Introduction
Cardiac endothelial cells (ECs) serve an essential
role in regulating blood perfusion, myocyte survival and
contractile performance. The endothelium in the myocardial
capillaries is in direct contact and communication with
cardiomyocytes through a paracrine signalling pathway (1–3).
Endothelial dysfunction due to ischaemia/reperfusion (I/R) has been
reported to attenuate the flow or no-reflow phenomenon (1,4–6). In addition, the diapedesis of
neutrophils from the endothelium into the interstitial myocardium
is a hallmark of inflammation elicited by I/R (7). The activated endothelium and
neutrophils release a variety of mediators to modulate cell injury,
including proteases, oxygen-derived free radicals and
pro-inflammatory cytokines (8).
Endothelial dysfunction has been suggested as a trigger of the
neutrophil amplification in I/R (8–10). In
I/R injury, evidence has demonstrated that ECs are the first cell
type to undergo apoptosis and to consequently migrate to
neighbouring myocytes (11,12). This would suggest that the
endothelium is a more sensitive and soluble apoptogenic mediator
from damaged endothelium that may diffuse into surrounding cardiac
myocytes (11). In addition, the
co-culture of ECs and cardiomyocytes under a hypoxia/reoxygenation
(H/R) injury was demonstrated to reduce the lactate dehydrogenase
(LDH) activity and increase the nitric oxide production, suggesting
that the endothelium may contribute to cardioprotection against an
H/R injury (13). Therefore, it is
hypothesised that maintaining endothelial function may be a
potential strategy to minimise I/R injury.
Secretory leukocyte protease inhibitor (SLPI) is an
inhibitor of serine protease regulating enzymatic activity and
synthesis (14). Previously, it has
been demonstrated that SLPI promotes early myocardial contraction,
as well as inhibiting post-ischaemic inflammation in cardiac
transplantation (15). Particularly
during IR injury, increased protease production and intracellular
Ca2+ overload promote the activation of
Ca2+-dependent protease activity (9,16). Thus,
inhibition of the secreted protease activity may be an attractive
target of SLPI, and may have a potential therapeutic effect in
limiting cellular injury during I/R. However, the effect of SLPI in
endothelial I/R injury remains unexplored.
Therefore, the aim of the present study was to
determine the effect of recombinant human SLPI (rhSLPI) treatment
against endothelial I/R injury in human umbilical vein ECs
(HUVECs). The cell viability and expression of various mediators,
such as lactase dehydrogenase (LDH) and reactive oxygen species
(ROS), were assessed following rhSLPI treatment at different time
points, including prior to ischaemia, during ischaemia and at the
onset of reperfusion, in the cultured ECs subjected to I/R.
Furthermore, the activation of cellular stresses, p38
mitogen-activated protein kinase (MAPK), protein kinase B (Akt), as
well as the expression levels of apoptosis-regulating proteins,
including B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X protein
(Bax) and cleaved caspase-3, were determined.
Materials and methods
Chemicals and reagents
rhSLPI was purchased from Sino Biological, Inc.
(Beijing, China). Medium 200 (M-200-500), Low Serum Growth
Supplement kit (S003K-LSGS) and trypsin-EDTA were purchased from
Thermo Fisher Scientific, Inc. (Gibco; Waltham, MA, USA). The LDH
liquid-UV assay kit was obtained from Human (Wiesbaden, Germany),
and MTT was purchased from Ameresco, Inc. (Solon, OH, USA).
Antibodies against total p38 (sc-728), phosphorylated-p38
(sc-17852-R), total Akt (sc-8312), phosphorylated-Akt (sc-293125),
Bax (sc-6236), Bcl-2 (sc-783), and cleaved caspase 3 (sc-56053),
β-actin (sc-130301), and the horseradish peroxidase-conjugated
secondary antibody (sc-2004) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Other chemicals were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell culture
HUVECs were obtained from Thermo Fisher Scientific,
Inc. (C01510C; Gibco) and were maintained in Medium 200
supplemented with the Low Serum Growth Supplement kit. Cells were
maintained at 37°C at an atmosphere with 5% CO2 and 95%
O2 until they reached 80% confluence prior to performing
any subsequent experiments.
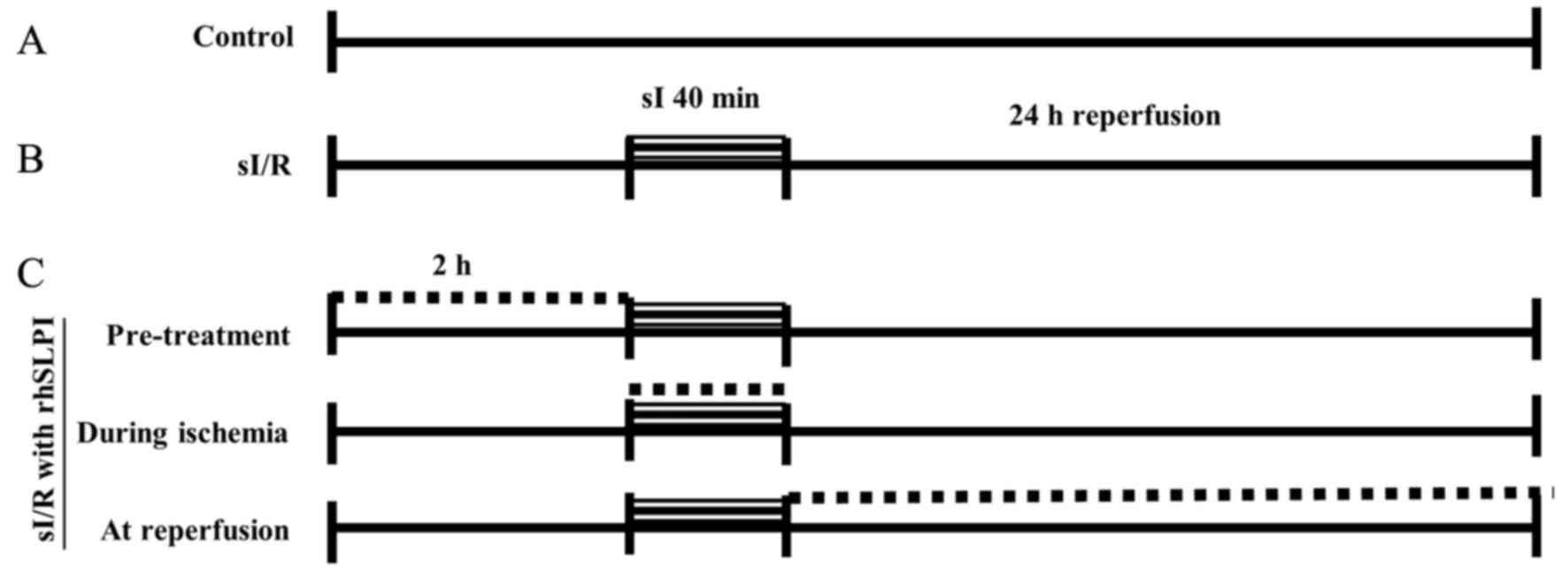
Treatment protocol
HUVECs were cultured in 96-well plates (at 10,000
cells/cm2) until 80% confluence was reached. Next, cells
were treated with various concentrations (0, 1, 10, 100 and 1,000
ng/ml) of rhSLPI at different time points, including 2 h prior to
ischaemia (pretreatment group), at the onset of ischaemia until end
of ischaemic period (during ischaemia group), and at the onset of
reperfusion until the end of reperfusion period (reperfusion
group). During these experiments, simulated ischaemia was performed
for 40 min, followed by 24 h of reperfusion. In the control group,
cells were incubated with control buffer, during the simulated
ischaemia. The different treatments are shown in more detail in
Fig. 1.
Simulated I/R (sI/R)
In HUVECs, simulated ischaemia was induced by
incubating the cells with a modified Krebs-Henseleit buffer
(containing 137 mM NaCl, 3.8 mM KCl, 0.49 mM MgCl2, 0.9
mM CaCl2 and 4.0 mM HEPES) supplemented with 20 mM
2-deoxyglucose, 20 mM sodium lactate and 1 mM sodium dithionite at
pH 6.5, as previously described (17). The buffer used in the control group
was composed of the Krebs-Henseleit buffer supplemented with 20 mM
D-glucose and 1 mM sodium pyruvate. For optimizing the simulated
ischemic duration to lead to cell death, cells were subjected to
simulated ischaemia at 37°C in an atmosphere with 5% CO2
for 40 min, then the simulated ischaemic buffer was removed and
replaced with complete medium for reperfusion and incubating at
37°C in 5% CO2 for 24 h. Cell viability was then
measured via MTT assay. For the simulated ischemic duration that
gave the strongest p38 MAPK activation, cells were exposed with
simulated ischaemic buffer for different durations (10, 20, 30, 40,
50, and 60 min). At each time point, the ischaemic buffer was
removed and the cellular protein sample was collected by adding
Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 2%
SDS, 0.01% Bromophenol blue). Protein samples were kept at −20°C
prior to western blot analysis.
Measurement of cell viability
HUVECs were cultured as mentioned earlier and the
cell viability was examined by an MTT assay. Briefly, subsequent to
removal of the culture media, 0.5 mg/ml MTT reagent was added and
incubated for 2 h at 37°C. Following incubation, the excess MTT
reagent was discarded and dimethyl sulfoxide (DMSO) was added to
dissolve the formazan crystals. The sample was then centrifuged at
500 × g at 4°C for 5 min. The optical density was determined from
supernatant fraction using a spectrophotometer at a wavelength of
490 nm, using DMSO as the blank group. The relative percentage of
cell viability was compared against the control group.
Measurement of cellular injury
The culture medium was collected from all
experimental groups following completion of the sI/R protocol and
kept at −20°C untill released-LDH activity was determined using the
LDH activity assay kit. Briefly, 10 µl culture medium was mixed
with 1,000 µl reaction buffer and incubated at 37°C for 5 min.
Next, 250 µl of substrate reagent was added, the solution was mixed
and the absorbance was read after 1 min at a wavelength of 340 nm.
The mean absorbance changes per min (ΔA/min) were used to calculate
the LDH activity with the following formula: LDH activity
(U/l)=ΔA/min ×20,000.
Determination of cellular ROS
For the determination of cellular ROS,
1×105 cells/ml HUVECs was cultured in a black 96-well
plate with complete medium at 37°C and 5% CO2 until 80%
confluence was reached. The culture media were removed and the
cells were washed once with PBS prior to incubating with complete
media containing 250 µM 6-carboxy-2′,7′-dichlorodihydrofluorescein
diacetate (carboxy-H2DCFDA) in a dark room for 30 min at
37°C. The medium containing carboxy-H2DCFDA was then
removed and the cells were washed with PBS. For rhSLPI treatment,
200 µl completed medium containing rhSLPI at the concentration
1,000 ng/ml was added and incubated for 1 h at 37°C. Subsequently,
250 µM H2O2 was applied to the cells and
incubated for 30 min at 37°C. The intracellular ROS production was
determined by measuring the fluorescence intensity using an EnSpire
Multimode plate reader (PerkinElmer, Inc., Waltham, MA, USA) with a
suitable set-up for detecting the signal with an excitation
wavelength of 498 nm and an emission wavelength of 522 nm.
Measurement of MAPK activation by
western blot analysis
The protein was extracted from the cells by adding
for Laemmli sample buffer, 100 ng protein of cell lysate was
separated on a 12% SDS-polyacrylamide gel by electrophoresis and
transferred to a polyvinylidene difluoride (PVDF) membrane. The
PVDF membrane was then probed with the appropriate primary antibody
by incubating at 4°C overnight with specific antibodies against
total p38, phosphorylated-p38, total-Akt and phosphorylated-Akt at
1:1,000 dilution. In addition, the expression levels of apoptotic
regulatory proteins were also determined by specific antibodies
against Bax, Bcl-2 and cleaved caspase 3 (1:1,000). Membranes were
subsequently washed using TBS/Tween-20 four times at room
temperature and incubated with to horseradish peroxidase
(HRP)-conjugated secondary antibody (1:2,000) for 1 h at room
temperature. The antibody-antigen complexes were visualised by
enhanced chemiluminescence using Luminata Crescendo Western HRP
substrate (Merck KGaA) and detected using the Gel Doc XR+ system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) Band densities were
quantified using Image Lab software version 5.2.1 (Bio-Rad
Laboratories, Inc.) and compared in order to provide information
concerning the relative abundance of the protein of interest.
Phosphorylated-p38 and -Akt were compared with total p38 and Akt,
respectively, whereas Bax, Bcl-2 and cleaved caspase 3 were
compared against β-actin as a reference protein.
Determination of cell morphology
HUVECs were grown on cell culture slides (SPL Life
Sciences, Pocheon, Korea) at a concentration of 1×105
cells/ml and cultured in complete medium at 37°C and 5%
CO2. Next, the cells were subjected to 40 min of
simulated ischaemia followed by 24 h of reperfusion (sI/R) in the
absence or presence of rhSLPI pretreatment or during ischaemia.
Cells on the culture slides were then washed with PBS and fixed
with a fixative agent (2% formaldehyde and 0.05% glutaraldehyde) at
room temperature for 30 min. The cells were permeabilised with 0.5%
Triton X-100 in PBS for 20 min and then stained with 50 µg/ml
FITC-phalloidin conjugate (Sigma-Aldrich; Merck KGaA) for 40 min in
a dark moist box for staining actin filaments. Subsequently, the
cell culture slides were washed with PBS prior to nuclear staining
with 0.01 µg/ml DAPI (Sigma-Aldrich; Merck KGaA) for 20 min. The
cell culture slides were then mounted by adding 20 µl of 50%
glycerol on a glass slide and sealing the edges with nail varnish.
These samples were kept in a dark chamber until visualised under a
fluorescence microscope (Carl Zeiss Jena GmbH, Jena, Germany).
Statistical analysis
All values are expressed as the mean ± standard
error of the mean. All comparisons were assessed for significance
using analysis of variance, followed by the Tukey-Kramer test when
appropriate. The statistical tests were performed using GraphPad
Prism version 5 software (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to be an indicator of a
statistically significant difference between the results.
Results
Optimisation of sI/R injury and
examination of cytotoxicity of rhSLPI treatment
HUVECs were subjected to various periods of
simulated ischaemia, followed by 24 h of reperfusion (sI/R). The
results demonstrated that simulated ischaemia reduced the cell
viability in a time-dependent manner (Fig. 2A). The sI/R at 40 min reduced the
percentage of cell viability by ~50%, and thus this treatment
duration was used in the sI/R protocol for determining the sI/R
injury-induced cell death in subsequent experiments. In addition,
HUVECs were treated with various concentrations of rhSLPI for 24 h
prior to measuring cell viability. The results indicated that the
cell viability was not affected by rhSLPI treatment and thus no
cytotoxicity was observed (Fig.
2B).
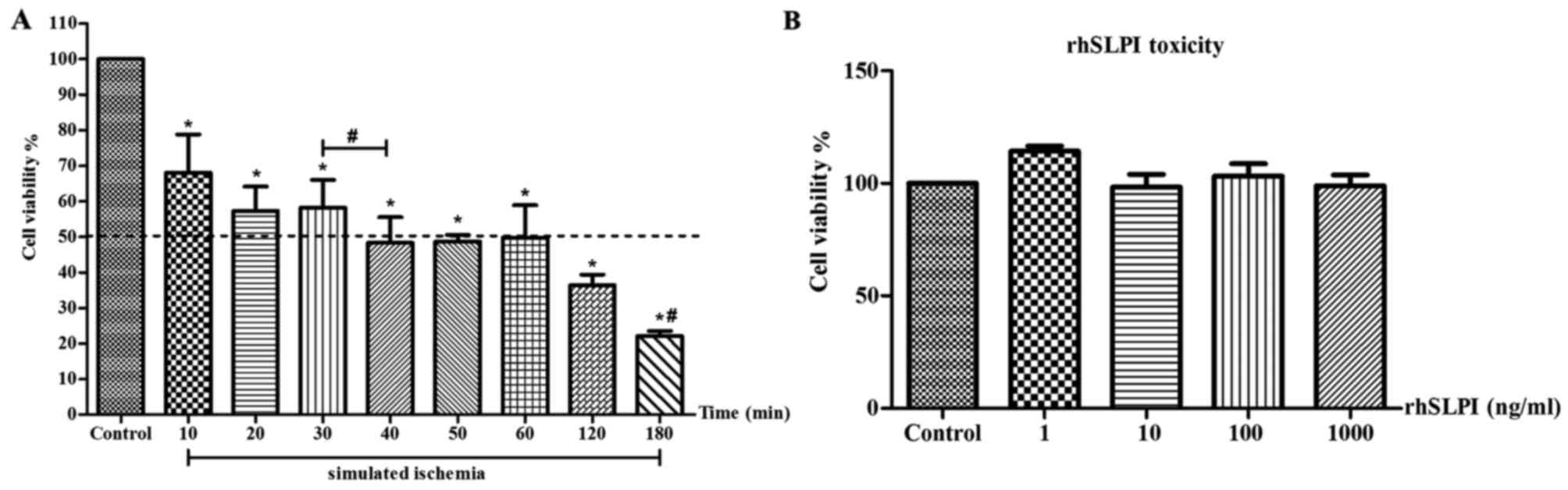 | Figure 2.Optimised time for simulated
ischaemia/reperfusion injury and investigation of rhSLPI
cytotoxicity. (A) Viability of HUVECs subjected to simulated
ischaemia for different durations (10, 20, 30, 40, 50, 60, 120 or
180 min) followed by 24 h reperfusion. Cell viability was measured
after reperfusion. (B) Viability of HUVECs after 24 h of rhSLPI
treatment at the concentrations of 1, 10, 100 or 1,000 ng/ml to
determine the cell toxicity. The results are expressed as the mean
± standard error of six experiments with independent cell
preparations. *P<0.05 vs. control group; #P<0.05
between groups. HUVECs, human umbilical vein endothelial cells;
rhSLPI, recombinant human secretory leukocyte protease
inhibitor. |
Treatment of rhSLPI prior to or during
simulated ischaemia protects against vascular EC death as a result
of sI/R injury
HUVECs were treated with various concentrations of
rhSLPI in three different time points, including pretreatment,
during the simulated ischaemia stage or at the onset of
reperfusion. The results revealed that pretreatment with rhSLPI and
treatment during the ischaemic period increased the cell viability
and reduced the released LDH activity compared with the untreated
sI/R group (Fig. 3). Among the
pretreatment groups, the results indicated that pretreatment with
rhSLPI at 1,000 ng/ml prior to sI/R significantly reduced the
ischaemia-induced cell death when compared with the untreated group
(65.23±4.8 vs. 47.16±1.8%, respectively; P<0.05; Fig. 3A). This concentration also
significantly lowered the released LDH activity when compared with
the untreated group (104±30.3 vs. 247.5±26.9 U/I, respectively;
P<0.05; Fig. 3D). Furthermore,
treatment with rhSLPI at 100 or 1,000 ng/ml during ischaemia
significantly increased the cell viability (67.88±4.2 and
60.74±2.7% vs. 46.26±7.8%, respectively; P<0.05; Fig. 3B). In terms of the released LDH
activity, only rhSLPI at 1,000 ng/ml was able to markedly reduce
the activity when compared with the untreated sI/R group
(57.50±10.3 vs. 247.5±26.9 U/I, respectively; P<0.05; Fig. 3E). By contrast, all the
concentrations of rhSLPI treatment at the onset of reperfusion did
not demonstrate a significant difference in cell viability or the
released LDH activity when compared with the untreated sI/R group
(Fig. 3C and F).
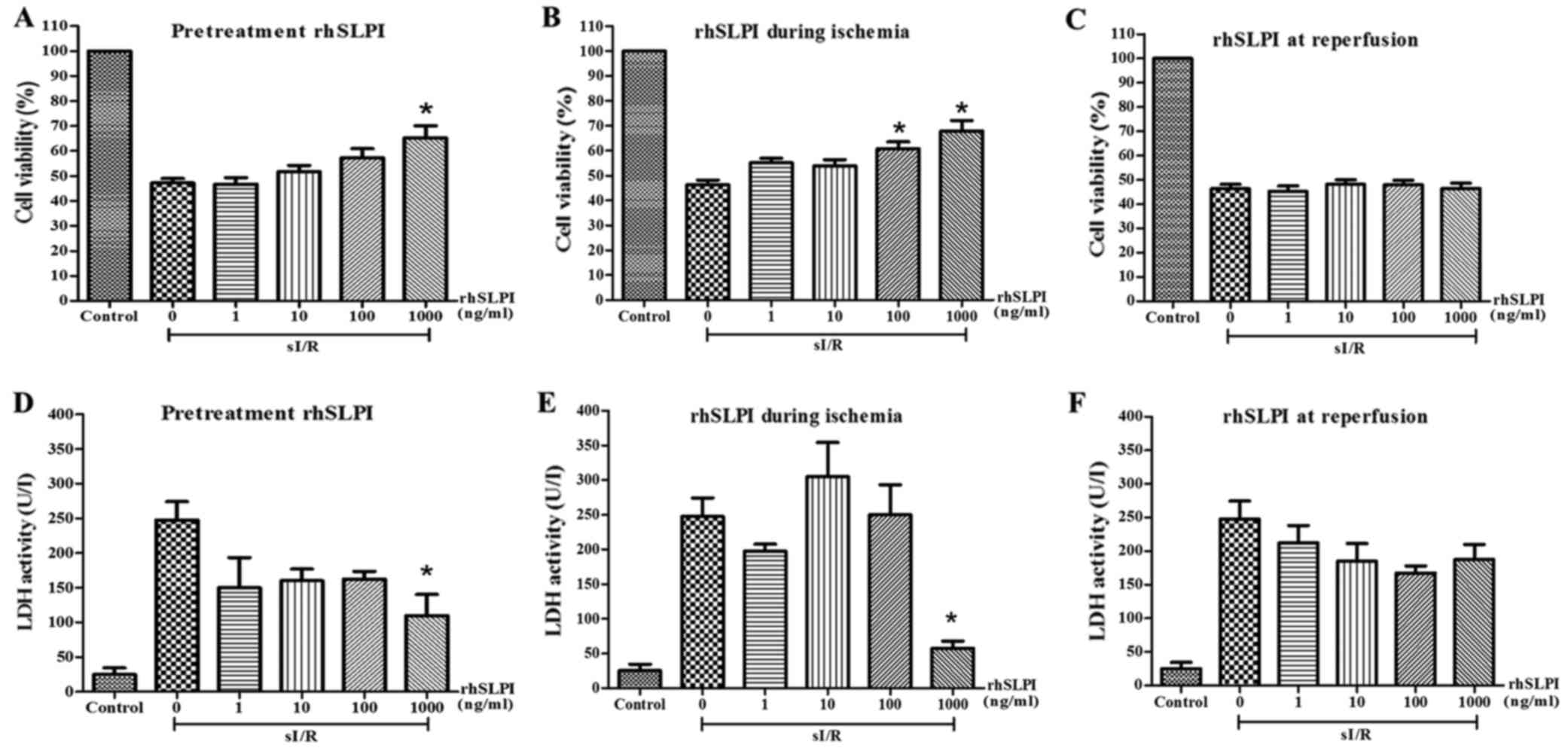 | Figure 3.Effect of rhSLPI on endothelial cell
viability. The cell viability was examined in HUVECs subjected to
sI/R and treated with rhSLPI at the concentrations of 1, 10, 100 or
1,000 ng/ml in the course of (A) 2 h before ischaemia, (B) for 40
min during ischaemia, or (C) at the onset of reperfusion for 24 h.
The medium of HUVECs treated with rhSLPI at different time points
was collected for determining cellular injury by LDH activity assay
when cells were treated (D) 2 h prior to sI/R, (E) during ischaemia
and (F) at the onset of reperfusion. The results are expressed as
the mean ± standard error of 4–6 experiments with independent cell
preparations. *P<0.05 vs. untreated sI/R group. HUVECs, human
umbilical vein endothelial cells; rhSLPI, recombinant human
secretory leukocyte protease inhibitor; sI/R, simulated
ischaemia/reperfusion; LDH, lactate dehydrogenase. |
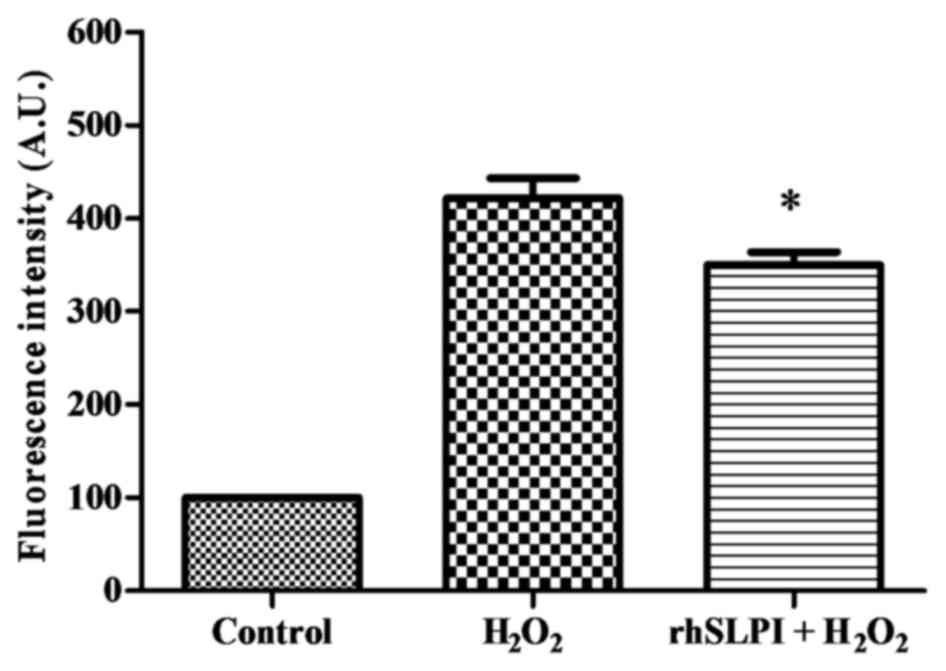
Effect of rhSLPI on intracellular ROS
production
To examine whether rhSLPI was able to reduce
intracellular ROS production, an H2O2
challenge was performed. The results demonstrated that treatment
using the H2O2 challenge resulted in a
significant increase in intracellular ROS production by ~4-fold
when compared with the control group (Fig. 4). However, pretreatment with 1,000
ng/ml rhSLPI was observed to significantly reduce the
H2O2-induced oxidative stress by ~17%
compared with the H2O2 group (Fig. 4).
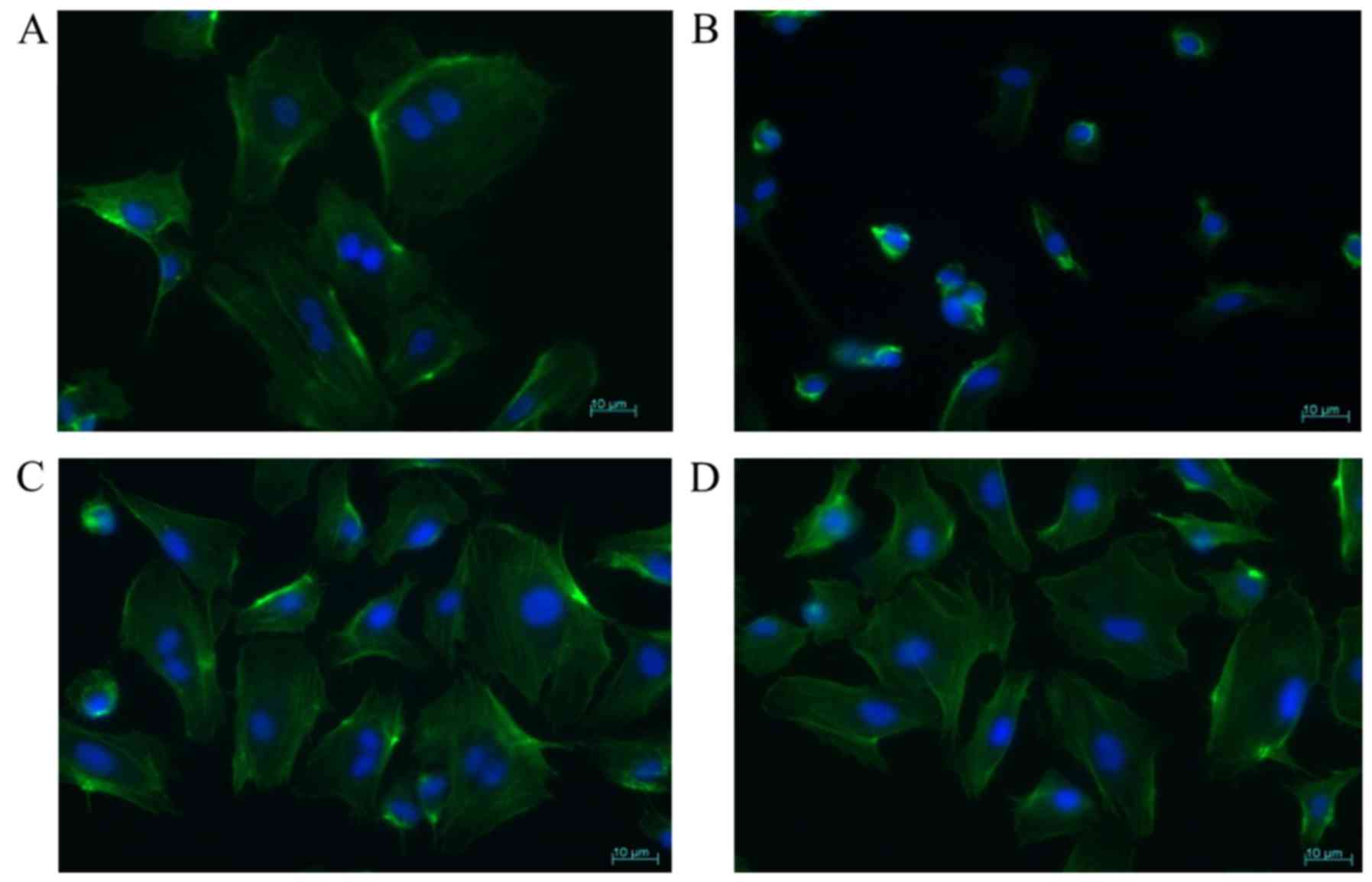
Effect of rhSLPI on cell
morphology
To determine whether the treatment of protease
inhibitor protected the cellular morphology and intracellular
integrity, the HUVECs were treated with rhSLPI and exposed to sI/R.
The results revealed that sI/R did not only cause cytoskeletal
destruction, but also reduced the cell size and altered the
cellular morphology when compared with the control cells (Fig. 5A and B). The disruption of the
cytoskeleton was preserved in the ECs treated with 1,000 ng/ml
rhSLPI prior to ischaemia, as well as cells treated prior to
ischaemia and stayed until the end of ischaemic period, as compared
with simulated ischaemia alone (Fig.
5B-D).
Treatment of rhSLPI activates Akt
phosphorylation and attenuates p38 MAPK activation
To determine cellular signalling in response to the
rhSLPI treatment in HUVECs during sI/R injury, including the
regulation of p38 MAPK and Akt, the cells were pretreated with
1,000 ng/ml rhSLPI for 2 h before simulated ischaemia. Western blot
assay was performed to analyse the phosphorylation of p38 MAPK and
Akt. Initially, the simulated ischaemic period was optimised to
determine the period required for marked activation of p38 MAPK.
The results revealed that simulated ischaemia for 20 min led to a
strong activation of p38 MAPK (Fig.
6A). Therefore, this treatment duration was then used to
determine the effect of rhSLPI on signal activation. The results
indicated that 20-min simulated ischaemia was able to strongly
activate the p38 MAPK phosphorylation. Furthermore, pretreatment
with 1,000 ng/ml rhSLPI significantly reduced the p38 MAPK
phosphorylation (Fig. 6B and C) and
activated Akt phosphorylation (Fig. 6B
and D), as compared with the simulated ischaemia alone.
However, pretreatment with 1,000 ng/ml rhSLPI had no significant
effect on the phosphorylation of p38 MAPK and Akt compared with
controls.
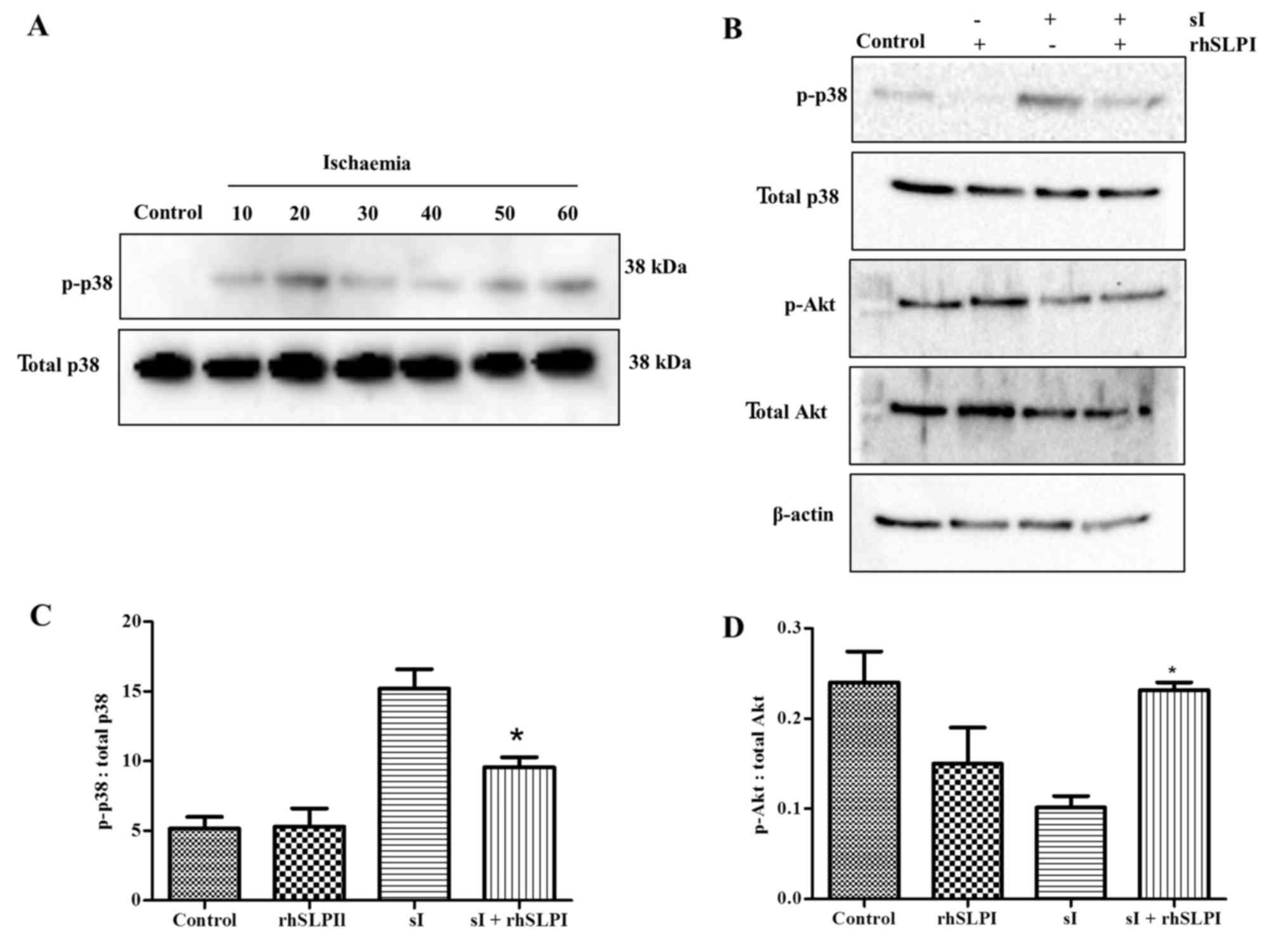 | Figure 6.(A) Optimised ischaemia duration for
MAPK activation in HUVECs. Cells were subjected to sI for 10, 20,
30, 40, 50 or 60 min, and the proteins expression of p38 MAPK was
determined by western blot analysis in three experiments with
independent cell preparations. (B) Effects of rhSLPI treatment on
MAPK activation. HUVECs were subjected to control medium,
pretreatment with 1,000 ng/ml rhSLPI without sI, 20 min of sI or
1,000 ng/ml rhSLPI pretreatment with 20-min sI. Endothelial
proteins were prepared for western blot analysis to detect the
expression levels of p38 and Akt. The quantified levels of (C) p38
and (D) Akt expressions were studied in 3–6 experiments with
independent cell preparations. *P<0.05 vs. sI. HUVECs, human
umbilical vein endothelial cells; rhSLPI, recombinant human
secretory leukocyte protease inhibitor; sI/R, simulated
ischaemia/reperfusion; sI, simulated ischaemia; Akt, protein kinase
B; MAPK, mitogen-activated protein kinase. |
Pretreatment of rhSLPI protects
vascular ECs by attenuation of apoptotic regulatory protein
activation
The EC regulatory pathway is a key determinant of
cell death, particularly necrosis and apoptosis in I/R injury;
thus, the effects of rhSLPI on the levels of apoptotic regulatory
proteins, including Bax, Bcl-2 and caspase 3, were determined. The
results of the present study revealed that pretreatment with 1,000
ng/ml rhSLPI significantly reduced the level of Bax protein
expression, the Bax:Bcl-2 ratio and the cleaved caspase-3
expression, with no marked changes observed in the levels of Bcl-2
expression (Fig. 7). This suggests
that rhSLPI pretreatment leads to a reduction in apoptotic
processes.
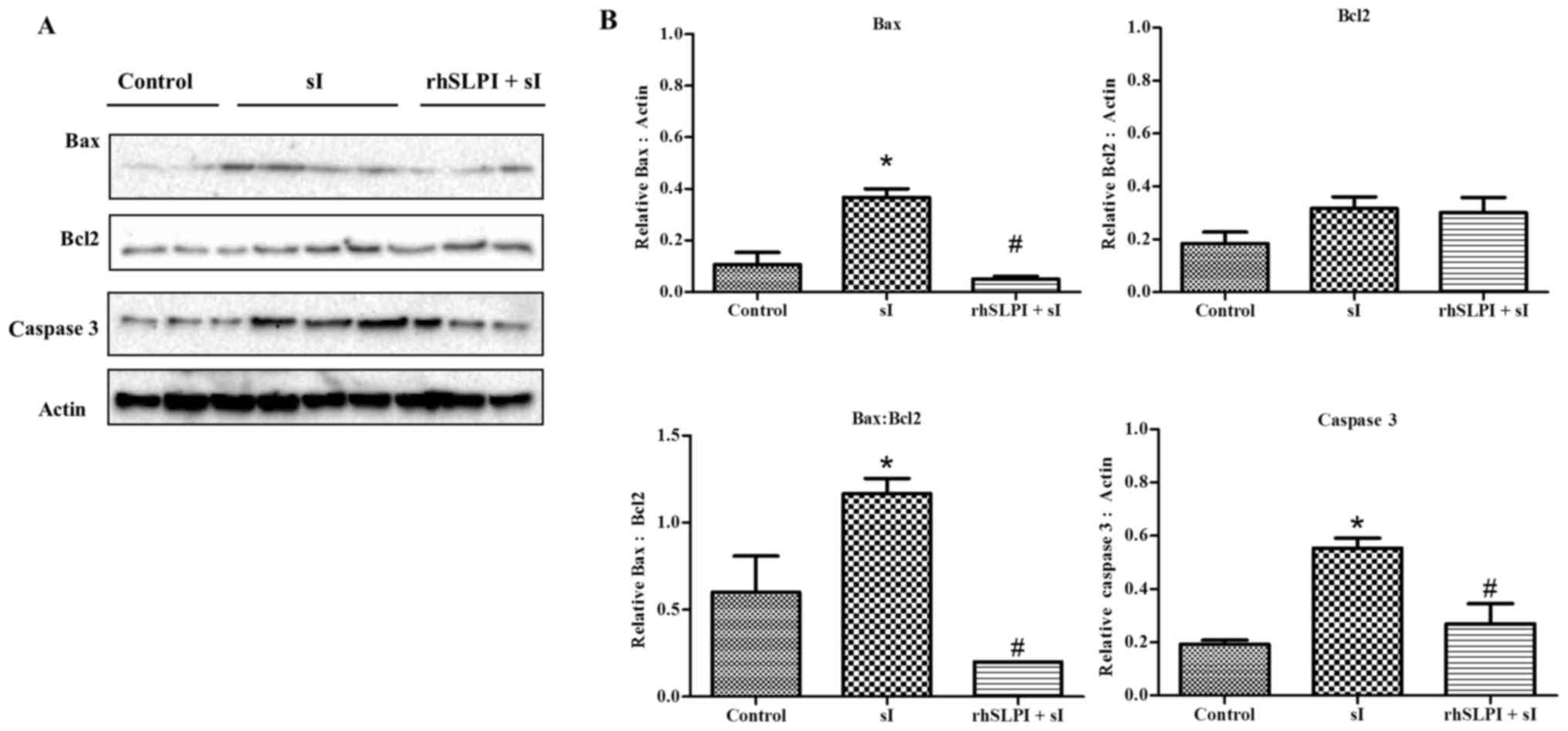 | Figure 7.(A) Effects of rhSLPI on the
activation of apoptotic regulatory proteins. HUVECs were subjected
to control medium incubation, 20-min sI, or 1,000 ng/ml rhSLPI
prior to ischaemia. Endothelial cell proteins were prepared for
western blot analysis to detect the expression levels of Bax, Bcl-2
and cleaved caspase 3. (B) Quantified levels of Bax, Bcl-2, Bax:
Bcl-2 ratio and cleaved caspase 3 were detected in 3–6 experiments
with independent cell preparations. *P<0.05 vs control group;
#P<0.05 vs sI group. HUVECs, human umbilical vein
endothelial cells; rhSLPI, recombinant human secretory leukocyte
protease inhibitor; sI/R, simulated ischaemia/reperfusion; sI,
simulated ischaemia; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein. |
Discussion
ECs are sensitive to necrosis and apoptosis during
I/R injury (11,12). The ativated endothelium is an area of
adhesion cascades involved in neutrophil extravasation (7,12,18),
aggravated inflammatory responses and accelerated microenvironment
susceptibility to lethal injury (19). Thus, an attempt to preserve the EC
integrity is an attractive therapeutic target in order to limit I/R
injury.
To the best of our knowledge, the present study
revealed for the first time that rhSLPI protects against in
vitro I/R injury in ECs. The results also demonstrated the
cytoprotective effect of rhSLPI treatment on ECs depending on the
time point at which treatment was given. Administration of rhSLPI
prior to or during ischaemia (for the entire period of ischaemia)
was observed to potentially promote EC survival following sI/R
injury, although treatment at a later stage upon the onset of
reperfusion did not have an evident protective effect (Fig. 3). In addition, pre-incubation with
rhSLPI manifested an inhibitory effect on
H2O2-induced intracellular ROS production
(Fig. 4), preserved the cytoskeletal
organisation, reduced the p38 MAPK activation, activated the
pro-survival kinase Akt, as well as reduced the expression levels
of apoptotic regulatory proteins.
The present study also examined the significance of
the period at which rhSLPI treatment should be provided in order to
achieve protection against the sI/R-induced EC injury. Early rhSLPI
administration was the most effective way to prevent deterioration
of the injury, including intracellular ROS production and
progression to apoptotic death. The most likely and effective
intervention depends on the timing of drug administration; it
should be given prior to reperfusion. However, the present study
demonstrated the cytoprotective effect of rhSLPI and the
significance of different time points of rhSLPI administration
in vitro; consequently, in vivo experiments in
further clinical studies are required to provide more clinically
relevant findings. Therefore, the in vitro nature
constitutes a limitation of the present study.
The endothelial protective ability of rhSLPI when
added prior to the reperfusion period that was demonstrated in the
present study is crucial for protection from cellular injury. When
treatment of rhSLPI was applied prior to or during ischaemia, the
protective effect remained over the reperfusion period. However, it
remains unclear whether the actual cytoprotective mechanism of
rhSLPI treatment is due to the inhibition of the protease activity
by rhSLPI itself or the interaction of rhSLPI with other molecules.
Previous studies reported that the phospholipid-binding protein
Annexin II is a receptor of SLPI, while human ECs also express the
Annexin II receptor (20,21). However, whether the direct effect of
rhSLPI on ECs occurs via interaction with phospholipid-binding
protein Annexin II was not investigated in the present study and
requires further investigation. In addition, the anti-protease
activity of SLPI may possibly attenuate the protease mediated cell
death and injury. However, the current study did not demonstrate
whether rhSLPI protect EC death was due to the inhibition of
protease activity. These associations remain unclear, constituting
a further limitation of the present study.
The development of I/R injury involves a variety of
mediators, including endothelial-derived ROS (8). The current findings demonstrated that
treatment with rhSLPI attenuated the intracellular ROS production,
suggesting the antioxidative property of SLPI in limiting the
endogenous ROS generation during I/R, thus leading to
cytoprotection against I/R injury. However, the potential
suppressive effect of rhSLPI on endothelial ROS production in terms
of the expression of specific effectors, including oxidative
scavenging enzymes, superoxide dismutase, catalase and glutathione
peroxidase, was not examined in the present study and needs to be
considered for further investigation.
Signal transduction serves a key role in I/R injury
and responses. Several protein kinases and cellular apoptotic
regulatory proteins have been known to serve critical roles in the
pathogenesis of I/R injury (7). The
p38 MAPK, Bax, Bcl-2, and caspase cascades are well defined as
mediators of cellular apoptotic in I/R injury (22). In the current study, it was observed
that rhSLPI attenuated p38 MAPK activation and pro-apoptotic Bax
protein expression, as well as promoted pro-survival Akt protein
activation under endothelial I/R injury (Figs. 6 and 7). These findings may explain the
cytoprotective effect of rhSLPI in ECs against I/R injury. A
limitation of the present study was that the results provided
restricted mechanistic insights on the function of rhSLPI in ECs,
since the protein levels of apoptotic factors Bcl-2 and caspase-3
may not provide sufficient information to confirm cellular
apoptotic. Therefore, an apoptosis-specific assay, such as TUNEL
assay or Annexin V staining, should be conducted in future
studies.
In conclusion, the present study is the first to
report that rhSLPI protected against I/R-induced EC injury through
the reduction of intracellular ROS production, attenuation of p38
MAPK, activation of pro-survival kinase Akt and reduced the levels
of certain apoptotic factors. Furthermore, the data in the current
study suggested that the therapeutic potential of rhSLPI in
protecting vascular ECs from I/R injury provides a more significant
clinical benefit when applied prior to reperfusion.
Acknowledgements
The present study was supported by the Naresuan
University Endowment Fund (grant nos. R2558C085, R2559C007 and
R2558B067), and the Royal Golden Jubilee PhD Program (grant no.
PHD/0043/2555; joined funding between the Thailand Research Fund
and Naresuan University) for PhD program scholarship.
References
|
1
|
Brutsaert DL: Cardiac
endothelial-myocardial signaling: Its role in cardiac growth,
contractile performance, and rhythmicity. Physiol Rev. 83:59–115.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Narmoneva DA, Vukmirovic R, Davis ME, Kamm
RD and Lee RT: Endothelial cells promote cardiac myocyte survival
and spatial reorganization: Implications for cardiac regeneration.
Circulation. 110:962–968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsieh PC, Davis ME, Lisowski LK and Lee
RT: Endothelial-cardiomyocyte interactions in cardiac development
and repair. Annu Rev Physiol. 68:51–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schulz R, Kelm M and Heusch G: Nitric
oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res.
61:402–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rezkalla SH and Kloner RA: No-reflow
phenomenon. Circulation. 105:656–662. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verma S, Fedak PW, Weisel RD, Butany J,
Rao V, Maitland A, Li RK, Dhillon B and Yau TM: Fundamentals of
reperfusion injury for the clinical cardiologist. Circulation.
105:2332–2336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lefer AM and Lefer DJ: Pharmacology of the
endothelium in ischemia-reperfusion and circulatory shock. Annu Rev
Pharmacol Toxicol. 33:71–90. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singhal AK, Symons JD, Boudina S, Jaishy B
and Shiu YT: Role of endothelial cells in myocardial
ischemia-reperfusion injury. Vasc Dis Prev. 7:1–14. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eltzschig HK and Collard CD: Vascular
ischaemia and reperfusion injury. Br Med Bull. 70:71–86. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scarabelli T, Stephanou A, Rayment N,
Pasini E, Comini L, Curello S, Ferrari R, Knight R and Latchman D:
Apoptosis of endothelial cells precedes myocyte cell apoptosis in
ischemia/reperfusion injury. Circulation. 104:253–256. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chatauret N, Badet L, Barrou B and Hauet
T: Ischemia-reperfusion: From cell biology to acute kidney injury.
Prog Urol. 24 Suppl 1:S4–S12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leucker TM, Ge ZD, Procknow J, Liu Y, Shi
Y, Bienengraeber M, Warltier DC and Kersten JR: Impairment of
endothelial-myocardial interaction increases the susceptibility of
cardiomyocytes to ischemia/reperfusion injury. PLoS One.
8:e700882013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Majchrzak-Gorecka M, Majewski P, Grygier
B, Murzyn K and Cichy J: Secretory leukocyte protease inhibitor
(SLPI), a multifunctional protein in the host defense response.
Cytokine Growth Factor Rev. 28:79–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schneeberger S, Hautz T, Wahl SM,
Brandacher G, Sucher R, Steinmassl O, Steinmassl P, Wright CD,
Obrist P, Werner ER, et al: The effect of secretory leukocyte
protease inhibitor (SLPI) on ischemia/reperfusion injury in cardiac
transplantation. Am J Transplant. 8:773–782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Powers SK, Murlasits Z, Wu M and Kavazis
AN: Ischemia-reperfusion-induced cardiac injury: A brief review.
Med Sci Sports Exerc. 39:1529–1536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jacquet S, Nishino Y, Kumphune S, Sicard
P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotten M and
Marber MS: The role of RIP2 in p38 MAPK activation in the stressed
heart. J Biol Chem. 283:11964–11971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen GY and Nuñez G: Sterile inflammation:
Sensing and reacting to damage. Nat Rev Immunol. 10:826–837. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong Q, Dai L, Wang Y, Zhang X, Li C,
Jiang S, Li Y, Ding Z and Liu L: HSPA12B attenuated acute
myocardial ischemia/reperfusion injury via maintaining endothelial
integrity in a PI3K/Akt/mTOR-dependent Mechanism. Sci Rep.
6:336362016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma G, Greenwell-Wild T, Lei K, Jin W,
Swisher J, Hardegen N, Wild CT and Wahl SM: Secretory leukocyte
protease inhibitor binds to annexin II, a cofactor for macrophage
HIV-1 infection. J Exp Med. 200:1337–1346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cesarman GM, Guevara CA and Hajjar KA: An
endothelial cell receptor for plasminogen/tissue plasminogen
activator (t-PA). II. Annexin II-mediated enhancement of
t-PA-dependent plasminogen activation. J Biol Chem.
269:21198–21203. 1994.PubMed/NCBI
|
|
22
|
Kumphune S, Surinkaew S, Chattipakorn SC
and Chattipakorn N: Inhibition of p38 MAPK activation protects
cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol.
53:1831–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|