Introduction
Cardiac arrest (CA) is a common cause of disability
and mortality and thus an important risk to human health. CA
initiates an ischemia/reperfusion scenario in the entire organism
with particular impact for the CNS. Oxidative stress,
excitotoxicity and neuro-inflammation contribute to the impairment
of the brain. Within the ischemic phase the deprivation of
substrate and oxygen causes dramatic restriction in mitochondrial
ATP production which essentially contributes to energetic deficits
in the brain. The duration of ischemia determines the extent of
damage. Reperfusion results in a dramatic increase in the
concentration of reactive oxygen species (ROS). Oxidation of
mitochondrial constituents can cause membrane permeabilization and
coupled to restriction in ATP production (1,2). As a
consequence, cell death may occur.
It has been shown that the early reperfusion phase
is of particular impact for the damage of neuronal cells.
Therefore, intervention concepts directed to the early phase of
cerebral reperfusion are promising for the reduction of early and
late impairments of neuronal cells after resuscitation (3,4).
Numerous efforts have been undertaken including hypothermia
(5), application of antioxidants
(3) or noble gases (6) and normoxic resuscitation (7) to improve the neurological outcome in
patients after CA. Although significant progress could be achieved
there is still a need for further improvement of the intervention
including the development of new drugs. According to the AHA's
Heart and Stroke Statistics (2017 update) (8), the annual incidence of out-of-hospital
CAs in the U.S. is ~111/100,000. Of those patients treated by
emergency medical services, only 23.8% survived. This motivated us
to study the effect of an extract from Gynostemma
pentaphyllum (GP) Makino on CA-depending brain injury. GP is
also known as Jiaogulan or xiancao, the ‘Herb of Immortality’. It
grows throughout Asian countries. It was first described in 1406 CE
by Zhu Xiao in the book Materia Medica for Famine as useful
survival food (9). Medical use of
Jiaogulan has been recorded in 1578 in Li Shi-Zehen's Compendium of
Materia Medica for the treatment of hematuria, edema in the pharynx
and neck, tumors, and trauma (10).
Today, Jiaogulan is appreciated as a medical plant with powerful
biological effects. At the International Conference of TCM in
Beijjing in 1991, Jiaogulan was declared to be one of 10 most
effective medicinal plants used for a variety of as much as 54
diseases, conditions and syndromes, e.g., cardiovascular disease,
hypertension, hepatitis, atherosclerosis, inflammation, and cancer.
It is composed of a complex mixture of about 80 gypenosides,
several amino acids, vitamins, and trace elements. Jiaogulan
appertains to a class of plants called adaptogens. These herbs help
the body to adapt to many forms of stress and imbalance. It exerts
a unique two-way action on the central nervous system, calming the
nerves when they are irritated and gently energizing them when they
are depressed (11). Further
beneficial effects attributed to GP are strong antioxidative
activities (12), glucose lowering
activities (13) and neuroprotective
activity (14).
In the past we studied effects of GP on isolated
mitochondria exposed to hypoxia/reoxygenation (15). There is increasing evidence that the
mitochondrial phospholipid cardiolipin (CL) is an important
component of oxidative phosphorylation mediating the transport of
electrons along the respiratory chain and supporting ATP synthesis
(16). Both the amount of CL and the
composition of its molecular species affect the function of
respiratory chain complexes and F0F1-ATPase
(17). It has been demonstrated that
oxidation and subsequent degradation of molecular CL species can
occur during ischemia/reperfusion (18,19). We
demonstrated a complete protection from permeabilization of the
mitochondrial membrane system by GP (15). Moreover, GP prevented functional
impairment of brain slices that were challenged with oxygen glucose
deprivation even when GP was administered with reperfusion
(20).
Indication for protective effects of GP in
vivo has been provided previously (21). The authors demonstrated an
attenuation of cognitive impairment at chronic cerebral
hypo-perfusion in rats due to GP administration. Data regarding
effects of GP on CA/resuscitation-mediated impairment of the CNS
are, however, missing, although CA is one of the top three causes
of death in the industrial world.
We tested the neuroprotective potential of GP in our
well-established asphyxia induced CA (ACA) model in rat (22–24). In
order to mimic the practical situation in health care in which
intervention starts at best with but mostly after resuscitation, we
applied GP simultaneously with resuscitation. In order to evaluate
ACA-induced damage of the brain we examined vital parameters, novel
object recognition as well as hippocampal cellular formation.
Further, we focused on the impairment of mitochondria by evaluating
amount and composition of CL, amount of mitochondria and activities
of respiratory chain complexes.
Materials and methods
ACA model-animals and intervention
protocol
Ethical approval for this study was granted
according to the requirements of the German Animal Welfare Act on
the Use of Experimental Animals and the Animal Care and Use
Committees of Saxony-Anhalt (permit number 42502-2-2-947 Uni MD).
Male rats (300–400 g; Institute's breeding population of inbred
Wistar rats; Harlan-Winkelmann, Borchen, Germany; altogether 162;
128 included, 34 excluded, see below) were housed under controlled
laboratory conditions (light cycle of 12 h light/12 h dark; lights
on at 6:00 a.m.; temperature, 20±2°C; air humidity, 55–60%) with
free access to water and chow. Every effort, including restriction
to one single GP dose, was made to minimize the amount of suffering
and the number of animals used in the experiments.
The study comprised of the following groups: i)
Sham-operated; ii) sham-operated with DMSO applied once at the
moment of spontaneous circulation re-establishment; iii)
sham-operated with GP applied once at the moment of spontaneous
circulation re-establishment; iv) ACA-treated; v) ACA-treated with
DMSO applied once at the moment of spontaneous circulation
re-establishment; vi) ACA-treated with GP applied once at the
moment of spontaneous circulation re-establishment; vii)
sham-operated with daily applied DMSO for 7 days; viii)
sham-operated with daily applied GP for 7 days; ix) ACA-treated
with daily applied DMSO for 7 days; and x) ACA-treated with daily
applied GP for 7 days.
Groups i-vi consisted of 18 animals each; 5 for
evaluation of mitochondrial parameters 6 h after ACA, 5 for
evaluation of mitochondrial parameters 24 h after ACA, 3 for
assessment of neurodegeneration 24 h after ACA, 5 for assessment of
neurodegeneration 7 days after ACA; groups vii-x consisted of 5
animals each; altogether 128 animals. DMSO groups were installed to
discriminate between GP effects and artificial effects of its
solubilizer DMSO.
Anesthesia was induced with 5% sevoflurane (Pfizer
GmbH, Berlin, Germany) in an oxygen/nitrous oxide mixture (40:60)
via facemask followed by endotracheal intubation with a modified
laryngoscope and a venous catheter and muscular relaxation with
vecuronium (1 mg/kg; Pfizer). Mechanical ventilation was performed
with intermittent positive pressure ventilation (IPPV). For drug
administration, blood sampling, and continuous blood pressure
monitoring both left femoral vessels were cannulated with
polyethylene catheters. After 5 min of room air ventilation and
baseline control, ACA was induced by an end-expiratory interruption
of IPPV on paralyzed rats for 6 min. ACA (defined as a
non-pulsatile blood pressure of less than 10 mmHg) was reached
within approximately 3 min (Fig.
1).
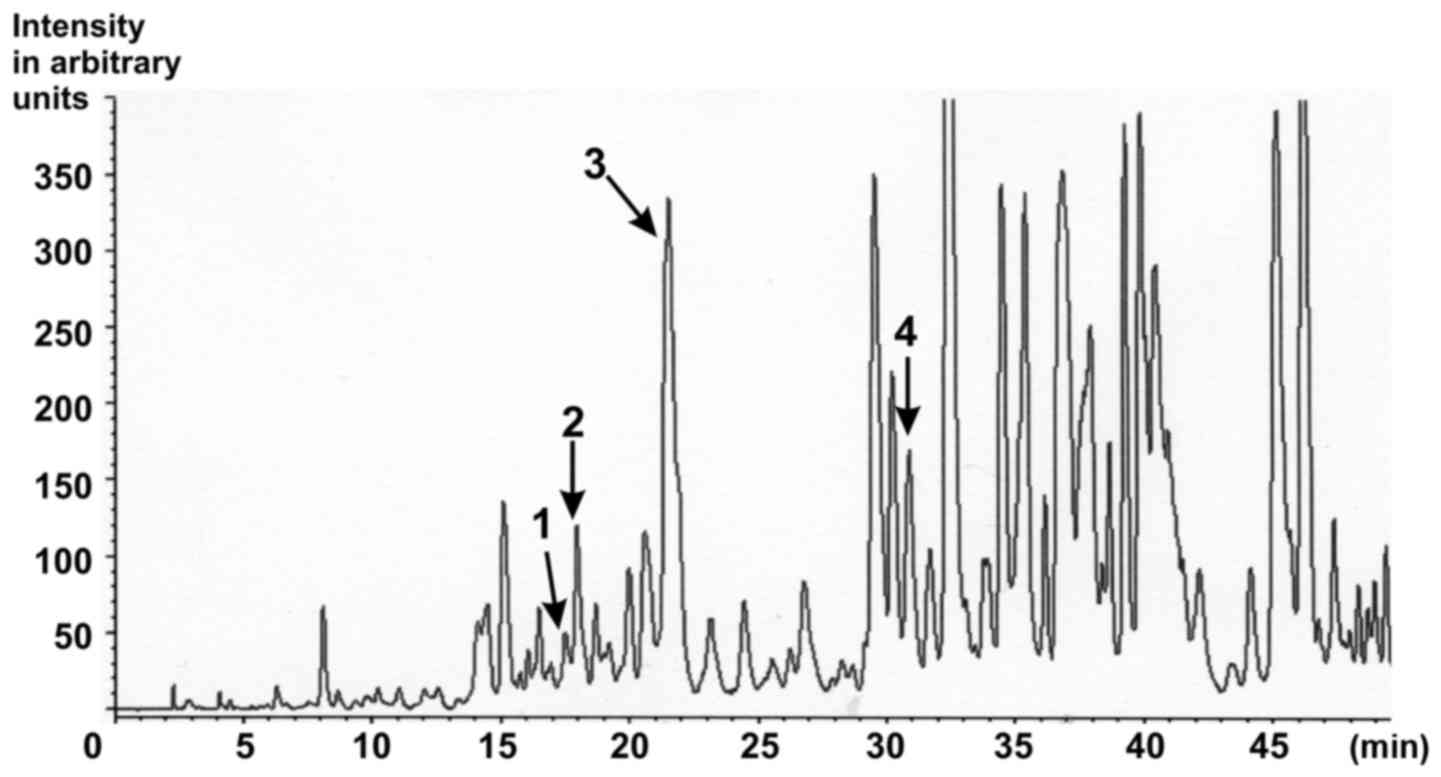 | Figure 1.Characterisation of the ethanolic
Gynostemma pentaphyllum extract. HPLC chromatogram of
saponins from G. pentaphyllum extract with MS detection.
Peak 1, Gypenoside LXIII (retention time, 16.373; content, 129.4
ppm); Peak 2, Gypenoside LXIII (retention time, 17.297; content,
316.4 ppm); Peak 3, Gypenoside IV (Ginsenoside Rb3;
retention time, 20.497; content, 5899.5 ppm); Peak 4, Gypenoside
VIII (Ginsenoside Rd; retention time, 29.813; content, 414.0 ppm)
[as published in (25)]. |
Resuscitation was performed by the administration of
epinephrine (i.v.; 1 µg/kg; Pfizer) and sodium bicarbonate (1
mEq/kg), restarting IPPV with 100% oxygen for 1 h, and manual
external chest compression (200/min). Return of spontaneous
circulation (ROSC) was defined as a pulsatile mean arterial
pressure (MAP) above 60 mmHg. Rats with no ROSC within 2 min were
excluded (altogether 34). The 2-min interval is our lab standard to
avoid non-homogeneous ACA periods leading to pathophysiological
differences and, subsequently, to the need for larger numbers of
animals. Vital parameters (ECG, blood pressure, temperature, airway
pressure) were monitored continuously during the first 30 min of
the post-resuscitation intensive-care phase. At 5, 15 and 30 min
after ROSC, arterial blood samples were collected and evaluated for
blood gases (pCO2, pO2), pH and glucose.
After sufficient spontaneous respiration was established (~30 min
post ROSC), catheters were removed, cannulated vessels were ligated
and incisions were surgically closed. Rats were extubated and
returned to their cages. Body temperature was maintained at
37±0.3°C during preparation, insult and the first 30 min post-ROSC.
Thereafter, normal body temperature was stably maintained within
normal range by placing the rats in an incubator cage for 24 h.
Gynostemma pentaphyllum (GP)/DMSO
intervention
The standardized GP powder, obtained by extraction
of dried aerial parts with 75% ethanol/25% water, was received from
Herbasin Co., Ltd. (Shenyang, China; batch no. 061001E074). The
extract composition was certified by the manufacturer. According to
the manufacturer, the extract contained a gypenoside content of
99.7%, a heavy metal contamination <10 ppm and no
microbiological contamination. In the HPLC-fingerprint performed by
the consumer, eight peaks corresponding to gypenosides were
detected (15). In particular,
Gypenoside LXIII, Gypenoside Rb3 and Gypenoside VIII
could be identified (Fig. 1)
(25). The retention times of
specific gypenosides were determined by using standard gypenosides
in separate experiments. The respective method has been described
in detail previously (25). Briefly:
Sample preparation: 5 mg of the dried ethanolic extract were
dissolved in 1 ml of ethanol, filtered over Millipore®
filtration unit, type 0.45 µm, and injected into the HPLC
apparatus. Injection volume: Gynostemma pentaphyllum
extract: 20.0 µl. HPLC parameter: Apparatus: MERCK HITACHI D-6000 A
Interface, MERCK HITACHI AS-2000 Autosampler, MERCK HITACHI L-6200
A Intelligent Pump. Separation column: LiChroCART® 250-4
LiChrospher® 100 RP-18 (5 µm) (Merck). Precolumn:
LiChroCART® 4-4 LiChrospher® 100 RP-18 (5 µm)
(Merck). Solvent: A: dist. Water (Millipore Ultra Clear UV
plus® filtered), B: acetonitrile (Fa. VWR). Gradient:
5–100% B in 60 min, total runtime: 60 min. Flow: 0.8 ml/min. A
single quadruple mass spectrometer (LC/MS) with electrospray
ionization (ESI) in negative mode was used for detection (26).
GP powder (60 mg) was dissolved in 0.5 ml DMSO and
diluted with PBS (phosphate-buffered saline) to a final volume of
10 ml. In pilot studies a volume of 200 µl of this solution
injected i.p. was found to be optimal for fast resorption and
distribution within the circulation.
The tolerability and efficacy of GP was tested in
vitro using both cultures of dispersed astrocytes and
hippocampal slices (20). A dosis of
60 µg/ml was found to be optimal in vitro. The applied 200
µl of the stock solution contained 1,200 µg GP. At an average
animal weight of about 350 g/a blood volume of about 20 ml that
resulted in the intended final concentration of 60 µg/ml.
As vehicle control a corresponding DMSO solution was
injected. First dose of GP/DMSO was administered at the time when
spontaneous circulation was re-established. In groups vii-x, GP or
DMSO were additionally administered every morning for the next 6
days.
Preparation of tissue homogenates
After the respective survival times, animals were
sacrificed by over-dosed anesthesia (isoflurane 10% in a in a 4.35
l sealed desiccator; Baxter, Unterschleissheim, Germany). Brains
were quickly removed and hippocampi were separated on ice, weighed,
minced using small scissors, transferred into ice-cold phosphate
buffer solution (PBS; pH 7.4; 10% tissue portion) and homogenized
at 4°C using a Potter-Elvehjem glass-Teflon homogenizer (10 strokes
at 600 rpm). The mitochondrial parameters citrate synthase
activity, NADH:cytochrome c oxidoreductase activity,
succinate:cytochrome c oxidoreductase activity and cardiolipin were
determined by using the homogenates. The data were related either
to mg protein of the homogenate or to the activity of the
mitochondrial marker enzyme citrate synthase.
Determination of citrate synthase
activity (EC 2.3.3.1)
Citrate synthase (CS) activity was assayed in
homogenates of hippocampi 6 and 24 h after ACA using a standard
procedure at 30°C (27). Before
running the assay samples were fivefold frozen and defrosted to
permeabilize membranes. Homogenates of 40 µg were evaluated. The
increase of CoA absorption was monitored at 412 nm with a Cary 100
Bio spectrophotometer (Varian, Darmstadt, Germany).
Determination of NADH:cytochrome c
oxidoreductase activity (EC 1.6.99.3)
The parameter was assayed in homogenates of
hippocampi 6 and 24 h after ACA. Samples (500 µl) of the respective
tissue homogenates were fivefold frozen and defrosted to
permeabilize membranes. A volume of 15 µl (40 µg protein) was used
to run the standard assay at 30°C. The reduction of cytochrome c
was followed by measuring the absorption at 550 nm with a Cary 100
Bio spectrophotometer (Varian). The rotenone-sensitive absorption
was used for quantification.
Determination of succinate:cytochrome
c oxidoreductase activity (EC 1.3.5.1)
The parameter was assayed in homogenates of
hippocampi 6 and 24 h after ACA. Samples (500 µl) of the respective
tissue homogenates were fivefold frozen and defrosted to
permeabilize membranes. A volume of 15 µl (40 µg protein) was used
to run the standard assay at 30°C. The reduction of cytochrome c
was followed by measuring the absorption at 550 nm with a Cary 100
Bio spectrophotometer (Varian). The antimycin A-sensitive
absorption was used for quantification.
Assessment of cardiolipin (CL)
CL was also analyzed in hippocampi 24 h after ACA.
Therefore, samples (see above) were frozen on dry ice and stored at
−80°C until CL analysis, steps of which were i) extraction of CL,
50 ng of tetra-myristoyl-CL [(C14:0)4-CL; Avanti Polar
Lipids Inc., Alabaster, AL, USA; internal standard] and 4.2 ml
chloroform/methanol (2/1, v/v) containing 0.05% butylated
hydroxytoluene (BHT; Avanti Polar Lipids Inc.) were added to 100 µl
of defrosted tissue homogenates. The lipid and aqueous phases were
separated by adding 800 µl of 0.01 M HCl, intensive shaking and
subsequent centrifugation. After centrifugation, the lipid phase
(lower phase) was collected and dried under nitrogen atmosphere and
acidified. Ice-cold methanol (2 ml), chloroform (1 ml) and 1 ml of
0.1 M HCl were added. The solution was intensively mixed. After 5
min of incubation on ice the samples were separated by the addition
of chloroform (1 ml) and 0.1 M HCl (1 ml). The chloroform/methanol
phase was recovered as CL-containing sample. Afterwards, the
samples were dried under nitrogen and dissolved in 0.8 ml
chloroform/methanol/water (50/45/5, v/v/v). After mixing and
filtering of the mixture over 0.2 µm PTFE membranes the samples
were ready for use. ii) HPLC-MS/MS analysis as described in detail
earlier (28).
Determination of protein
The protein content was determined according to the
Bradford method (29) using bovine
serum albumin as the standard.
Assessment of brain degeneration
After survival times of 24 h and 7 days,
anaesthetized rats were sacrificed by transcardial perfusion with
4% 0.1 M phosphate-buffered paraformaldehyde (PFA; pH 7.4;
Millipore, Darmstadt, Germany). The brains were quickly removed,
post-fixed in the same fixative at 4°C overnight, cryoprotected in
30% sucrose in 0.4% PFA (pH 7.4) for 2 days, and rapidly frozen at
−20°C. Slice preparation and staining procedure were performed as
described (24). The following
mixtures of primary antibodies (diluted in 10% fetal calf serum and
0.3% Triton-X 100 in PBS) were used: (i) Mouse monoclonal anti-NeuN
(neuronal nuclei antibody; Chemicon, Billerica, USA; 1:100) and
polyclonal rabbit anti-glial fibrillary acidic protein (GFAP;
Progen, Heidelberg, Germany; 1:500), and ii) monoclonal mouse
anti-MAP2 (microtubule-associated protein 2; Covance, Münster,
Germany; 1:1,000); and rabbit polyclonal anti-ionized calcium
binding adaptor molecule 1, (IBA1; Abcam, Cambridge, UK; 1:1,000).
The mixture of the secondary antibodies consisted of goat
anti-mouse Alexa 488 (green; Invitrogen, Carlsbad, USA; 1:500) and
donkey anti-rabbit Cy3 (red; Dianova, Hamburg, Germany; 1:500) and
was diluted in 1% normal goat serum and 0.3% Triton-X in PBS.
Using an AxioImager.M1 fluorescence microscope
(Zeiss, Jena, Germany; with a Plan-Neofluar fluorescein/rhodamine
objectives) slices were evaluated. For MAP2 analysis, five
alternating slices/animal were scanned image field by image field
(objective ×40/0.75) to compose an image of the complete
hippocampus (1,388×900 pixel; AxioVision software ‘Panorama’,
Zeiss). After that a standard evaluation window (500×300 pixel),
including the complete hippocampal CA1 region, was selected
manually and the respective staining intensity was quantified using
ImageJ software (http:/rsbweb.nih.gov/ij/).
For quantification of pyknotic (24 h
post-intervention) and normal NeuN-positive (7 days
post-intervention) cells of the CA1 region, the AxioVision z-stack
software (Zeiss) was used to get a composition image from 5–8
single images (objective ×40/0.75) taken at different focal
distances of the slice. All clearly recognizable
pyknotic/hyperchromatic cells and NeuN-positive cells were counted
unbiased.
GFAP and IBA1 immunostainings of the hippocampal CA1
region were only descriptively analyzed.
For each antibody, microscopic settings and the
exposure time of the fluorescence channels were set on the basis of
control slices and kept equal for the corresponding
preparation.
Novel object recognition test
After 7 days of survival, rats underwent the object
recognition test in an open field (clear Plexiglas, 50 cm wide ×25
cm high) combined with a video tracking system (VPC-FH1; Sanyo
Electric, Moriguchi, Japan). The experiment started with an
adaptation trial without object in the open field, followed by two
sessions, a sampling and a test trial, each 6 min long. In the
sampling session, rats explored one object (A1, glass bottle 12 cm
tall, 5 cm wide). The test session was conducted 120 min later by
allowing rats to explore object A (now A2) together with a novel
object (B, metal box, 5 cm tall ×5 cm wide). Thereby object A was
placed at the same place as in the first session. Each session was
video recorded for later analysis. Three independent experimenters,
blinded to group treatment, scored each behavioral test.
Exploration was defined as sniffing or touching the object with the
nose. Longer exploration duration of object B vs. object A2 can be
interpreted as evidence for an intact recognition memory (30). Moreover, global habituation
(comparison of the total time spent in exploring object A1 during
the sampling session to the summarized total time spent in
exploring objects A2 and B in the test phase) was determined. Data
are presented as mean ± SD.
Statistical analysis
All quantitative data are presented as the mean ± SD
per animal. The means (the exact n is given in the respective
figure legends) were analyzed with the non-parametric
Kruskal-Wallis test and the Dunn's multiple comparison post-hoc
test using Graph Pad Prism 6 (GraphPad Software Inc., La Jolla, CA,
USA). For intra-group differences of sham- and ACA-animals one-way
ANOVA was performed. Differences of groups with equal treatment
regimens but different survival times were analyzed with the
Wilcoxon-Mann-Whitney-test. In either case P≤0.05 was considered to
indicate a statistically significant difference.
Results
Effect of ACA and DMSO/GP on vital
parameters
The preparation of animals including anesthesia via
facemask, trachea intubation, cannulation of the left femoral
vessels and determination of baseline parameters required 22±5 min.
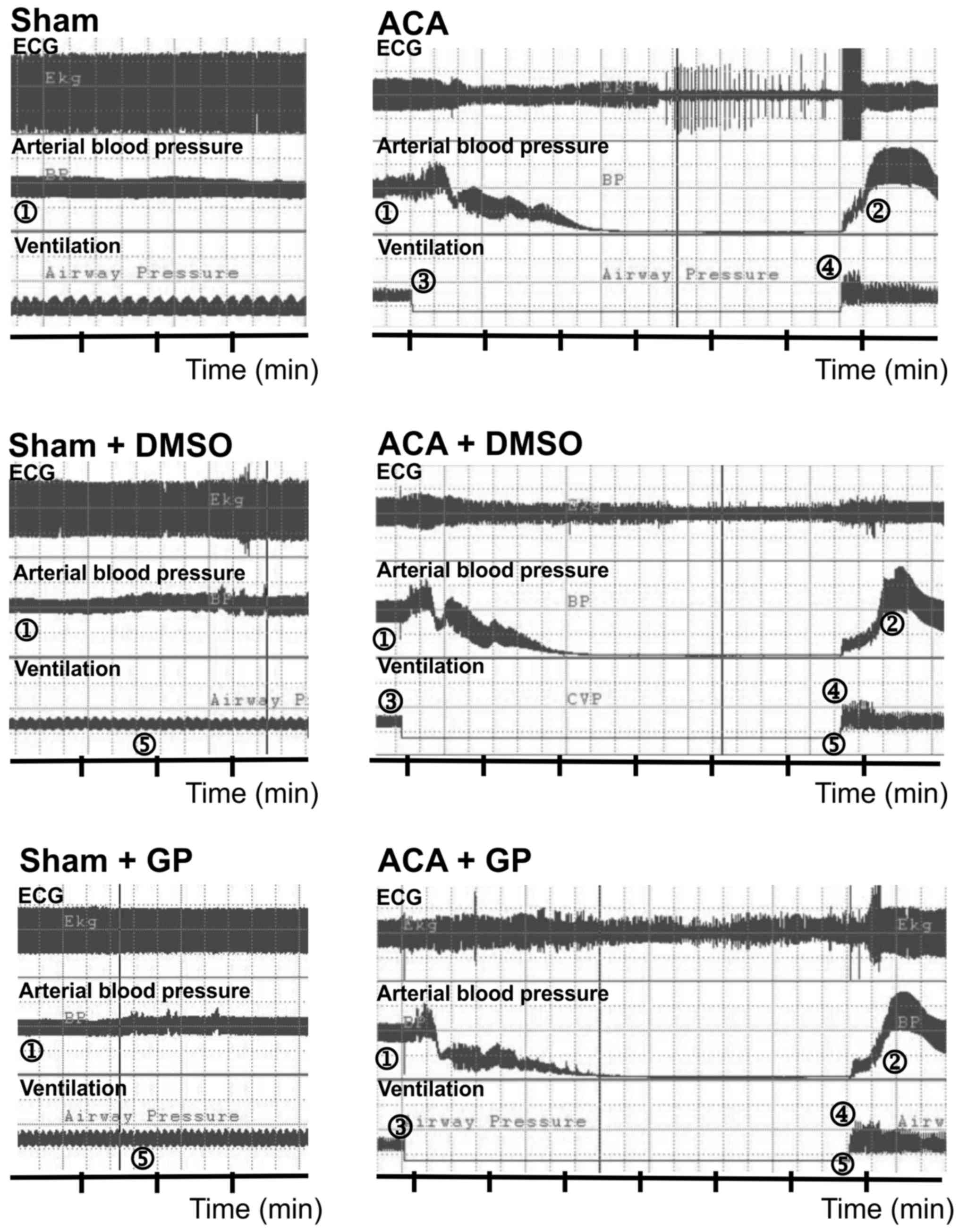
Fig. 2 illustrates the recording of
ECG, arterial blood pressure and ventilation. In sham-operated
animals, DMSO/GP intervention did not influence ECG and blood
pressure pattern. In ACA animals, arterial blood pressure dropped
to null line after 170±28 sec of asphyxiation without statistical
difference between the three ACA groups. Shortly after stop of
ventilation, the ECG signal amplitude became suppressed with
remarkable fluctuation. It proceeded to asystole within 180±50 sec.
The recording was aggravated by typical technical transients. When
6 min of asphyxiation were completed, resuscitation was initiated.
Thereby, ROSC was achieved within 30±24 sec. Independent of DMSO/GP
intervention, resume of ECG activity started with an initial
pattern resemblant to burst-suppression.
The time course of the basic vital parameters is
displayed in Fig. 3. ACA induced a
transient increase of MAP (hypertensive in tendency 1 min after ACA
but, not significant, P=0.07 returning to normal values afterwards;
Fig. 3A), a significant increase of
heart rate (Fig. 3B), of arterial
carbon dioxide tension (pCO2; Fig. 3C) and of blood glucose values
(Fig. 3E), whereas pH levels were
significantly reduced (Fig. 3F). In
the presence of GP, the ACA-mediated increase of pCO2
(Fig. 3C) and blood glucose
concentration (Fig. 3E) was
prevented. DMSO was able to normalize both ACA-mediated increases
too (Fig. 3C and E).
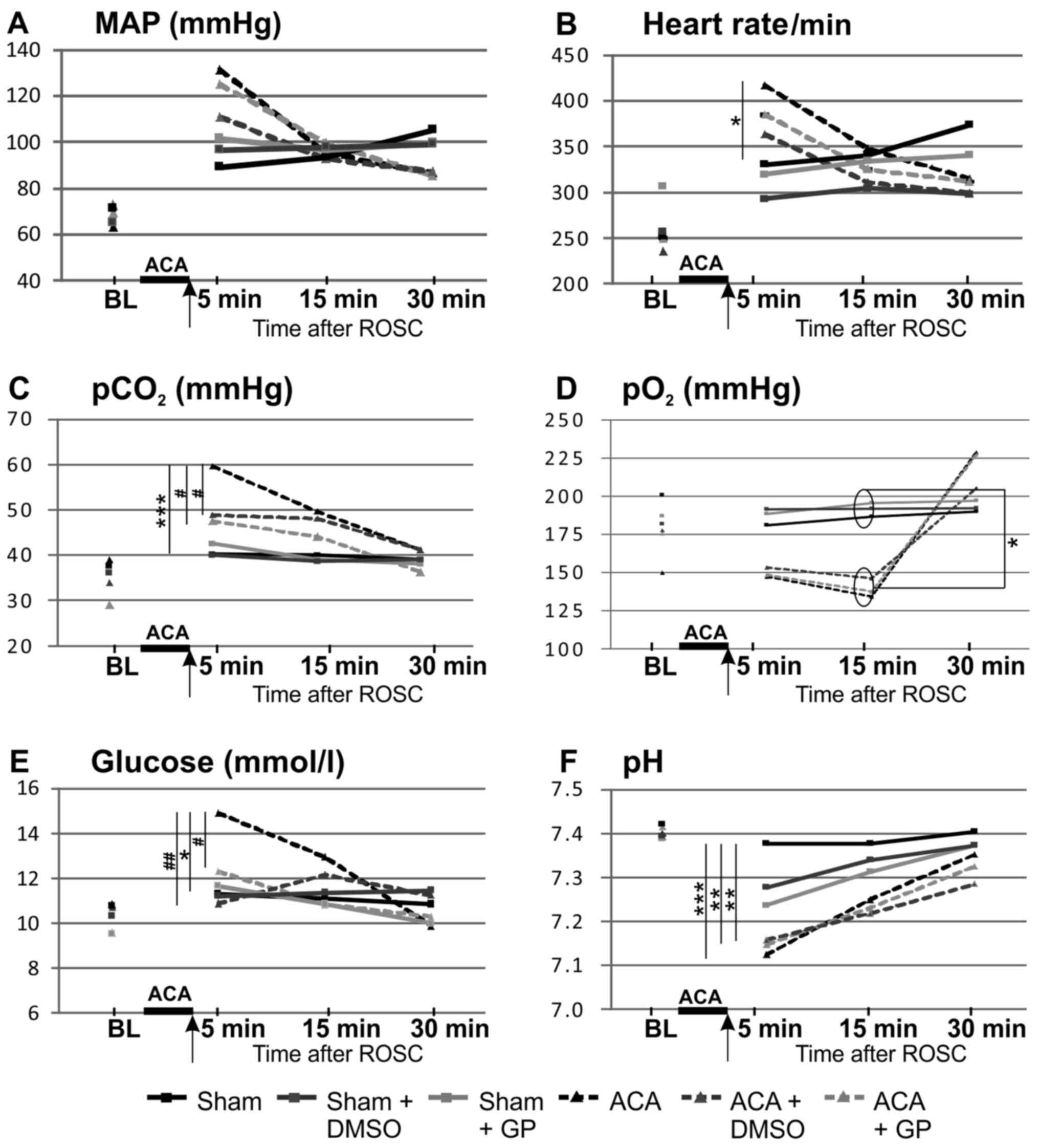 | Figure 3.Pre- and post-resuscitation
physiological parameters: (A) MAP, (B) heart rate, (C)
pCO2, (D) pO2, (E) glucose and (F) pH. Arrow,
time point of DMSO/GP intervention. Data: Mean ± SD with n≥15;
Kruskal-Wallis/Dunn's post-hoc test; significant differences sham
vs. ACA: *P<0.05 and ***P<0.001 within the ACA groups;
#P<0.05 and ##P<0.005 within the sham
groups, yet no significant differences were found. BL, base line;
MAP, mean arterial pressure; pCO2, arterial carbon
dioxide tension; pO2, arterial oxygen tension, ACA,
asphyxia cardiac arrest; GP, Gynostemma pentaphyllum; ROSC,
return of spontaneous circulation.; DMSO, dimethyl sulfoxide. |
Preparation of the animals did not affect these
vital parameters; all parameters were within the physiological
ranges (baseline). The arterial pO2 of ACA-animals was
increased after 30 min as consequence of ventilation with 100%
oxygen (Fig. 3D). The body
temperature (tympanal and rectal) was not affected within the 30
min-monitoring period after resuscitation (data not shown).
Effect of ACA and DMSO/GP on amount of
mitochondria and activity of respiratory chain complexes
We used the activity of CS as marker for the amount
of mitochondria in order to study changes in mitochondrial mass. In
sham animals, 6 h after treatment (early response) increased values
of CS were found in animals exposed to DMSO as the vehicle control
(Fig. 4A). However, in combination
with GP no effect was observed (Fig.
4A). Twenty four hours after treatment (late response), no
effect of DMSO could be detected in sham animals (Fig. 4B). In ACA treated animals, DMSO did
not cause changes in CS activity at all (Fig. 4A, B). GP caused increase in CS
activity only in ACA animals at both time points (Fig. 4A, B).
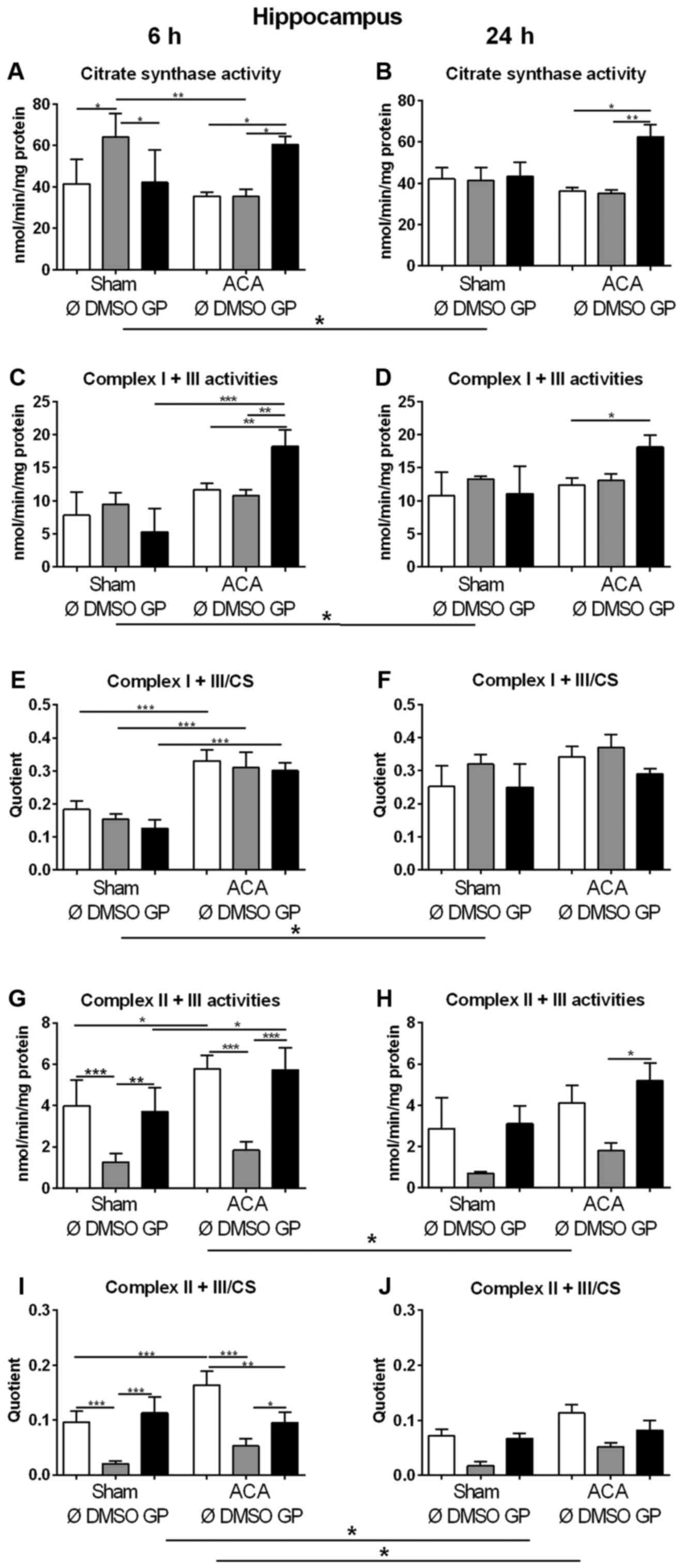 | Figure 4.Effect of ACA on mitochondrial
content and respiratory chain complex activities. (A and B) Citrate
synthase (CS) activities and specific activities of the respiratory
chain complexes I + III (C and D) and II + III (G and H) were
determined in hippocampus after 6 h (A, C, E, G and I) and 24 h (B,
D, F, H and J). GP and the vehicle control DMSO were administered
with resuscitation. Specific respiratory chain complex activities
were calculated by relating the data to CS activities (E, F, I and
J). Data are expressed as mean ± SD from 5 animals. Significance of
differences was tested with Kruskal-Wallis/Dunn's post-hoc test;
(*P<0.05; **P<0.005, ***P<0.001). ACA, asphyxial cardiac
arrest; GP, Gynostemma pentaphyllum; DMSO, dimethyl
sulfoxide |
In order to explore the effect of ACA on the
expression of respiratory chain complexes in mitochondria the
activities of NADH-cytochrome c oxidoreductase (complex I + III)
and succinate:cytochrome c oxidoreductase (complex II + III) were
analyzed. For clear interpretation of the data, the complex
activities were related to protein content of homogenate samples
(Fig. 4C, D, G, H) or to CS
activities representing the mitochondrial amount in the homogenate
(Fig. 4E, F, I, J). When complex I +
III activities were related to mg protein of homogenate samples
similar results of GP as for citrate synthase activities were
obtained (Fig. 4C, D). The specific
complex I + III activities (related to CS activity) displayed no
singular DMSO or GP effect. 6 h after treatment, we detected
increased specific activities of complex I + III of all ACA-treated
animals in comparison to the respective sham operated groups
(Fig. 4E). Twenty four h after
treatment, the specific complex I + III activities of all sham
animals were in tendency elevated possibly indicating a stress
response to anesthesia (Fig.
4F).
The analysis of complex II + III activities revealed
that DMSO caused decrease in complex activity in sham operated and
in ACA-treated animals 6 h post ACA as a side effect (Fig. 4G, I). This effect declined with
survival time (Fig. 4H, J). ACA
alone caused increase in complex II + III activity 6 h after
treatment (Fig. 4G, I) that was not
detected in the presence of GP. Moreover, GP neutralized the side
effect of DMSO (Fig. 4G, I).
Content and composition of the
mitochondrial phospholipid CL
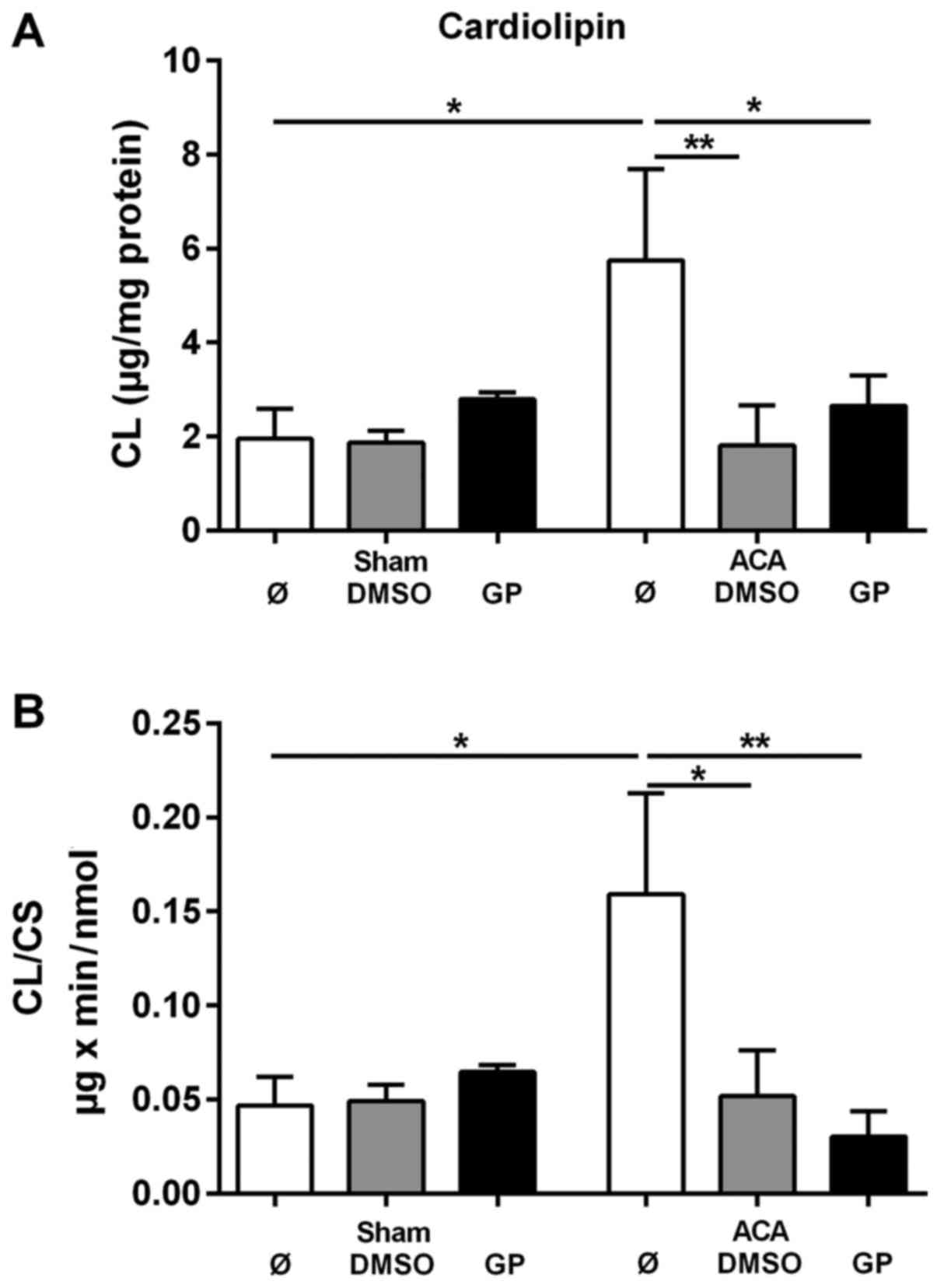
To evaluate the CL tissue content the amount of CL
was related to mg protein of homogenate. The corresponding data are
presented in Fig. 5A. For the
determination of the specific mitochondrial content of CL the data
were related to the activity of the mitochondrial marker enzyme CS.
These data are presented in Fig. 5B.
ACA caused significant increase in both the tissue content of CL
and in the mitochondrial CL content. The stimulation of CL
synthesis by ACA was prevented by the administration of DMSO and
GP, respectively. We did not find any change in the composition of
molecular CL species under the conditions of investigation (data
not shown).
Histological outcome of hippocampal
CA1 region 24 h post ACA
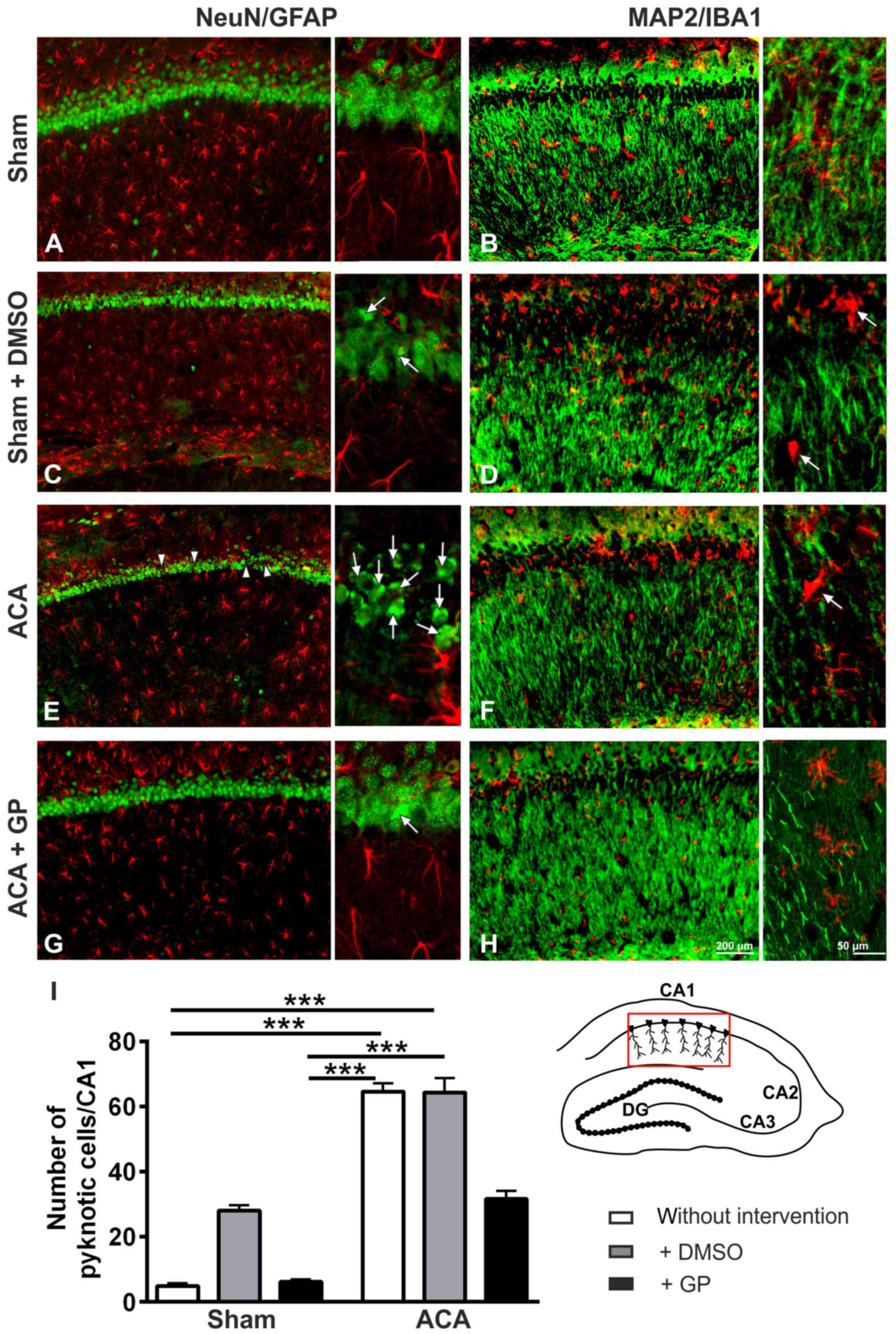
In sham animals, the NeuN-immunostaining showed
intact hippocampal CA1 neurons (Fig.
6A). The MAP2-immunostained fiber net (Fig. 6B) was well developed. The patterns of
GFAP-positive astroglia (Fig. 6A) or
IB1-positive microglia (Fig. 6B)
conformed to norms. In DMSO-treated sham animals, a couple of
pyknotic cells was seen in the CA1 pyramidal cell layer (Fig. 6C, arrows). Pyknotic cells are
characterized by size-reduction and/or condensation usually
associated with hyperchromatosis, that has been demonstrated by
NeuN immunostained nuclei. It indicated cell death induction by
DMSO, which was confirmed by MAP2 staining, offering an ongoing
loosening up of the nerve fiber net (Fig. 6D). The pattern of astroglia (Fig. 6C) and microglia (Fig. 6D) showed signs of activation. The
respective staining patterns of GP-treated sham animals were
identical to those of the untreated group (data not shown).
In the hippocampal CA1 pyramidal cell layer of ACA
treated animals (Fig. 6E), a massive
increase of pyknotic cells (arrows) as well as first cell losses
(arrowheads) were found. The respective MAP2-positive fiber nets
revealed distinct signs of disruption (Fig. 6F). Yet, astroglia activation was not
evidenced by GFAP immunostaining (Fig.
6E). Activation of microglia was, however, clearly
demonstrable; the cell bodies became more rounded and the
originally ramified branches became shorter and stockier (arrow in
Fig. 6F). The respective staining
patterns of DMSO-treated ACA-animals were identical to those of the
untreated ACA group (data not shown). In case of GP administration,
neuroprotective tendencies were found: lower number of pyknotic
neurons (arrows in Fig. 6G) and less
disruption of the MAP2-positive fiber net (Fig. 6H). Semi-quantification of pyknotic
NeuN-positive cells is given in Fig.
6I.
Histological outcome of hippocampal
CA1 region 7 days post-ACA
In untreated sham-operated animals, NeuN (Fig. 7A) as well as MAP2 (Fig. 7B) immunostaining revealed intact
hippocampal CA1 neurons with a dense fiber network. A consistent
standard pattern of GFAP-positive astroglia (Fig. 7C) and IB1-positive microglia
(Fig. 7D) was also evident. In both,
onetime (shown in Fig. 7E-H) or
multiple (not shown) DMSO-treated sham-animals, the NeuN-stained
CA1 pyramidal cell layer showed a spotty cell loss (arrows in
Fig. 7E). These areas offered a
narrow accumulation of IBA1-positive microglia cells (arrows in
Fig. 7H). MAP2 (Fig. 7F) and GFAP (Fig. 7G) immunostainings revealed, however,
regular patterns. In GP treated sham-animals, there were no signs
of abnormal neuronal viability or cell pattern, irrespective of
whether GP was applied once or multiple (Fig. 7I-L, demonstrated for onetime applied
GP). That indicates that GP was able to counteract cell stress
mediated by the vehicle DMSO.
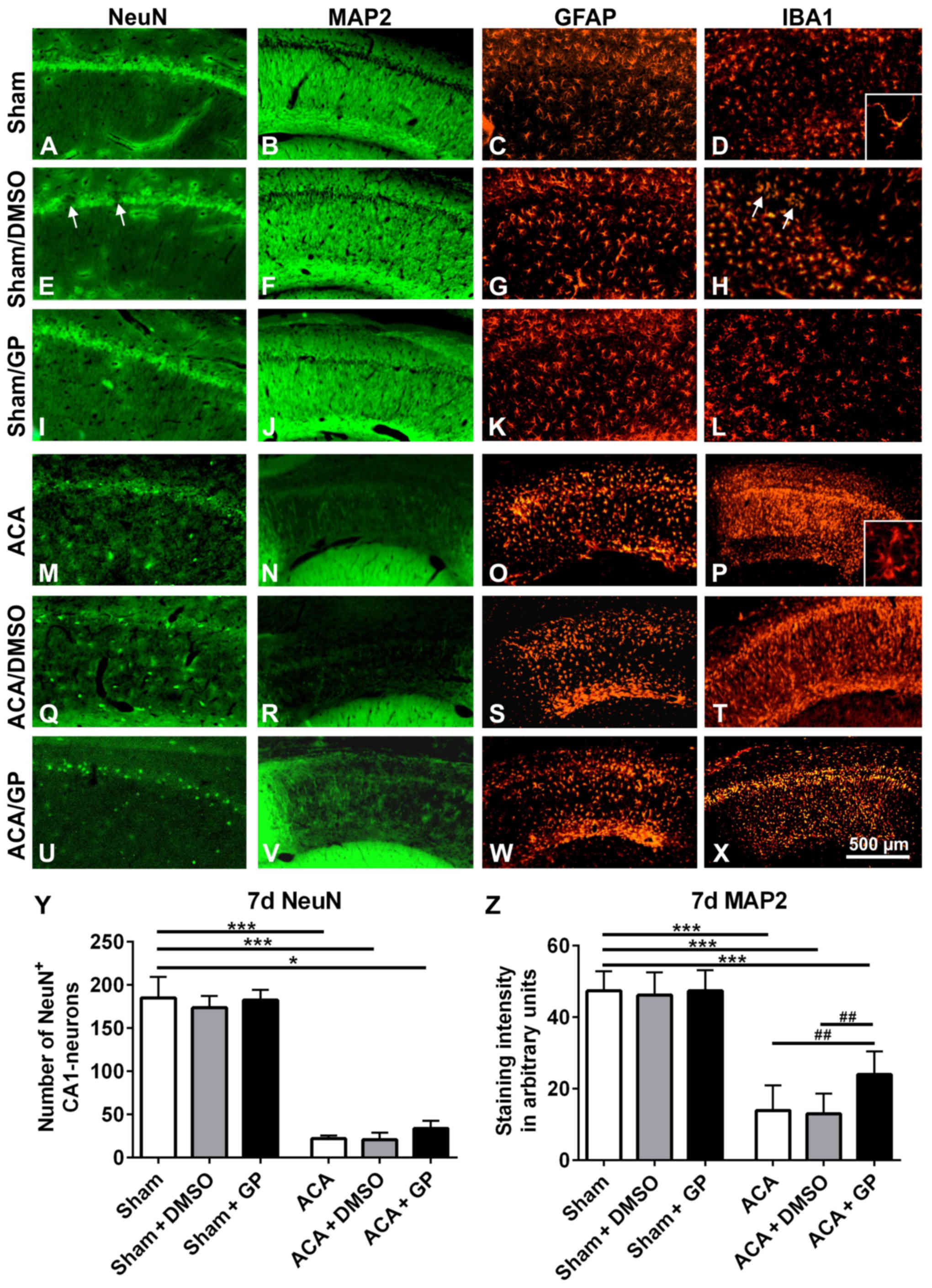 | Figure 7.Representative fluorescence images of
rat hippocampal CA1 pyramidal cell layer 7 days post-intervention.
In untreated sham-operated animals, the CA1 pyramidal cell line (A)
NeuN, the respective fiber network (B) MAP2, the astroglia pattern
(C) GFAP, and the microglia shape (D) IBA1 were normal.
Intervention with onetime applied DMSO induced a spotty loss of
pyramidal cells (arrows in E). The respective fiber network (F) and
the astroglia shape (K) showed, however, no abnormalities. IBA1
staining offered a limited microglia accumulation in the region of
damaged neurons (arrows in H). In GP treated sham-animals, the CA1
pyramidal cell line (I), the respective fiber network (J), the
astroglia pattern (K), and the microglia shape (L) were again
properly formed. ACA induced a massive loss of CA1 pyramidal cells.
The neuronal cell loss was indicated by the distinct reduction of
NeuN immunofluorescence signal (M) as well as by the massive
reduction of MAP2 staining of the respective nerve fiber network
(N). Consistently, significant activation of astroglia, indicated
by massive upregulation of GFAP staining (O), and activation of
microglia, indicated by upregulation of IBA1 immunofluorescence
signal (P) were seen. Thereby, microglia conversed from ramified to
amoeboid cell morphology (insert D vs. insert P). (Q-T) Onetime
applied DMSO treatment did not further amplify these ACA-induced
signs of neurodegeneration. (U-X) Onetime applied GP counteracted
these ACA-induced processes to some extent. NeuN-positive CA1
neurons were partly preserved (U) and also some MAP2-positive nerve
fibers survived (V). Patterns of astroglia (W) and microglia (X)
were, however, equally sever pronounced as in ACA and ACA-DMSO
animals. (Y) Semi-quantification of NeuN-positive CA1 neurons. (Z)
Semi-quantification of MAP2-stained CA1 fibers. Data are mean ± SD;
Kruskal-Wallis/Dunn's post-hoc test (*P<0.05; ***P<0.001) and
one-way ANOVA for intra-group differences of ACA-animals
(##P<0.005); all with n=5/group, ACA, asphyxia
cardiac arrest; GP, Gynostemma pentaphyllum; MAP2,
microtubule-associated protein 2; NeuN, neuronal nuclei; GFAP,
glial fibrillary acidic protein; IBA1, ionized calcium binding
adaptor molecule 1; DMSO, dimethyl sulfoxide; the given bar applies
for all pictures. |
In ACA-animals, the hippocampal NeuN-stained CA1
pyramidal cells were massively degenerated (Fig. 7M). In parallel, MAP2 immunostained
nerve fibers showed a massive failure (Fig. 7N). Activation of GFAP-positive
astroglia (Fig. 7O) and
IBA1-positive microglia (Fig. 7P)
was clearly evident. That was demonstrated by enhanced
immunoreactivity of the resident glia cells. Moreover, microglia
migration as well as conversion from ramified to amoeboid cell
morphology took place (insert Fig.
7D vs. insert 7P). DMSO intervention, irrespective of its
application regime, did not further impair these patterns (Fig. 7Q-T; demonstrated for onetime applied
DMSO). GP, independently of its application regime, was able to
counteract these degenerative changes to some degree; the band of
NeuN-positive CA1 neurons seemed to be partly retained (Fig. 7U, demonstrated for onetime applied
GP) and some of MAP2-positive fibers survived (Fig. 7V). Activation of astroglia (Fig. 7W) as well as microglia (Fig. 7X) was as high as in ACA and ACA-DMSO
animals. Obviously, the strong neurodegenerative potential of ACA
limited the GP-mediated neuroprotection. Semi-quantification is
given for NeuN-positive CA1 neurons in Fig. 7Y and for MAP2-stained CA1 fibers in
Fig. 7Z.
Novel object recognition test
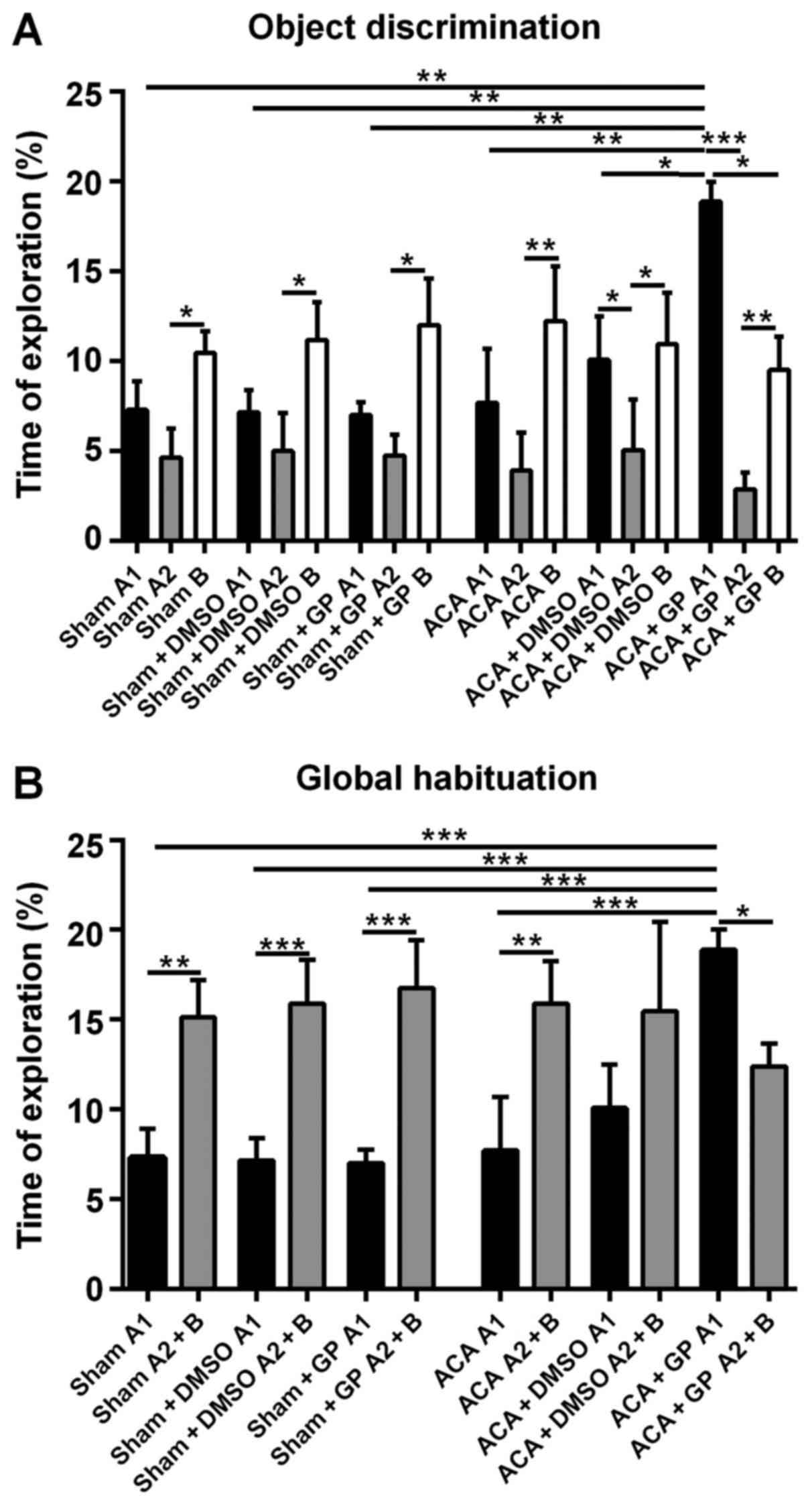
Sham-operated rats, independent of
non-/DMSO-/GP-treatment, were able to discriminate between a
‘known’ and a novel object; the exploration time of object B at
simultaneous presence of object A (A2) was roughly twice as long
(Fig. 8A). Moreover, the totalized
exploration time for A2 plus B in the test trial was significantly
higher than for object A alone (A1) in the sampling trial (Fig. 8B).
The recognition ability of ACA-stressed animals
without DMSO- or GP-intervention was, actually unexpected,
unchanged when compared with sham-animals. The exploration time
difference (B vs. A2) was even more pronounced (Fig. 8A). Now, this pattern was moderately
affected by DMSO and dramatically by GP. DMSO-treated animals
seemed to be better in the recognition of the ‘known’ object A (A1
vs. A2 significantly different; P<0.05). This effect of better
recognition, even more distinctive (P<0.001), was also found in
GP-treated rats. Additionally, the sampling behavior of the
GP-treated ACA-stressed animals was extremely pronounced. They
explored object A1 significantly longer than animals from all other
groups (Fig. 8A). And the GP-treated
animals were the only group with significant shorter (P<0.05)
exploration time for the novel object B when compared with the
exploration time of object A at the first session.
As demonstrated in Fig.
8B, the GP-effect in ACA-animals was strong enough to
completely reverse the significant difference of the global
habituation pattern. In the test trial (object A2 + B), the
exploration time of GP-treated ACA-animals was significantly lower
(P<0.05) then the time they spend to explore object A1 in the
first session. DMSO treatment of ACA-animals reduced the difference
between exploration times when both sessions were compared, but it
was not able to reverse the difference of the global habituation
pattern.
Discussion
This study was undertaken to test the
neuroprotective effects of an ethanolic extract from GP applied
simultaneously with resuscitation after ACA. To simulate this
clinical relevant situation we used our expensive animal model of
ACA (24). This experimental
approach is appropriate to study clinical relevant the consequences
of ACA in brain (3,31).
We applied an ethanolic extract from GP and used
DMSO instead of ethanol as the solvent. This decision was founded
on our previous work on brain slices reporting effects of GP on
evoked potentials in hippocampus in rats exposed to transient
oxygen/glucose deprivation (15,20). In
this model it has been demonstrated by others that ethanol caused a
dramatic decrease in evoked potentials especially in the
hippocampal CA1 region due to modulation of GABA and NMDA receptors
(32,33). DMSO doesn't cause such detrimental
effects (34,35). With respect to other potential side
effects we used the lowest possible DMSO concentration that we
estimated to be lower than 0.5% DMSO within the circulation. At
this concentration no side effect was found in vitro
(36) and also in vivo
(37). Nevertheless, we got a series
of DMSO effects which will be discussed in the respective
chapters.
The observed hyperglycaemia during the first 30 min
after reperfusion is assumed to result from suppressed insulin
secretion and is typical for critical care patients (38). It has been shown that GP stimulates
insulin release from islets (39).
Since disturbance of glucose homeostasis is a well-known cause of
neurodegeneration (reviewed in (40)
the prevention of hyperglycaemia by GP should possess
neuroprotective potency. A second hallmark of pathophysiological
alterations after ACA in this period of reperfusion is hypercapnia
that has been detected also under our conditions. Hypercapnia
contributes to the impairment of brain tissue due to mitochondrial
dysfunction (41). In our
experiments, GP prevented both post-ischemic hyperglycemia and
hypercapnia.
Like GP, DMSO was able to prevent the ACA-mediated
increase of pCO2 and blood glucose concentration but in
contrast to GP no neuroprotective potency was observed in the
presence of DMSO. In contrast to GP, DMSO was shown to suppress
insulin secretion from rat pancreatic islet cells (42). Hence, the anti-hyperglycemic effect
of DMSO in the ACA animals should be attributed to other
mechanisms, e.g., its ability to enhance GLUT4 translocation from
intracellular compartments to the plasma membrane. That increases
cellular glucose uptake with subsequent reduction of blood glucose
levels (43). Together with the
ability of DMSO to increase gluconeogenesis a high-glucose-state
would be established. High glucose, however, is able to induce
extracellular ROS increase and heme oxygenase-1 expression from
astrocytes resulting in neuronal apoptosis (44). The neuroprotective potency of GP
suggested that GP is able to reverse the effect of its solvent DMSO
by its stimulating effect on insulin release (39) and by its capability to attenuate heme
oxygenase-1 expression significantly (45).
Reduced hypercapnia by administration of GP and DMSO
could be interpreted as a consequence of their anti-hyperglycemic
potency which is known to be effective in attenuating post-ischemic
hypercapnia (46). Interestingly it
was shown that DMSO is able to mimic the neurotoxic effects of
hypoxia-hypercapnia, (47) thus
counteracting its own anti-hypercapnic/neuroprotective potency.
Again, GP seemed to be able to reverse this detrimental DMSO
effect. A possible target could be aquaporin 4. Its inhibition,
inducible also by gypenosides (48),
was helpful in a rat model of hypoxia-hypercapnia-induced brain
damage (49).
The hypothesis that GP possesses neuroprotective
potency in ACA-dependent brain injury was supported by the
histological analysis of the brain. GP, administered once
simultaneous with resuscitation, protected the vulnerable
hippocampal CA1 region from massive cell injury. Neuroprotection
was detected 24 h after resuscitation but also 7 d post asphyxia.
Thus, the neuroprotective potency of GP turned out to be
sustainable. It could be assumed that the observed neuroprotection
was caused by an orchestrated action of the major components of the
GP extract, Gypenoside LXIII, Gypenoside Rb3 or
Gypenoside VIII. The effectiveness of at least Gypenoside VIII
[Ginsenoside Rd; (36,45)] and Gypenoside Rb3
(50,51) against ischemic neurodegeneration is
well-documented.
Intact hippocampal structure is required for normal
cognitive competences in humans. Therefore, it can be speculated
that ACA affects the cognitive potential of patients after
transient CA due to neurodegeneration [reviewed by (52)]. This encouraged us to examine
attention and recognition skills after ACA by using the novel
object recognition test. It is known that under stress conditions
hippocampal neurons are challenged by elevated glucocorticoid and
glutamate concentrations that are associated with changed cognition
abilities (53,54). Thereby, CA1 hippocampal neurons are
major targets of corticoids (55),
whereby mineralocorticoid and glucocorticoid receptors are
co-localized in this cell type (54). Mineralocorticoid receptors are
relevant for this stress response by switching from low affinity
for corticosterone under normal conditions to high activity at high
corticosteroid levels (56). High
levels of corticosterone cause increase in exploratory activity of
rats (57) paralleled by
glutamate-depending increase in working memory. Facilitated working
memory has been described when rodents were exposed to acute stress
(58). In our experiments the rats
failed to develop better performance of object recognition as
response to ACA, although ACA should be a massive stressor. Also,
the exploration time remained unaffected. Similar results were
reported by others (59–61). We would suggest that ACA caused
interruption of the physiological response circuit due to
degeneration of the respective neurons. When GP was administered
the animals responded to ACA with better performance of object
recognition and increased exploration time. Obviously, the here
demonstrated neuroprotective potency of GP led to an at least
partial survival of the physiological response circuits needed for
an adequate stress response. Such a positive effect of GP in case
of cognition deficits has been described by others too. Zhang et
al demonstrated that GP improved cognition impairment induced
by chronic cerebral hypoperfusion in rats by suppressing oxidative
stress and astrocytic activation (21,62).
Hong et al (63) and Joh
et al (64) showed that
gypenosides are able to ameliorate scopolamine-induced learning
deficits in mice. As a possible mechanism they mentioned an
increased BDNF expression via the CREB signaling pathway.
Impairment of mitochondria is part of the
pathomechanism of ischemia/reperfusion injury including
neurodegeneration (65,66). Although neurodegeneration in
hippocampus has been observed we did not find a decrease in complex
activities of the respiratory chain when evaluated 6 h after
resuscitation. Instead, increased specific activities of complex I
+ III and complex II + III were detected as a sign of increased
energy demand after ACA. From this we conclude that damage of
mitochondria could occur earlier with subsequent recovery of
mitochondria. This idea fits well with the observed suppression of
cell death and stimulation/induction of citrate synthase by GP.
The defense/protection adaption seems to be
completed after 24 h since mitochondrial activities of the
respiratory chain complexes widely normalized within this period of
time. Initiation of mitochondrial biosynthesis as stress response
supports glutamate release and activation of glutamate receptors by
providing sufficient ATP for these energy consuming processes
(67). Although respiratory chain
complex activities normalized within 24 h we still detected
increased CL contents. This particular stress response in
hippocampus may contribute to the promotion of mitochondrial ATP
synthesis under these conditions. Our data suggest that the vehicle
DMSO prevents stimulation of CL synthesis but is not toxic for CL
itself. However, a toxic effect of DMSO we observed with respect to
complex II + III activity. DMSO had been shown to possess
neurotoxic effects similar to ischemia/reperfusion (47). In our experiments, the administration
of GP compensated the toxic DMSO effect with respect to complex II
+ III activity even when solved in DMSO at identical concentration.
In line with this finding is the report that GP can stimulate
complex I, II and IV (68).
Alternatively, GP may directly counteract DMSO effects.
In the presence of GP we found likewise diminished
CL contents after ACA treatment. This situation basically differs
from the effect of DMSO. We found no indication for any toxic GP
effect under sham conditions. The combination of ACA and GP
administration resulted in elevated citrate synthase activity
reflecting stimulation of mitochondrial biogenesis. Under this
condition increased demand of ATP can be matched by the higher
quantity of mitochondria. It is reasonable to assume that in this
situation no stimulation of CL synthesis is required for further
increase in mitochondrial ATP generation.
This result emphasized the importance of the early
post-resuscitation phase for brain injury and concomitantly for
interventional strategies. Because of the structure of the solvent
DMSO and gypenosides, it is reasonable to assume that important
constituents of the ethanolic GP extract can cross blood brain
barrier and cellular membrane systems. The resorption of GP by
intestine had been found to operate quickly. GP effects had been
observed at least 5 min after i.p. injection. Similar kinetics had
been reported by others (69).
We have shown that 6 min of ACA causes early
hyperglycaemia and hypercapnia followed by damage of neurons in the
hippocampus. This subsequently leads to the inability of cognitive
responses in rats. The mechanism of the impairment of neurons
includes modifications of mitochondria. Since most of these effects
could be attenuated by the administration of GP with resuscitation
our results designate the early reperfusion phase as a promising
therapeutic window. Moreover, the results underline the need of a
careful handling of the popular vehicle DMSO, in some cases even
used as control (70,71).
Acknowledgements
The technical assistance of Susanne Bonifatius,
Leona Bück, Stefanie Holze, Daniela Peter and Elke Wölfel Silke
Niemann is gratefully acknowledged.
Glossary
Abbreviations
Abbreviations:
|
ACA
|
asphyxial cardiac arrest
|
|
CA
|
cornu ammonis
|
|
CL
|
cardiolipin
|
|
complex I + III
|
NADH:cytochrome c oxidoreductase
|
|
complex II + III
|
succinate:cytochrome c
oxidoreductase
|
|
DMSO
|
dimethyl sulfoxide
|
|
ECG
|
electrocardiogram
|
|
GFAP
|
glial fibrillary acidic protein
|
|
GP
|
Gynostemma pentaphyllum
|
|
IBA1
|
ionized calcium binding adaptor
molecule 1
|
|
IPPV
|
intermittent positive pressure
ventilation
|
|
MAP
|
mean arterial pressure
|
|
MAP2
|
microtubule-associated protein 2
|
|
NeuN
|
neuronal nuclei antibody
|
|
PBS
|
phosphate-buffered saline
|
|
PFA
|
phosphate-buffered
paraformaldehyde
|
|
ROSC
|
return of spontaneous circulation
|
References
|
1
|
Sims NR and Anderson MF: Mitochondrial
contributions to tissue damage in stroke. Neurochem Int.
40:511–526. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schild L and Reiser G: Oxidative stress is
involved in the permeabilization of the inner membrane of brain
mitochondria exposed to hypoxia/reoxygenation and low micromolar
Ca2+. FEBS J. 272:3593–3601. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang L, Applegate PM, Gatling JW, Mangus
DB, Zhang J and Applegate RL II: A systematic review of
neuroprotective strategies after cardiac arrest: From bench to
bedside (part II-comprehensive protection). Med Gas Res. 4:102014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mangus DB, Huang L, Applegate PM, Gatling
JW, Zhang J and Applegate RL II: A systematic review of
neuroprotective strategies after cardiac arrest: From bench to
bedside (Part I-Protection via specific pathways). Med Gas Res.
4:92014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sjoberg F and Singer M: The medical use of
oxygen: A time for critical reappraisal. J Int Med. 274:505–528.
2013. View Article : Google Scholar
|
|
6
|
Dell'anna AM, Scolletta S, Donadello K and
Taccone FS: Early neuroprotection after cardiac arrest. Curr Opin
Crit Care. 20:250–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Llitjos JF, Mira JP, Duranteau J and
Cariou A: Hyperoxia toxicity after cardiac arrest: What is the
evidence? Ann Intensive Care. 6:232016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke statistics-2017 Update: A Report
From the American Heart Association. Circulation. 135:e146–e603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng JG: Investigation of the plant
jiaogulan and its analogous herb, Wulianmei. Zhong Cao Yao.
21:4241990.
|
|
10
|
Mishra RN and Joshi D: Jiao Gu Lan
(Gynostemma pentaphyllum): The Chinese Rasayan-Current
Research Scenario. Int J Res Pharm Biom Sci. 2:1483–1502. 2011.
|
|
11
|
Chen J: Antistress action of Gynostemma
pentaphyllum. Chinese Tradit Patent Med. 11:31–32. 1989.
|
|
12
|
Shang L, Liu J, Zhu Q, Zhao L, Feng Y,
Wang X, Cao W and Xin H: Gypenosides protect primary cultures of
rat cortical cells against oxidative neurotoxicity. Brain Res.
1102:163–174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Norberg A, Hoa NK, Liepinsh E, Van Phan D,
Thuan ND, Jörnvall H, Sillard R and Ostenson CG: A novel
insulin-releasing substance, phanoside, from the plant
Gynostemma pentaphyllum. J Biol Chem. 279:41361–41367. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang P, Niu L, Gao L, Li WX, Jia D, Wang
XL and Gao GD: Neuroprotective effect of gypenosides against
oxidative injury in the substantia nigra of a mouse model of
Parkinson's disease. J Int Med Res. 38:1084–1092. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schild L, Roth A, Keilhoff G, Gardemann A
and Brödemann R: Protection of hippocampal slices against
hypoxia/hypoglycemia injury by a Gynostemma pentaphyllum
extract. Phytomedicine. 16:734–743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Cardiolipin and mitochondrial function in health and
disease. Antioxid Redox Signal. 20:1925–1953. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mileykovskaya E and Dowhan W:
Cardiolipin-dependent formation of mitochondrial respiratory
supercomplexes. Chem Phys Lipids. 179:42–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakahara I, Kikuchi H, Taki W, Nishi S,
Kito M, Yonekawa Y, Goto Y and Ogata N: Changes in major
phospholipids of mitochondria during postischemic reperfusion in
rat brain. J Neurosurg. 76:244–250. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ji J, Baart S, Vikulina AS, Clark RS,
Anthonymuthu TS, Tyurin VA, Du L, St Croix CM, Tyurina YY, Lewis J,
et al: Deciphering of mitochondrial cardiolipin oxidative signaling
in cerebral ischemia-reperfusion. J Cereb Blood Flow Metab.
35:319–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schild L, Cotte T, Keilhoff G and
Brodemann R: Preconditioning of brain slices against hypoxia
induced injury by a Gynostemma pentaphyllum
extract-stimulation of anti-oxidative enzyme expression.
Phytomedicine. 19:812–818. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang GL, Deng JP, Wang BH, Zhao ZW, Li J,
Gao L, Liu BL, Xong JR, Guo XD, Yan ZQ and Gao GD: Gypenosides
improve cognitive impairment induced by chronic cerebral
hypoperfusion in rats by suppressing oxidative stress and
astrocytic activation. Behav Pharmacol. 22:633–644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Keilhoff G, John R, Langnaese K, Schweizer
H and Ebmeyer U: Triggered by asphyxia neurogenesis seems not to be
an endogenous repair mechanism, gliogenesis more like it.
Neuroscience. 171:869–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Keilhoff G, Schweizer H, John R, Langnaese
K and Ebmeyer U: Minocycline neuroprotection in a rat model of
asphyxial cardiac arrest is limited. Resuscitation. 82:341–349.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Keilhoff G, Titze M, Esser T, Langnaese K
and Ebmeyer U: Constitutive and functional expression of YB-1 in
microglial cells. Neuroscience. 301:439–453. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schild L, Chen BH, Makarov P, Kattengell
K, Heinitz K and Keilhoff G: Selective induction of apoptosis in
glioma tumour cells by a Gynostemma pentaphyllum extract.
Phytomedicine. 17:589–597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kao TH, Huang SC, Inbaraj BS and Chen BH:
Determination of flavonoids and saponins in Gynostemma
pentaphyllum (Thunb.) Makino by liquid chromatography-mass
spectrometry. Anal Chim Acta. 626:200–211. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moellering H and Gruber W: Determination
of citrate with citrate lyase. Anal Biochem. 17:369–376. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martens JC, Keilhoff G, Halangk W,
Wartmann T, Gardemann A, Päge I and Schild L: Lipidomic analysis of
molecular cardiolipin species in livers exposed to
ischemia/reperfusion. Mol Cell Biochem. 400:253–263. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ennaceur A and Delacour J: A new one-trial
test for neurobiological studies of memory in rats. 1: Behavioral
data. Behav Brain Res. 31:47–59. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kandiah P, Ortega S and Torbey MT:
Biomarkers and neuroimaging of brain injury after cardiac arrest.
Semin Neurol. 26:413–421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schummers J and Browning MD: Evidence for
a role for GABA(A) and NMDA receptors in ethanol inhibition of
long-term potentiation. Brain Res Mol Brain Res. 94:9–14. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Steffensen SC, Nie Z, Criado JR and
Siggins GR: Ethanol inhibition of N-methyl-D-aspartate responses
involves presynaptic gamma-aminobutyric acid(B) receptors. J
Pharmacol Exp Ther. 294:637–647. 2000.PubMed/NCBI
|
|
34
|
Albertson TE and Joy RM: Increased
inhibition in dentate gyrus granule cells following exposure to
GABA-uptake blockers. Brain Res. 435:283–292. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Laaris N, Good CH and Lupica CR:
Delta9-tetrahydrocannabinol is a full agonist at CB1 receptors on
GABA neuron axon terminals in the hippocampus. Neuropharmacology.
59:121–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang G, Xia F, Zhang Y, Zhang X, Cao Y,
Wang L, Liu X, Zhao G and Shi M: Ginsenoside Rd is efficacious
against acute ischemic stroke by suppressing microglial
proteasome-mediated inflammation. Mol Neurobiol. 53:2529–2540.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bakar B, Kose EA, Sonal S, Alhan A, Kilinc
K and Keskil IS: Evaluation of the neurotoxicity of DMSO infused
into the carotid artery of rat. Injury. 43:315–322. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nurmi J, Boyd J, Anttalainen N,
Westerbacka J and Kuisma M: Early increase in blood glucose in
patients resuscitated from out-of-hospital ventricular fibrillation
predicts poor outcome. Diabetes Care. 35:510–512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lokman EF, Gu HF, Mohamud Wan WN and
östenson CG: Evaluation of antidiabetic effects of the traditional
medicinal plant gynostemma pentaphyllum and the possible
mechanisms of insulin release. Evid Based Complement Alternat Med.
2015:1205722015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hassan M, Sehgal SA and Rashid S:
Regulatory cascade of neuronal loss and glucose metabolism. CNS
Neurol Disord Drug Targets. 13:1232–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Douglas RM, Ryu J, Kanaan A, Del Carmen
Rivero M, Dugan LL, Haddad GG and Ali SS: Neuronal death during
combined intermittent hypoxia/hypercapnia is due to mitochondrial
dysfunction. Am J Physiol Cell Physiol. 298:C1594–C1602. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sakonju I, Taura Y, Inayoshi Y, Suzuki T,
Takimoto K, Nakaichi M and Nakama S: Cryopreservation of isolated
rat islets of Langerhans in the presence of ethylene glycol or
dimethyl sulfoxide: Evaluation of toxicity and the dynamic pattern
of subsequent insulin release in vitro. Cryobiology. 33:354–362.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Berenguer M, Zhang J, Bruce MC, Martinez
L, Gonzalez T, Gurtovenko AA, Xu T, Le Marchand-Brustel Y and
Govers R: Dimethyl sulfoxide enhances GLUT4 translocation through a
reduction in GLUT4 endocytosis in insulin-stimulated 3T3-L1
adipocytes. Biochimie. 93:697–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang CM, Lin CC and Hsieh HL:
High-glucose-derived oxidative stress-dependent heme oxygenase-1
expression from astrocytes contributes to the neuronal apoptosis.
Mol Neurobiol. 54:470–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ye R, Zhao G and Liu X: Ginsenoside Rd for
acute ischemic stroke: translating from bench to bedside. Expert
Rev Neurother. 13:603–613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim YB, Gidday JM, Gonzales ER, Shah AR
and Park TS: Effect of hypoglycemia on postischemic cortical blood
flow, hypercapnic reactivity and interstitial adenosine
concentration. J Neurosurg. 81:877–884. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu LS, Fan YY, Ye G, Li J, Feng XP, Lin K,
Dong M and Wang Z: Curcumin alleviates brain edema by lowering AQP4
expression levels in a rat model of hypoxia-hypercapnia-induced
brain damage. Exp Ther Med. 11:709–716. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou Y, Li HQ, Lu L, Fu DL, Liu AJ, Li JH
and Zheng GQ: Ginsenoside Rg1 provides neuroprotection against
blood brain barrier disruption and neurological injury in a rat
model of cerebral ischemia/reperfusion through downregulation of
aquaporin 4 expression. Phytomedicine. 21:998–1003. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu H, Qi GL, Wang J, Chen L, Deng Z, Zhao
YS, Lei SS and Zhu XQ: Aquaporin 4 inhibition decreased synthesis
of cytokines by acetazolamide in the hippocampus of rats with
pentrazol-induced chronic epilepsy. Genet Mol Res. 15:2016.
View Article : Google Scholar
|
|
50
|
Jiang S, Miao B, Song X and Jiang Z:
Inactivation of GABA(A) receptor reduces ginsenoside Rb3
neuroprotection in mouse hippocampal slices after oxygen-glucose
deprivation. J Ethnopharmacol. 133:914–916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhu JR, Tao YF, Lou S and Wu ZM:
Protective effects of ginsenoside Rb(3) on oxygen and glucose
deprivation-induced ischemic injury in PC12 cells. Acta Pharmacol
Sin. 31:273–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Green CR, Botha JA and Tiruvoipati R:
Cognitive function, quality of life and mental health in survivors
of our-of-hospital cardiac arrest: A review. Anaesth Intensive
Care. 43:568–576. 2015.PubMed/NCBI
|
|
53
|
Popoli M, Yan Z, McEwen BS and Sanacora G:
The stressed synapse: The impact of stress and glucocorticoids on
glutamate transmission. Nat Rev Neurosci. 13:22–37. 2012.
|
|
54
|
Takeda A and Tamano H: Proposed
glucocorticoid-mediated zinc signaling in the hippocampus.
Metallomics. 4:614–618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Joels M: Functional actions of
corticosteroids in the hippocampus. Eur J Pharmacol. 583:312–321.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
de Kloet ER, Karst H and Joels M:
Corticosteroid hormones in the central stress response:
Quick-and-slow. Front Neuroendocrinol. 29:268–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sutt S, Raud S, Abramov U, Innos J, Luuk
H, Plaas M, Kõks S, Zilmer K, Mahlapuu R, Zilmer M and Vasar E:
Relation of exploratory behaviour to plasma corticosterone and Wfs1
gene expression in Wistar rats. J Psychopharmacol. 24:905–913.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yuen EY, Liu W, Karatsoreos IN, Ren Y,
Feng J, McEwen BS and Yan Z: Mechanisms for acute stress-induced
enhancement of glutamatergic transmission and working memory. Mol
Psychiatry. 16:156–170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hoge J and Kesner RP: Role of CA3 and CA1
subregions of the dorsal hippocampus on temporal processing of
objects. Neurobiol Learn Mem. 88:225–231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vago DR, Bevan A and Kesner RP: The role
of the direct perforant path input to the CA1 subregion of the
dorsal hippocampus in memory retention and retrieval. Hippocampus.
17:977–987. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Piterkin P, Cole E, Cossette MP, Gaskin S
and Mumby DG: A limited role for the hippocampus in the modulation
of novel-object preference by contextual cues. Leam Mem.
15:785–791. 2008. View Article : Google Scholar
|
|
62
|
Zhang G, Zhao Z, Gao L, Deng J, Wang B, Xu
D, Liu B, Qu Y, Yu J, Li J and Gao G: Gypenoside attenuates white
matter lesions induced by chronic cerebral hypoperfusion in rats.
Pharmacol Biochem Behav. 99:42–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hong SW, Yang JH, Joh EH, Kim HJ and Kim
DH: Gypenoside TN-2 ameliorates scopolamine-induced learning
deficit in mice. J Ethnopharmacol. 134:1010–1013. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Joh EH, Yang JW and Kim DH: Gypenoside
LXXIV ameliorates scopolamine-induced learning deficit in mice.
Planta Med. 76:793–795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hou ST and MacManus JP: Molecular
mechanisms of cerebral ischemia-induced neuronal death. Int Rev
Cytol. 221:93–148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Szeto HH: Mitochondria-targeted
cytoprotective peptides for ischemia-reperfusion injury. Antioxid
Redox Signal. 10:601–619. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Osborne DM, Pearson-Leary J and McNay EC:
The neuroenergetics of stress hormones in the hippocampus and
implications for memory. Front Neurosci. 9:1642015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yu H, Guan Q, Guo L, Zhang H, Pang X,
Cheng Y, Zhang X and Sun Y: Gypenosides alleviate myocardial
ischemia-reperfusion injury via attenuation of oxidative stress and
preservation of mitochondrial function in rat heart. Cell Stress
Chaperones. 21:429–437. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen DJ, Hu HG, Xing SF, Gao YJ, Xu SF and
Piao XL: Metabolic profiling of Gynostemma pentaphyllum
extract in rat serum, urine and faeces after oral administration. J
Chromatogr B Analyt Technol Biomed Life Sci. 969:42–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nejad KH, Gharib-Naseri MK, Sarkaki A,
Dianat M, Badavi M and Farbood Y: Effects of ellagic acid
pretreatment on renal functions disturbances induced by global
cerebral ischemic-reperfusion in rat. Iran J Basic Med Sci.
20:75–82. 2017.PubMed/NCBI
|
|
71
|
Filippone SM, Samidurai A, Roh SK, Cain
CK, He J, Salloum FN, Kukreja RC and Das A: Reperfusion Therapy
with Rapamycin Attenuates Myocardial Infarction through Activation
of AKT and ERK. Oxid Med Cell Longev. 2017:46197202017. View Article : Google Scholar : PubMed/NCBI
|