Introduction
Persistent Müllerian duct syndrome (PMDS) is a rare
form of internal male pseudohermaphroditism characterized by
Müllerian duct (MD) derivatives in the genotypes and phenotypes of
males with a 46,XY karyotype (1,2).
Normally, Sertoli cells begin to produce anti-Müllerian hormone
(AMH) during week 7 of gestation, causing MD regression. However,
MDs remain in patients with PMDS, due to a deficiency of AMH or a
defect in the AMH type II receptor (AMHR-II) (1). PMDS is complex and anatomically
variable, which makes diagnosis challenging. Patients with PMDS are
categorized into three types, according to the position of the
testes and uterus: i) The female type occurs in 60–70% of patients,
in which the bilateral testes and epididymis are connected to the
fallopian tubes in the abdomen, the bilateral testes are in
analogue positions to the ovaries and their inguinal sacs remain
empty; ii) the Uteri Inguinal type occurs in 20–30% of patients
that exhibit a testis in the hernia sac, or a scrotal testis with a
contra lateral testis located in the abdomen; and iii) the
transverse testicular ectopia makes up the smallest group as it is
only classified in ~10% of patients where the two testes are
present in the same hernia sac along with the uterus and uterine
tubes (3,4).
The incidence of PMDS is low, with only 150 cases
documented globally (5). Only ~13
cases have been diagnosed in China, all without the use of genetic
analysis (6,7). Due to a lack of distinctive clinical
features in its early stages, PMDS is typically diagnosed in
children when they are tested for other diseases, including
cryptorchidism or inguinal hernia, or in adults when they are
tested for sterility or oligospermia. The impact on fertility in
adults with PMDS diagnosed and treated during childhood remains
unknown. However, according to two case reports, delayed diagnosis
at adulthood may cause infertility (8,9). The
correct diagnosis of PMDS relies on a systematic endocrine
assessment and analysis of mutations in the genes coding for AMH
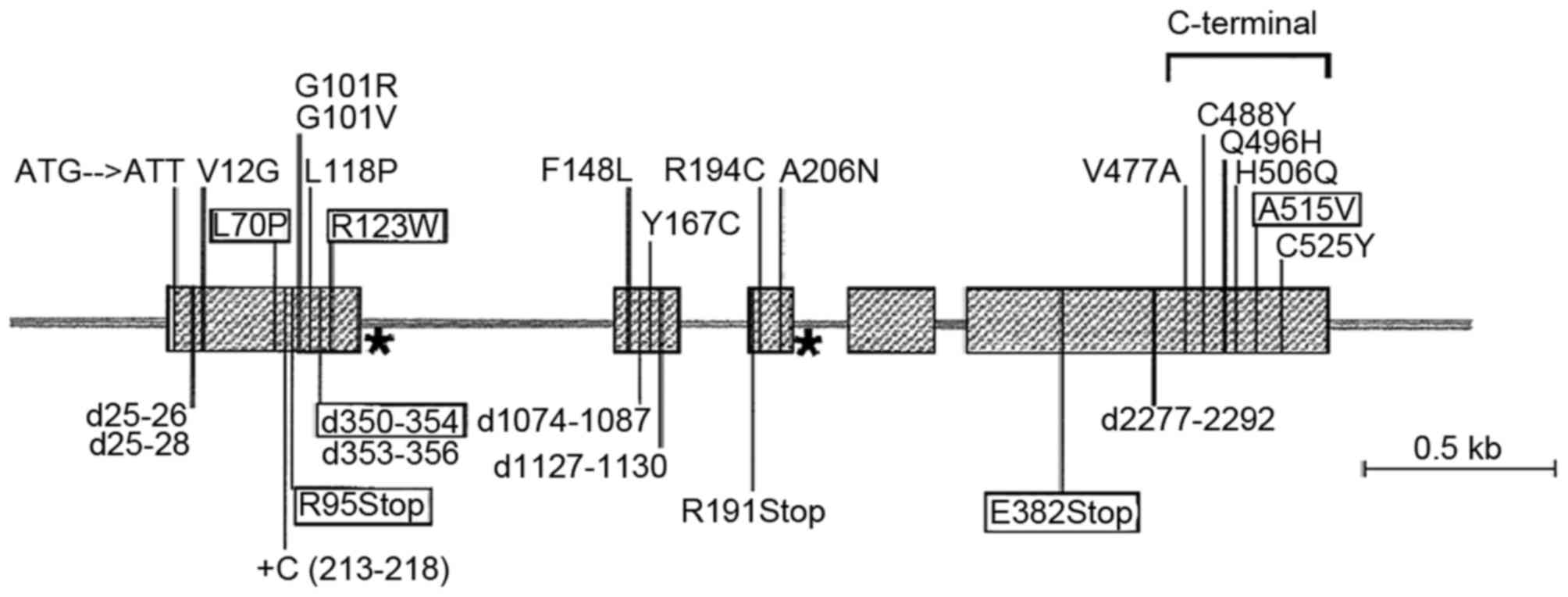
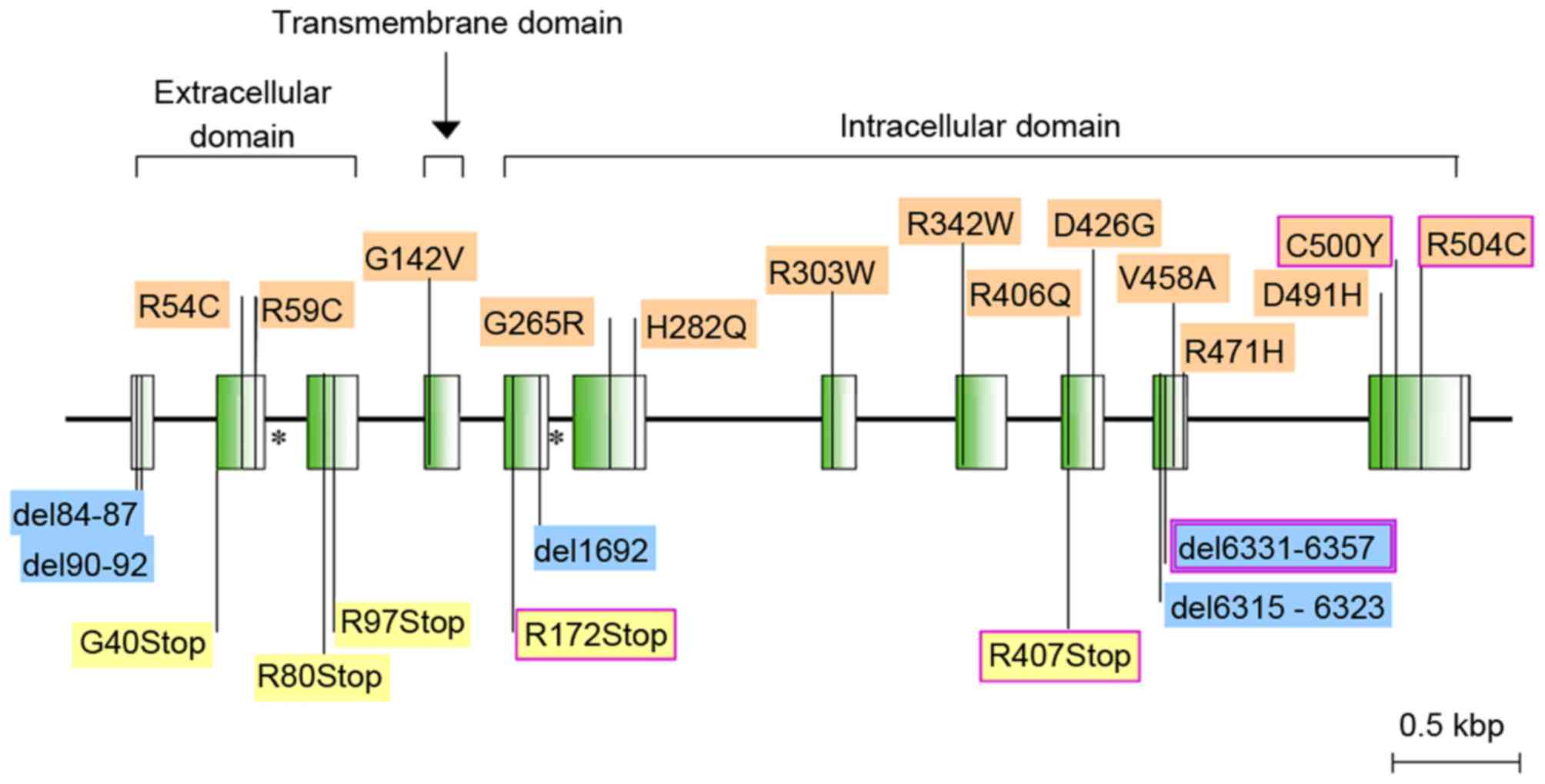
(Fig. 1) and AMHR-II (Fig. 2). At present, the treatment of PMDS
consists of repairing the external genitalia, which keeps MD
structures in the abdomen intact (4). The current study documents the case of
a 17-month-old patient diagnosed with PMDS, and presents a review
of the relevant literature on the diagnosis and treatment of
PMDS.
Case report
A 17-month-old male was admitted to Beijing
Children's Hospital (Beijing, China) in March 2016 presenting with
an empty right scrotum and left inguinal mass. The patient was a
full term baby with normal labor and had no siblings. The parents
were non-consanguineous with no history of infertility and the
patient had normal intelligence. The results of the physical
examinations were: Height, 79 cm; weight, 10 kg; proportionate
anatomy, no malformation of the head or limbs; pubic hair, Tanner
stage I (10); penis size, 4.3×1.5
cm; and a normal meatus. However, the two sides of the scrotum were
small with few folds, the right scrotum was empty and the top of
the left scrotum reached the testis and another soft tissue with no
tenderness.
A peripheral venous blood sample (2 ml) was taken
from the patient, and cultured for 72 h at 37°C with RPMI-1640
Medium (5 ml; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Dividing blood cells were then obtained and 0.1 ml colchicine
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added to stop
cell division. Hypotonic solution was then used to induce
cytolysis. The cells were fixed on glass slides with methanol and
glacial acetic acid for 20 min at room temperature, and their
structures and quantities were observed under a microscope at ×100
magnification with an oil immersion lens following giemsa staining
for 8 min at room temperature. A further 4 blood samples (2 ml)
were collected from the median cubital vein, and further serum
tests were performed following centrifuging blood samples at 2,200
× g for 7 min at 37°C. Samples were analysed via chemiluminescense
and radioimmunoassays, using a Cortisol Radioimmunoassay kit
(0304103D4101-1), Adrenocorticotropic Hormone Radioimmunoassay kit
(0304103D4001; both Roche Applied Science, Penzberg Germany) and
Hepatic kit (AU5800; Beckman Coulter, Inc., Brea, CA, USA). The
laboratory results were: Karyotype, 46,XY; sex determining region Y
protein +; normal liver and kidney function and normal electrolyte,
adrenocorticotropic hormone and cortisol levels. The results of the
sex hormone tests indicated that the patient was pre-pubescent
(Table I). A pelvic ultrasound
indicated that two testicular solid hypoechoic masses 1.2×0.5×0.6
and 1.4×0.5×0.8 cm large were present at the entrance of the deep
inguinal ring and a normal blood flow signal was detected. There
were no testicular masses identified during the examination of the
right scrotum, inguinal region, inguinal ring or abdomen. A
hyperechoic strip measuring ~1.3×0.7×2.2 cm was present behind the
left side of the bladder and a uterine hyperechoic line was
identified inside, which suggested the presence of a dysplastic
uterus behind the left side of the bladder. The result suggests a
dysplastic uterus behind. Genetic testing was performed in Beijing
Key Laboratory for Genetics of Birth Defects by Sanger sequencing
of AMHR-II, which indicated a compound heterozygous nucleotide
variation. This was performed using KAPA HiFi HotStart ReadyMix
(KAPA Biosystems, Inc., Wilmington, MA, USA) and the following
primers: c.1184(E9) to c.1185(E9), forward
5′-TGTGTCAACAGTTGTAGCAATACC-3′, and reverse
5′-GACTGTACCACCCATCCTTACC-3′; and c.1388G>A(E10), forward
5′-ACCCCTAGGACTAACTGATACCC-3′ and reverse
5′-TGAACCAAGGAGTGATGGGAGATA-3′.
 | Table I.Clinicopathological characteristics of
the patient. |
Table I.
Clinicopathological characteristics of
the patient.
| Clinicopathological
characteristics | Results | Normal ranges |
|---|
| Age (months) | 17 | – |
| Height (cm) | 79 | – |
| Testicular volume
(ml) | 2/2 | – |
| AST (U/l) | 32.4 | 5.0–40.0 |
| ALT (U/l) | 14.8 | 5.0–40.0 |
| Cortisol (µg/ml) | 5.84 | 5.0–25.0 |
| ACTH (pg/ml) | 13.5 | 0–46 |
| LH (IU/l) | 0.12 | ≤4.1 |
| FSH (IU/l) | 0.74 | ≤5.5 |
| T (ng/dl) | <20.0 |
0-21.2 |
| E2 (pg/ml) | 28.4 |
0-20.0 |
| PRL (ng/ml) |
4.23 | 0.6–29.0 |
| PGN (ng/ml) | <0.2 | ≤1.3 |
| hCG (IU/l) | <1.0 | <2.5 |
| AMH (ng/ml) | >23 | 1.43–11.60 |
| INH-B (pg/ml) | 54.8 | Normal sperm
production, 75–350 |
|
|
| Suspicious
spermatogenesis disturbance, 50–80 |
|
|
| Spermatogenesis
disturbance, <50 |
The c.1184 (E9) to c.1185 (E9) CT deletion mutant
gene originated from the father of the patient. This mutation led
to a frameshift change resulting in a truncated protein. The
c.1388G>A (E10) mutant site was derived from the patient's
mother and caused a change in the p.463, R>H, resulting in the
alteration of protein structure, inducing a conformational change
in AMHR-II. This change led to a decrease in AMHR-II binding
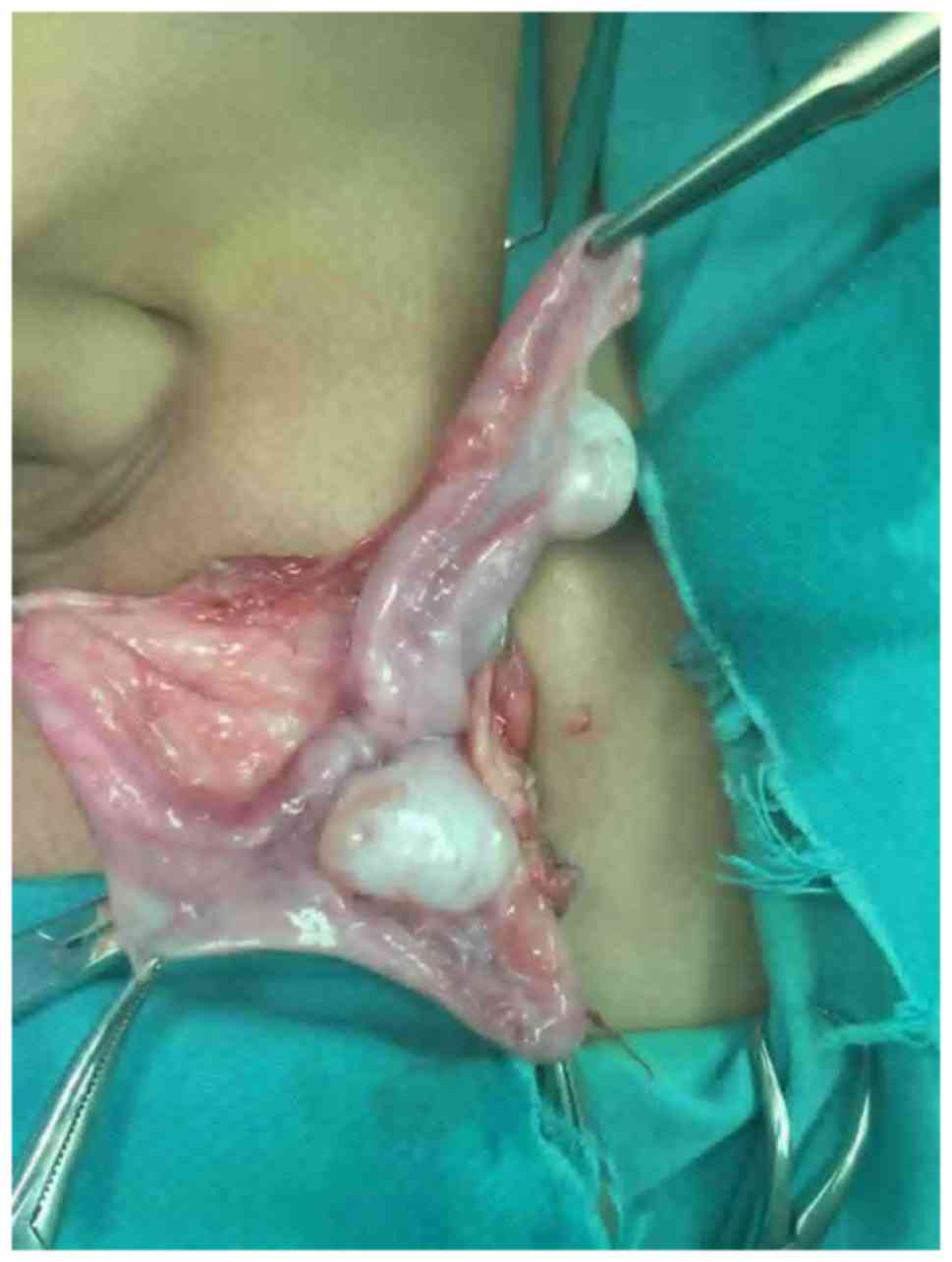
capacity. This patient underwent a left transverse testicular
ectopia orchidopexy and during this, a similar type of uterine
tissue was identified between the two testes (Fig. 3).
Discussion
PMDS was first described by Nilson (11) in 1939 as a rare type of male
pseudohermaphroditism characterized by the presence of MD
derivatives in genotypic and phenotypic males with a 46,XY
chromosome pattern (1,2,5). It is
caused by a deficiency in AMH or a defect in AMHR-II (12–14). The
patterns of PMDS inheritance include X linkage, autosomal dominant
and autosomal recessive inheritance (14). ‘Persistent Müllerian duct syndrome’
was used as a search term for case reports and reviews with the
PubMed database (www.ncbi.nlm.nih.gov/pubmed), covering 45 articles in
foreign languages, and 5 articles in Chinese. In total, there have
been ~150 cases of PMDS reported in adults, the majority in the US,
Europe and the Middle East (5). In
China, 11 pediatric patients were diagnosed with PMDS during
laparoscopic treatment at the Children's Hospital of Fudan
University (Shanghai, China) (6)
without the use of genetic analysis. Two adult patients were
misdiagnosed with ‘34 years of right inguinal hernia’ and ‘>20
years of left inguinal hernia with an empty right hemiscrotum’,
respectively, at the First Affiliated Hospital of Nanjing Medical
University (Nanjing, China) (7).
These misdiagnoses may have been caused by the lack of
under-virilism in these two adult patients; however, underdeveloped
uteruses were identified during surgical examination and then the
patients were subsequently diagnosed with PMDS. The incidence of
PMDS is low and there is a number of possible reasons for this: i)
Little is known about PMDS and therefore cryptorchidism surgery is
rarely considered to be indicative of PMDS when the patients
exhibit a normal virilism phenotype; and ii) physicians may lack
understanding and knowledge of the structure of MDs (15). There have been four cases of PMDS
diagnosed in infants (4,16), however, to the best of our knowledge
there have been no studies that have conducted genetic analysis
prior to diagnosis. The current study described the clinical data
and genetic analysis of a 17-month-old patient diagnosed with
PMDS.
The internal genital canal consists of MDs and
Wolffian ducts prior to sex differentiation during the first 7
weeks of gestation. The sex differentiation of a normal male
depends on testosterone, dihydrotestosterone and AMH (17). AMH is a member of the transforming
growth factor β superfamily with a molecular weight of 140 kDa and
is the first hormone to be synthesized by Sertoli cells (5,18). Its
physiological function is to induce MD regression during male sex
differentiation. Due to the existence of the sex-determining region
Y (SRY) gene in males, undifferentiated gonads develop into testes
and immature Sertoli cells begin to secrete AMH to induce the
regression of the MDs at the end of week 7 of gestation. AMH
expression is not dependent on gonadotropins in the embryo stage;
it is activated by SRY9 and then splicing factor-1 (SF-1), GATA
binding protein 4 and Wilms tumor 1 to increase transcription
(19). Following activation of the
hypothalamic-pituitary-gonadal axis, AMH upregulates follicle
stimulating hormone (FSH) and downregulates androgen (19). Gonadotropin secretion is low in the
week following birth and reaches a peak within 6 months (mini
puberty). Sertoli cell proliferation is stimulated by FSH and
increases the size of the testes and AMH secretion, which causes
AMH levels to peak. Luteinizing hormone then stimulates Leydig
cells to produce testosterone, stimulating testis descent (20,21).
Following the peak, AMH levels decrease as FSH levels also decrease
to baseline levels. The secretion of testosterone increases
following activation of the hypothalamic-pituitary-gonadal axis and
in response, the androgen receptor is expressed in Sertoli cells
(22). Androgens bound to androgen
receptors then suppress transcription of SF-1, which leads to a
decrease in AMHF expression (19,23).
Tumor necrosis factor-α produced by testicular meiosis of
spermatogenic cells activates nuclear factor (NF)-κB in Sertoli
cells. The activated NF-κB then enters the nucleus and binds to
SF-1, the promoter of AMH, to inhibit AMH expression (24).
The development of a normal male requires
testosterone, which is secreted by Leydig cells, to facilitate the
development of the epididymis, sperm duct and seminal vesicles
(17). A deficiency in AMH or a
defect in AMHR-II in male embryos leads to the development of PMDS.
The secretion and function of testosterone are not influenced
during PMDS and therefore, the development of the derivatives of
the Wolffian ducts and external genital differentiation occurs
normally. Therefore, patients with PMDS exhibit a 46,XY chromosome
pattern and MDs are present, although they exhibit a normal male
phenotype. The cryptorchidism phenomenon of PMDS may be explained
by decreased levels of insulin-like 3 (INSL-3), or because the
Müllerian duct construct mechanically pulls the testes, thereby
preventing testicular descent. INSL-3 levels were not detected in
the current study; however, it is hypothesized that mechanical
pulling by the MD construct may be the reason for
cryptorchidism.
PMDS is primarily caused by mutations in the AMH and
AMHR-II genes (25,26). The AMH gene is located on chromosome
19p13 and is 2.75 kbp long (8), with
mutations occurring in ~45% of patients with PMDS. The AMHR-II gene
is located on chromosome 12q13 and is 8.7 kbp long (8) with mutations occurring in ~40% of
patients with PMDS (25,27). The cause of PMDS is unclear in ~15%
of patients, as AMH or AMHR-II gene mutations cannot be detected.
PMDS may be caused by a mutation in the AMH transduction pathway in
these patients (18,25).
The majority of mutations in the AMH gene have been
identified in patients from the Mediterranean area and Saudi
Arabia. These are mostly missense mutations and 81% are homozygous
(8,28). Mutations may occur in any region of
the gene apart from exon 4, however, they most commonly arise in
exon 1 (8). It has been demonstrated
that the site of recurrent mutations may affect AMH processing and
secretion (8). A total of 26 AMHR-II
mutations have been identified to date and the majority has been
identified in patients from Northern Europe and the US, with 45% of
mutations being homozygous (8). The
most common mutation of AMHR-II that accounts for 47% of AMHR-II
mutations is del6331-6357, which is located on exon 10 and results
in the deletion of 27bp (8,28). This mutation affects the kinase of
AMHR-II, as a deletion may cause the early occurrence of a stop
codon, which leads to receptor truncation following the
transmembrane domain (8). To the
best of our knowledge, no genetic analyses have been performed in
Chinese patients.
In the current study, a novel compound heterozygous
variation was identified in the patient using genetic analysis. In
the mutant gene, there was a deletion of CT from c.1184 (E9) to
c.1185 (E9) that originated in the father of the patient and led to
a frame shift, resulting in the translation of a truncated protein.
The mutation site c.1388G>A (E10) was inherited from the mother
of the patient which led to a change in p.463, R>H, altering the
protein structure and causing a decrease in the level of its
binding. These two mutations have been identified as pathogenic by
the Protein Variation Effect Analyzer (provean.jcvi.org/index.php) and SIFT (sift.jcvi.org) (29).
Therefore, although they have not been reported previously, their
pathogenicity may be explained.
Based on clinical expression, hormone detection and
testicular biopsy are essential in the diagnosis of PMDS in adults;
however, there are fewer cases of PMDS diagnosed in children due to
naturally high levels of AMH in prepubescent boys, which makes
diagnosis more challenging (17,30).
There are no specific clinical symptoms, hormones or ultrasonic
tests for PMDS; therefore diagnosis is challenging for
pediatricians, particularly if they lack experience. It is also
difficult to distinguish PMDS from partial testicular dysfunction
(31). Therefore, further genetic
mutation analysis is required when diagnosing PMDS in children. The
patient in the current case report presented with an empty right
scrotum and left inguinal mass, which differs from the symptoms
identified in adult with PMDS, which include sterility,
oligospermia and inguinal hernia. The results of the physical
examination indicated an empty right scrotum, the top of the left
scrotum reached the testis, a normal sized external genital organ
and no hypospadias. In addition, AMH levels were increased whereas
levels of other hormones tested were within normal ranges for a
pre-pubescent boy. It should be noted that testosterone levels in
adults diagnosed with PMDS are usually within normal ranges. During
ultrasonic examination of the testis and pelvis, a left transverse
testicular ectopia and uterine hyperechoic line were identified
behind the left side of the bladder and the MD structure inside the
pelvic cavity was examined, supporting the diagnosis of PMDS. A
karyotyping test was subsequently performed to determine the
chromosomal sex and the results determined that the patient's
chromosome karyotype was 46, XY. Based on the symptoms of the
patient, it was suspected that the patient had PMDS. Genetic
analysis was then conducted with permission from the patient's
parents and a mutation in the AMHR-II gene was identified,
confirming the diagnosis of PMDS. Left transverse testicular
ectopia orchidopexy also confirmed diagnosis and during surgery,
there was a similar type of uterine like tissue identified between
the two testes.
Remnants of MD and ectopic testis must be managed
for effective treatment of PMDS. The primary aim of treatment is to
maintain the patient's fertility, as well as prevent or detect the
malignant transformation of the MDs and ectopic testis early on. It
has been demonstrated that abnormalities in the number of germ
cells of convoluted seminiferous tubules in patients with
cryptorchidism begins at the age of 1 year and the damage becomes
permanent after the age of 2. Undescended testes in the inguinal
canal and abdomen hinder the production of sperm; therefore an
orchidopexy is required by the age of 2 (32). In cases of cryptorchidism, the testes
typically descend spontaneously for those <2 years old,
therefore it is recommended that orchidopexy only be conducted in
patients aged >2 years.
It has been demonstrated that there is an
improvement in the fertility of patients with intra-abdominal
testis that undergo surgery prior to puberty. If the cord is long
enough, an orchidopexy may be performed so that the spermatic cord
brings the testicle down to the scrotum and the testicle can be
fixed there. Follow-up should then be performed following surgery.
If the cord is too short or separation of one of the arteries is
unsuccessful in creating a longer cord, the testicle may be placed
under the skin in the groin region and gonadotropin treatment may
be used so to promote testicular descent. If testicles remain
undescended, orchiectomy should not be considered. If the testicle
was located in the groin, it is easier to be observed in case
malignant transformation and other complications. Fertility is
maintained in only 14% of patients with intra-abdominal testis who
are diagnosed and treated following puberty (32). The malignant transformation rate is
also increased compared with those treated prior to puberty,
therefore, an orchiectomy may be considered. However, this is a
serious procedure and it is necessary to discuss this with children
and their parents due to the requirements of lifelong androgen
treatment following this procedure in childhood (31,33).
This procedure is also controversial due to the low malignancy rate
for ectopic testis in the inguinal hernia sac; therefore an
orchidopexy may be performed (9,34). A
lifetime of androgen replacement therapy is required when testicle
atrophy occurs, the testicles become cancerous or following removal
of the testes (33). If malignant
transformation has already occurred, removal of the testes must be
performed, in addition to radiation therapy and chemotherapy
(25). The overall incidence of
malignant transformation is 15% (17,30). The
higher the position of the testes, the higher the risk of malignant
transformation in patients with PMDS with cryptorchidism, however,
there is no difference in the malignant transformation rate between
the two sides of the ectopic testes (32). The results of previous reviews
demonstrate that malignant transformation occurs 6–18 years
following cryptorchidopexy. However, there is no correlation
between onset of malignant transformation and surgical treatment or
patient age (35,36). This indicates that the risk of
malignant transformation is congenital and does not depend on
external causes.
Treatment of the remnants of MD remains
controversial. Previous studies have demonstrated that children
with PMDS require removal of MD remnants due to malignant tumors
identified in Müllerian remnants and the MD being connected with
the seminal vesicle causing urinary tract infections, periodic
hematuria, stones and urination disorders (25,27).
However, other studies have suggested retaining Müllerian remnants
to prevent damage to the vas deferens and disruption of collateral
blood supply to the testes (25,27).
A long-term management strategy includes assessment
of testicular function and monitoring of malignant transformation
at an earlier stage (35,36). A total of 30 cases of malignant
transformation have been identified in patients with PMDS (2). The majority of malignant tumors are
identified in ectopic testes and few originate from the Müllerian
remnants. Only one patient has been reported to develop
adenocarcinoma of endocervical origin (37) and one other patient developed clear
cell adenocarcinoma (38).
Testicular tumors include seminomas, embryonic cell carcinoma, yolk
sac tumors and teratomas, of which seminomas are the most common
(2). In China, there has only been
one case of malignant seminoma reported (39). The long-term growth, development and
fertility of patients with PMDS must be considered prior to removal
of non-functional MD structures.
In conclusion, PMDS is a rare disease. Due to the
lack of distinct clinical features at an early stage, PMDS is
usually diagnosed when children are tested for other diseases,
including cryptorchidism and inguinal hernia. The early diagnosis
and treatment of PMDS in children is therefore challenging. Correct
diagnosis depends on a systematic endocrine assessment and genetic
analysis. The case presented in the current study furthers
knowledge of the gene profile of PMDS. However, removal of MD
structures from patients with PMDS as a treatment strategy remains
controversial and further studies investigating its impact are
required.
Acknowledgements
The present study was funded by The National Key
Research and Development Program of China (grant no.
2016YFC0901505) and Beijing Municipal Science and Technology
Funding (grant no. Z151100003915103).
References
|
1
|
Smith-Harrison LI, Patel MS, Smith RP and
Schenkman NS: Persistent Mullerian duct structures presenting as
hematuria in an adult: Case report of robotic surgical removal and
review of the literature. Urol Ann. 7:544–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barad AK, Bharath NM, Narayana V, Raja VO
and Jambula PR: Persistent Mullerian duct syndrome with Embryonal
cell carcinoma along with Ectopic cross fused kidney. J Clin Diagn
Res. 10:PD07–PD08. 2016.PubMed/NCBI
|
|
3
|
Clarnette TD, Sugita Y and Hutson JM:
Genital anomalies in human and animal models reveal the mechanisms
and hormones governing testicular descent. Br J Urol. 79:99–112.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shalaby MM, Kurkar A, Zarzour MA, Faddan
AA, Khalil M and Abdelhafez MF: The management of the persistent
Mullerian duct syndrome. Arab J Urol. 12:239–244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nerune SM, Hippargi SB, Mestri NB and
Mehrotra NM: Persistent Mullerian duct syndrome with ovarian
endometriosis-a rare case report. J Clin Diagn Res. 10:ED14–ED15.
2016.PubMed/NCBI
|
|
6
|
Shen J and Bi YL: Laparoscopic management
of Persistent Müllerian duct combined with cystoscope. J Clin
Pediat Surg. 12:107–109. 2013.
|
|
7
|
Ju XB, Qian LX, Zhang W, Wu HF, Xu ZQ and
Sui YG: Persistent Müllerian duct syndrome: report of two cases.
Chin J Surg. 45:863–864. 2007.
|
|
8
|
di Clemente N and Belville C:
Anti-Mullerian hormone receptor defect. Best Pract Res Clin
Endocrinol Metab. 20:599–610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fonkalsrud EW: Current management of the
undescended testis. Semin Pediatr Surg. 5:2–7. 1996.PubMed/NCBI
|
|
10
|
Marshall WA and Tanner JM: Variations in
the pattern of pubertal changes in boys. Arch Dis Child. 45:13–23.
1970. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nilson O: Hernia Uteri Inguinalis beim
manne. Aeta Chir Scand. 83:2311939.
|
|
12
|
Palanisamy S, Patel ND, Sabnis SC,
Palanisamy N, Vijay A and Chinnusamy P: Laparoscopic hysterectomy
with bilateral orchidectomy for Persistent Mullerian duct syndrome
with seminoma testes: Case report. J Minim Access Surg. 11:273–275.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohapatra M and Subramanya YS: Persistent
Müllerian duct syndrome of mixed anatomical variant (combined male
and female type) with mixed germ cell tumor of left intra-abdominal
testis. Indian J Pathol Microbiol. 59:212–215. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mohammadi Sichani M, Heidarpour M, Dadkhah
A and Rezvani M: Persistent mullerian duct syndrome with an
irreducible inguinal hernia. Urol J. 6:298–300. 2009.PubMed/NCBI
|
|
15
|
Dekker HM, de Jong IJ, Sanders J and Wolf
RF: Persistent Mullerian duct syndrome. Radiographics. 23:309–313.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kamble RS, Gupta RK, Gupta AR, Kothari PR,
Dikshit KV and Kesan KK: Laparoscopic management of transverse
testicular ectopia with persistent mullerian duct syndrome. J Minim
Access Surg. 11:213–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Renu D, Rao BG, Ranganath K and Namitha:
Persistent mullerian duct syndrome. Indian J Radiol Imaging.
20:72–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Belville C, Josso N and Picard JY:
Persistence of Müllerian derivatives in males. Am J Med Genet.
89:218–223. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matuszczak E, Hermanowicz A, Komarowska M
and Debek W: Serum AMH in physiology and pathology of male gonads.
Int J Endocrinol. 2013:1289072013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grinspon RP and Rey RA: Anti-müllerian
hormone and sertoli cell function in paediatric male hypogonadism.
Horm Res Paediatr. 73:81–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang F, Zhang H, Liu QX, et al: A clinical
study on the detection of serum anti-Mullerian hormone and inhibin
B in 1400 cases of healthy children aged 0 to 5 years. Chin J
Practical Pediatric. 31:450–454. 2016.
|
|
22
|
Teixeira J, Maheswaran S and Donahoe PK:
Müllerian inhibiting substance: An instructive developmental
hormone with diagnostic and possible therapeutic applications.
Endocr Rev. 22:657–674. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rey R, Lukas-Croisier C, Lasala C and
Bedecarrás P: AMH/MIS: What we know already about the gene, the
protein and its regulation. Mol Cell Endocrinol. 211:21–31. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aksglaede L, Sorensen K, Boas M, Mouritsen
A, Hagen CP, Jensen RB, Petersen JH, Linneberg A, Andersson AM,
Main KM, et al: Changes in anti-Müllerian hormone (AMH) throughout
the life span: A population-based study of 1027 healthy males from
birth (cord blood) to the age of 69 years. J Clin Endocrinol Metab.
95:5357–5364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patil V, Muktinaini S, Patil R and Verma
A: Persistent mullerian duct syndrome: A case report. Indian J
Surg. 75 Suppl 1:S460–S462. 2013. View Article : Google Scholar
|
|
26
|
Hoshiya M, Christian BP, Cromie WJ, Kim H,
Zhan Y, MacLaughlin DT and Donahoe PK: Persistent Mullerian duct
syndrome caused by both a 27-bp deletion and a novel splice
mutation in the MIS type II receptor gene. Birth Defects Res A Clin
Mol Teratol. 67:868–874. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manjunath BG, Shenoy VG and Raj P:
Persistent mullerian duct syndrome: How to deal with the mullerian
duct remnants-a review. Indian J Surg. 72:16–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Imbeaud S, Belville C, Messika-Zeitoun L,
Rey R, di Clemente N, Josso N and Picard JY: A 27 base-pair
deletion of the anti-müllerian type II receptor gene is the most
common cause of the persistent müllerian duct syndrome. Hum Mol
Genet. 5:1269–1277. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng J, Lin R, Zhang W, Liu G, Sheng H,
Li X, Zhou Z, Mao X and Liu L: Phenotype and molecular
characteristics in 45 Chinese children with 5α-reductase type 2
deficiency from South China. Clin Endocrinol (Oxf). 83:518–526.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prakash N, Khurana A and Narula B:
Persistent Müllerian duct syndrome. Indian J Pathol Microbiol.
52:546–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang SH, Jiang DP and Li ZZ: Research
progress of persistent Müllerian duct syndrome. Chin J Pediatr
Surg. 37:73–76. 2016.
|
|
32
|
Shao FM, Mao QL, Yang HM and Hou LJ:
Persistent Mullerian duct syndrome: A report of two cases and
literature review. Chin J Pediateic Surg. 32:468–470. 2011.
|
|
33
|
Odi TO, Abdur-Rahman LO and Nasir AA:
Persistent Mullerian duct syndrome: A case report and review of the
literature. Afr J Paediatr Surg. 7:191–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Batata MA, Whitmore WF Jr, Chu FC, Hilaris
BS, Loh J, Grabstald H and Golbey R: Cryptorchidism and testicular
cancer. J Urol. 124:382–387. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Manassero F, Cuttano MG, Morelli G,
Salinitri G, Spurio M and Selli C: Mixed germ cell tumor after
bilateral orchidopexy in persistent Mullerian duct syndrome with
transverse testicular ectopia. Urol Int. 73:81–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Melman A, Leiter E, Perez JM, Driscoll D
and Palmer C: The influence of neonatal orchiopexy upon the testis
in persistent Mullerian duct syndrome. J Urol. 125:856–858. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thiel DD and Erhard MJ: Uterine
adenosarcoma in a boy with persistent mullerian duct syndrome:
First reported case. J Pediatr Surg. 40:e29–e31. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shinmura Y, Yokoi T and Tsutsui Y: A case
of clear cell adenocarcinoma of the müllerian duct in persistent
mullerian duct syndrome: The first reported case. Am J Surg Pathol.
26:1231–1234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu DX, Fang WH and Jiang S: Persistent
Müllerian duct syndrome with transverse testicular ectopia (report
of 2 cases). Chin J Urol. 23:678–680. 2002.
|