes injured by hypoxia/reoxygenation. Further studies
have reported that carnosic acid attenuated the overproduction of
intracellular ROS, as well as the calcium overload, which reveals
that the cardioprotective effect of carnosic acid is associated
with the mitochondria-mediated apoptosis pathway. The present
experimental results demonstrated that carnosic acid improved the
dysfunction of mitochondria in H9c2 cardiomyocytes through
suppressing the collapse of MMP and the mPTP opening, which are
pivotal events in apoptosis. At the same time, carnosic acid
directly inhibited the apoptosis of H9c2 cardiomyocytes injured by
hypoxia/reoxygenation via downregulation of Capase-3 and Bax, and
upregulation of Bcl-2.
As the major phytochemicals in the genera of
Rosmarinus and Salvia, carnosic acid possesses
various beneficial bioactivities (11). Previous investigation has revealed
carnosic acid may attenuate the isoproterenol-induced myocardial
injury in a mouse model through preventing oxidative stress and
apoptosis (17). In the present
study, the investigations performed further elucidated the
protective effects of carnosic acid on myocardial injury in
vitro, which is reported in H9c2 cardiomyocytes for the first
time.
In conclusion, the results of the current study
revealed the cardioprotective effects of carnosic acid and the
potential underlying mechanisms in vitro. These findings
provided evidence for further evaluations in vivo that may
assist in the development of novel therapeutic approaches for
myocardial infarction.
References
|
1
|
Writing Group Members, . Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Executive summary: Heart disease and
stroke statistics-2016 update: A report from the American Heart
Association. Circulation. 133:447–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hausenloy DJ and Yellon DM: Targeting
myocardial reperfusion injury-the search continues. N Engl J Med.
373:1073–1075. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pagliaro P, Moro F, Tullio F, Perrelli MG
and Penna C: Cardioprotective pathways during reperfusion: Focus on
redox signaling and other modalities of cell signaling. Antioxid
Redox Signal. 14:833–850. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma V, Bell RM and Yellon DM: Targeting
reperfusion injury in acute myocardial infarction: A review of
reperfusion injury pharmacotherapy. Expert Opin Pharmacother.
13:1153–1175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B,
Liu XD, Zhang B, Liu ZH, Zhai ME, Jin ZX, et al: Berberine protects
rat heart from ischemia/reperfusion injury via activating
JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 37:354–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Q, Shen L, Wang Z, Jiang HP and Liu LX:
Tanshinone IIA protects against myocardial ischemia reperfusion
injury by activating the PI3K/Akt/mTOR signaling pathway. Biomed
Pharmacother. 84:106–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao Y, Jia P, Shu W and Jia D: The
protective effect of lycopene on hypoxia/reoxygenation-induced
endoplasmic reticulum stress in H9C2 cardiomyocytes. Eur J
Pharmacol. 774:71–79. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mena P, Cirlini M, Tassotti M, Herrlinger
KA, Dall'Asta C and Del Rio D: Phytochemical profiling of
flavonoids, phenolic acids, terpenoids, and volatile fraction of a
rosemary (Rosmarinus officinalis L.) extract. Molecules.
21:pii: E1576. 2016. View Article : Google Scholar
|
|
9
|
Exarchou V, Kanetis L, Charalambous Z,
Apers S, Pieters L, Gekas V and Goulas V: HPLC-SPE-NMR
characterization of major metabolites in Salvia fruticosa
Mill. extract with antifungal potential: Relevance of carnosic
acid, carnosol, and hispidulin. J Agric Food Chem. 63:457–463.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baliga MS, Jimmy R, Thilakchand KR,
Sunitha V, Bhat NR, Saldanha E, Rao S, Rao P, Arora R and Palatty
PL: Ocimum sanctum L (Holy Basil or Tulsi) and its
phytochemicals in the prevention and treatment of cancer. Nutr
Cancer. 1:26–35. 2013. View Article : Google Scholar
|
|
11
|
de Oliveira MR: The dietary components
carnosic acid and carnosol as neuroprotective agents: A mechanistic
view. Mol Neurobiol. 53:6155–6168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ou J, Huang J, Wang M and Ou S: Effect of
rosmarinic acid and carnosic acid on AGEs formation in vitro. Food
Chem. 221:1057–1061. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lipton SA, Rezaie T, Nutter A, Lopez KM,
Parker J, Kosaka K, Satoh T, McKercher SR, Masliah E and Nakanishi
N: Therapeutic advantage of pro-electrophilic drugs to activate the
Nrf2/ARE pathway in Alzheimer's disease models. Cell Death Dis.
7:e24992016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moore J, Yousef M and Tsiani E: Anticancer
effects of rosemary (Rosmarinus officinalis L.) extract and
rosemary extract polyphenols. Nutrients. 8:pii: E731. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maione F, Cantone V, Pace S, Chini MG,
Bisio A, Romussi G, Pieretti S, Werz O, Koeberle A, Mascolo N and
Bifulco G: Anti-inflammatory and analgesic activity of carnosol and
carnosic acid in vivo and in vitro and in silico analysis of their
target interactions. Br J Pharmacol. 174:1497–1508. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung KJ, Min KJ, Park JW, Park KM and Kwon
TK: Carnosic acid attenuates unilateral ureteral
obstruction-induced kidney fibrosis via inhibition of Akt-mediated
Nox4 expression. Free Radic Biol Med. 97:50–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sahu BD, Putcha UK, Kuncha M, Rachamalla
SS and Sistla R: Carnosic acid promotes myocardial antioxidant
response and prevents isoproterenol-induced myocardial oxidative
stress and apoptosis in mice. Mol Cell Biochem. 394:163–176. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Zhu X, Chen M, Ge Q, Shen Y and Pan
S: Resveratrol protects PC12 cells against OGD/R-induced apoptosis
via the mitochondrial mediated signaling pathway. Acta Biochim
Biophys Sin (Shanghai). 48:342–353. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duan ZZ, Li YH, Li YY, Fan GW, Chang YX,
Yu B and Gao XM: Danhong injection protects cardiomyocytes against
hypoxia/reoxygenation- and H2O2-induced
injury by inhibiting mitochondrial permeability transition pore
opening. J Ethnopharmacol. 175:617–625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marunouchi T and Tanonaka K: Cell death in
the cardiac myocyte. Biol Pharm Bull. 38:1094–1097. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: Preconditioning,
postconditioning and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Uchiyama T, Otani H, Okada T, Ninomiya H,
Kido M, Imamura H, Nogi S and Kobayashi Y: Nitric oxide induces
caspase-dependent apoptosis and necrosis in neonatal rat
cardiomyocytes. J Mol Cell Cardiol. 34:1049–1061. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu H, Shi L, Qi G, Zhao S, Gao Y and Li Y:
Gypenoside protects cardiomyocytes against ischemia-reperfusion
injury via the inhibition of mitogen-activated protein kinase
mediated nuclear factor kappa B pathway in vitro and in vivo. Front
Pharmacol. 7:1482016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang SW, Liu Y, Wang F, Qiang J, Liu P,
Zhang J and Xu JW: Ilexsaponin a attenuates
ischemia-reperfusion-induced myocardial injury through
anti-apoptotic pathway. PLoS One. 12:e01709842017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding H, Han R, Chen X, Fang W, Liu M, Wang
X, Wei Q, Kodithuwakku ND and Li Y: Clematichinenoside (AR)
attenuates hypoxia/reoxygenation-induced H9c2 aardiomyocyte
apoptosis via a mitochondria-mediated signaling pathway. Molecules.
21:pii: 683. 2016. View Article : Google Scholar
|