Introduction
Tuberous sclerosis complex (TSC) is an autosomal
dominant genetic disease with an incidence of 1 in 6,000 and its
symptoms include seizures, mental retardation, skin lesions and the
formation of hamartomas in multiple organs, including the heart,
brain, eye and kidney (1). Mutations
in one of the two tumor suppressor genes TSC1-9q34 and TSC2-16p13.3
are responsible for TSC (2). The
absence of clinical features during the neonatal period makes the
diagnosis of TSC difficult. Neonatal ultrasound and cerebral
magnetic resonance imaging (MRI) may be used to detect hamartomas
in the heart and brain (3).
Additionally, genetic mutation analysis means that the risk of a
couple conceiving a child with TSC can be determined prior to
pregnancy (4). Shepherd et al
(5) analyzed 355 cases of TSC and
reported that the mortality rate is 13.8%.
Medication and surgery are the major treatment
methods for TSC (6,7). Vigabatrin, an antiepileptic medicine,
was approved in 2009 and recommended as a first-line drug for TSC-
associated infantile spasms by 2012 (8). In addition, adrenocorticotropic hormone
was approved to treat infantile spasms in 2010 (9). Everolimus is applied for brain
(subependymal giant cell astrocytoma) and kidney (renal
angiomyolipoma) tumor treatment in children with TSC (10) and has also been demonstrated to be
effective for TSC-epilepsy treatment (11). In 2017 votubia was recommended by the
European Commission as a treatment for refractory partial-onset
seizures in patients with TSC (12).
Canpolat et al (13)
demonstrated that rapamycin effectively controls epilepsy without
causing any marked side effects in children with TSC.
The current study describes a case of multiple
cardiac rhabdomyomas confirmed by routine echocardiogram screening
in week 31 of pregnancy. The infant experienced seizures in the
neonatal period (2 weeks of age), which is earlier than previously
reported cases in which the onset was 1 month of age (14). Genetic mutation analysis revealed a
novel mutation in TSC2. The clinical presentation and final outcome
of neonatal TSC is discussed in the current study.
Case report
The mother of the infant in the current case report
was a 29-year-old female (gravida 2, para 0) who experienced an
uncomplicated pregnancy until week 31 of gestation. A routine
echocardiography examination revealed multiple substantial
hyperechoic masses in the ventricular wall of the fetus, suggesting
a diagnosis of cardiac rhabdomyomas. A male infant weighing 3 kg
was successfully delivered in week 39 of pregnancy. At 23 days old,
the infant was admitted to the Emergency Department of the
Children's Hospital, Zhejiang University School of Medicine
(Hangzhou, China) in January 2016. The infant had undergone a
series of active seizures in the week prior to admittance. Seizure
episodes were characterized by paroxysmal jittery limbs followed by
the passing of urine. Each episode lasted 5–10 sec and was not
accompanied by fever. The parents of the infant were healthy and
had no history of TSC. The patient's family denied a history of
epilepsy, mental retardation and behavioral problems.
Physical examination revealed a small number of
hypomelanotic macules on the skin on the chest and back of the
infant. The results of the neurological (including physiological
reflex) and cardiovascular (heart rate, 124 bpm; blood pressure,
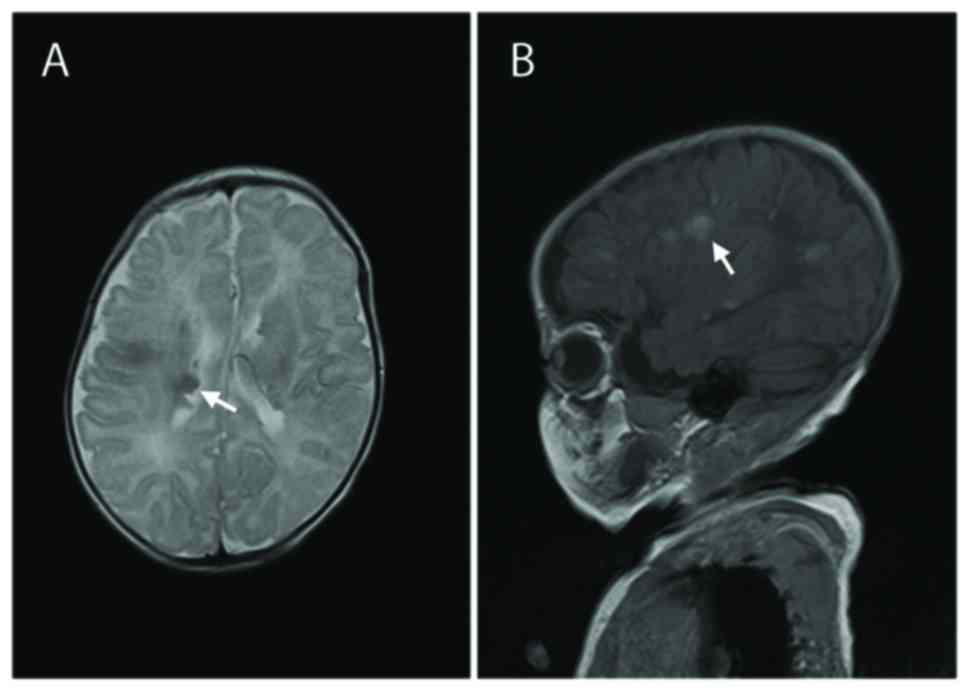
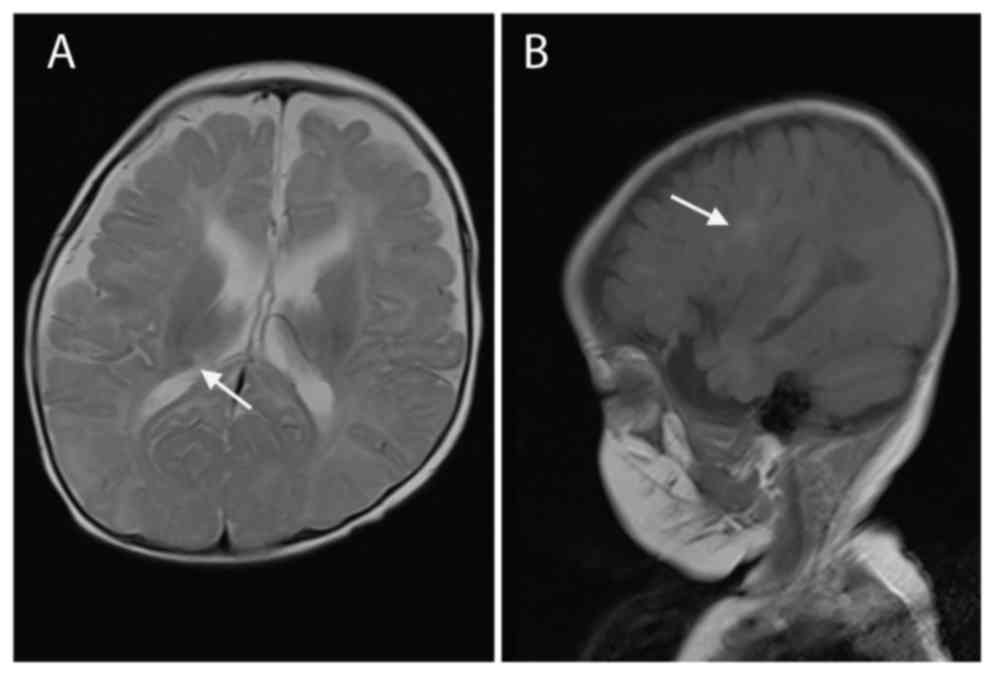
74/40 mmHg) examinations were normal. MRI scans of the brain
identified subependymal nodules and subcortical tubers (Fig. 1), which are two distinct features of
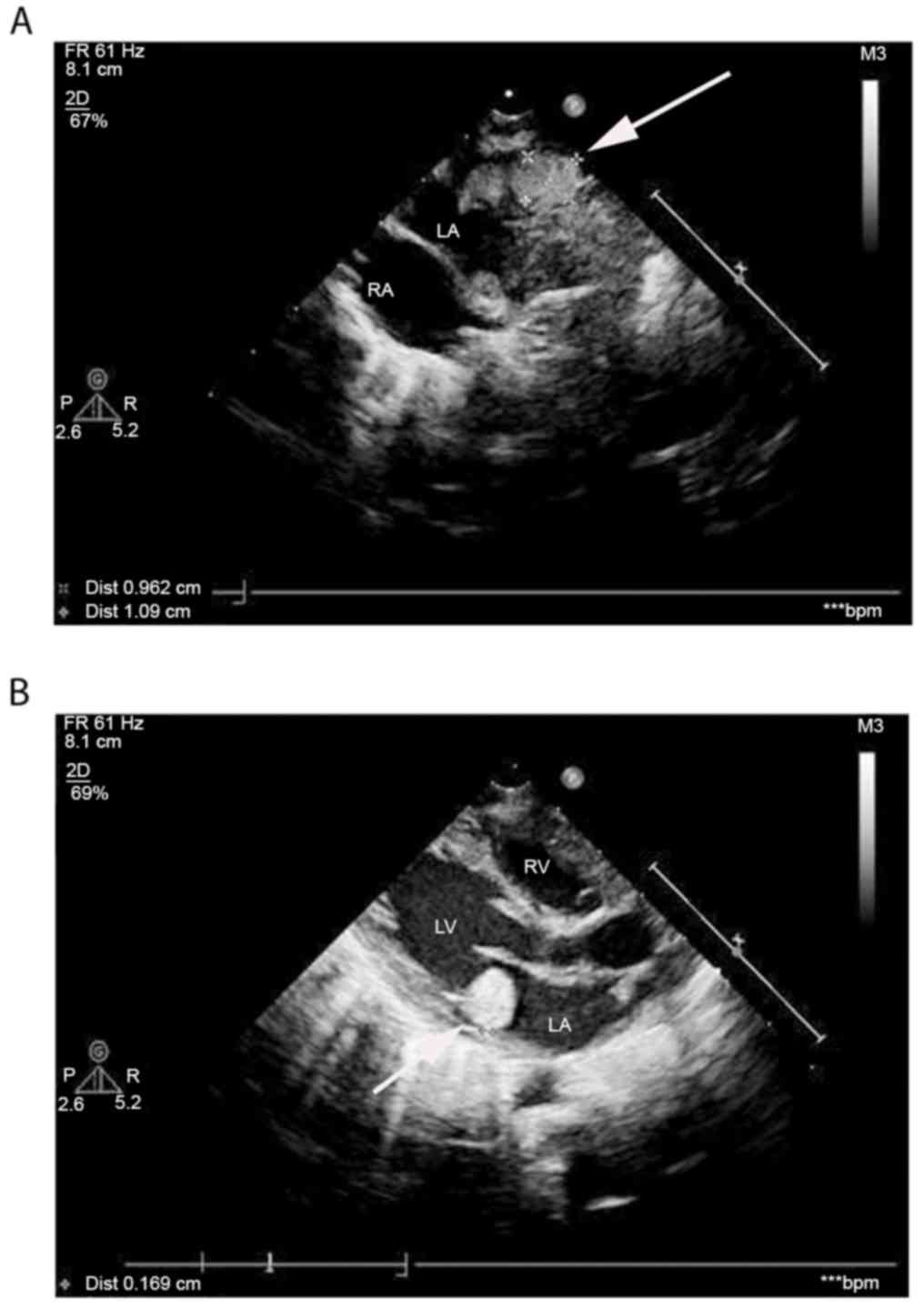
TSC. Echocardiography revealed hyperechoic masses with clear
borders and uniform echoes, measuring 1.06×0.89 and 1.77×1.68 cm in
the left atrium and the outlet of the right ventricle,
respectively, verifying the presence of intracardial tumors, which
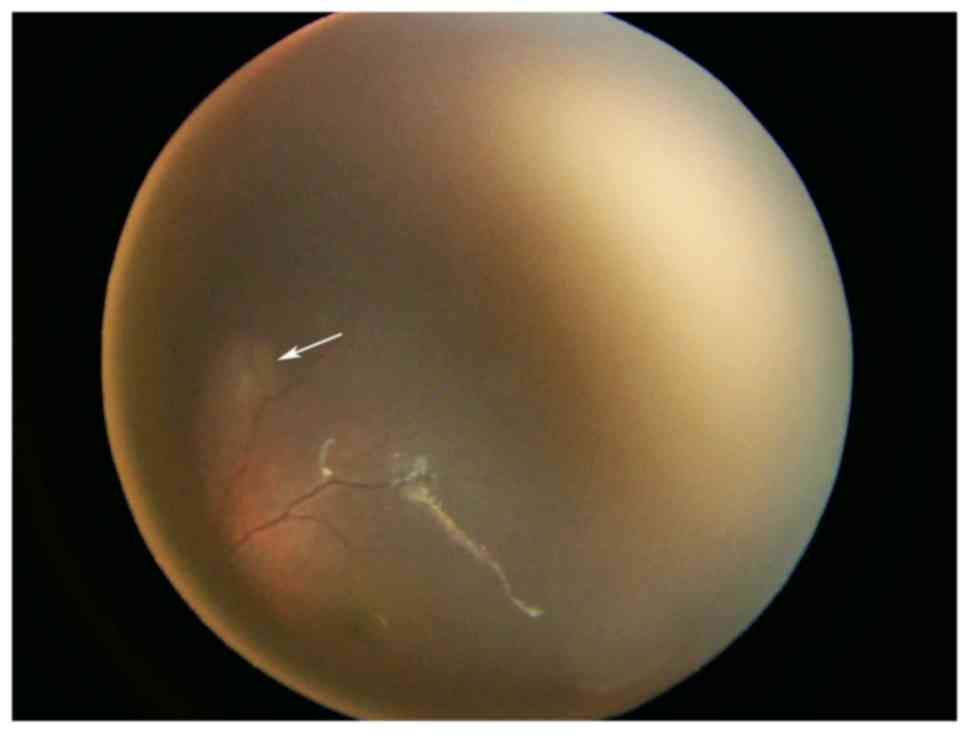
indicated the presence of cardiac rhabdomyomas (Fig. 2). Ophthalmological examination
clearly identified multiple nodules (Fig. 3). A detailed abdominal ultrasound did
not identify any signs of TSC. Electrocardiogram revealed a sinus
rhythm without cardiac arrhythmia and video electroencephalographic
monitoring did not prompt a diagnosis of epileptic seizures.
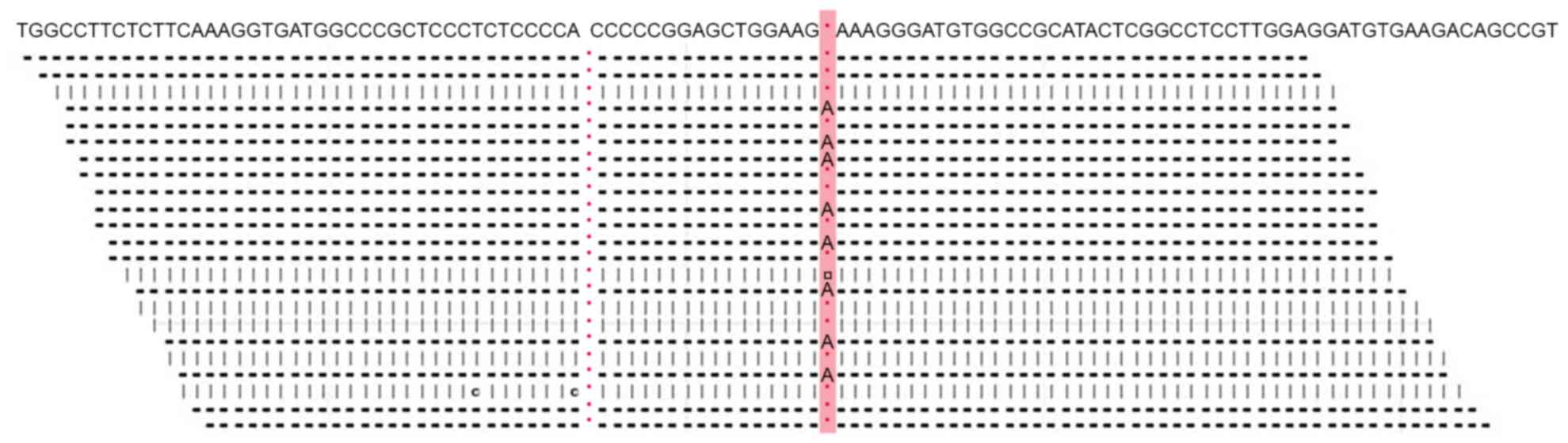
Mutation analysis of the TSC1 and TSC2 genes confirmed a diagnosis
of TSC. A TSC2c.1642_1643insA [or TSC2p.K549fsX589; TSC2 normal
gene reference is from NP_0,01070651.1 (https://www.ncbi.nlm.nih.gov/protein/116256350/)]
frameshift mutation was identified, which terminated the
translation of the encoded protein (Fig.
4). No mutations were identified in the TSC1 sequence.
Following diagnosis of TSC, the patient received
antiepileptic drugs including Topamax® (3 mg/kg/day;
Xian-Janssen Pharmaceutical Ltd., Xi'an, China),
Depakin® (30 mg/kg/day; Sanofi S.A., Paris, France) and
nitrazepam (1 mg/kg/day; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). However, this treatment did not alter the frequency of
seizures. At the age of 3 months, the infant underwent treatment
with 1 mg/(m2/day) rapamycin orally to treat resistant
epileptic seizures and the blood concentration of rapamycin was
maintained at 5–10 µg/l, as measured using the Rapamycin ARCHITECT
Sirolimus Reagent kit (cat. no. 72003M800) and the Abbott Architect
i1000 (both Abbott Pharmaceutical Co., Ltd., Lake Bluff, IL, USA).
During this period of 3 months, seizures and results of an
electroencephalogram (EEG) were recorded every month and the
results of the routine blood and urine tests, and liver and kidney
function tests (including glutamic-pyruvic transaminase, normal
range at 8–40 U/l and serum creatinine, normal range at 15–77
µmol/l) were normal. Echocardiography performed on the infant at
the age of 6 months identified regression of multiple cardiac
rhabdomyomas and MRI of the brain revealed a decrease in the size
of cerebral lesions (Fig. 5). The
results of the EEG were normal and the frequency of seizures
decreased. However, the patient continued to exhibit mental
retardation.
Discussion
A thorough search of literature published since 1990
(384 published manuscripts, including 98 reviews) was performed
using PubMed (https://www.ncbi.nlm.nih.gov/pubmed) with the
following key words: Tuberous sclerosis complex, newborn, neonates,
neonatal, infants. Data from 36 infants aged <4 weeks who were
diagnosed with TSC were included in the review. The clinical
manifestations of TSC were present prior to birth in 8 patients
(22.22%), at birth in 13 patients (36.11%) and by 4 weeks of age in
7 patients (19.44%). Information regarding onset of TSC was not
available for the remaining 8 patients included in the current
review (22.22%). TSC was diagnosed at birth in 7 patients (19.44%)
and by 4 weeks of age in 29 patients (80.56%). A greater number of
males than females (ratio, 1.4:1) were diagnosed with TSC. There
was a family history of TSC in 2 patients (5.55%). The most common
features at onset of TSC were cardiac rhabdomyomas (36.11%),
seizures (19.44%), arrhythmia (16.67%) and skin lesions (13.89%)
followed by renal cyst, opisthotonus, feeding difficulties and
respiratory distress. A total of 34 neonates underwent brain MRI or
computed tomography imaging and 2 neonates underwent a brain biopsy
following mortality. Cortical tubes, subependymal nodules and
subependymal giant cell astrocytomas were identified in 26
(72.22%), 28 (77.78%) and 5 (13.89%) patients, respectively.
Retinal hamartomas were detected in 5 neonates. The overall
survival rate of neonates with TSC was 81% (21/26) and the duration
of follow-up varied from 1 month to 4 years.
Cardiac rhabdomyomas were the most common initial
symptom detected in neonates with TSC included in the current
review and were identified in 13 neonates (36.11%). Cardiac
rhabdomyomas are the most prevalent heart tumors in neonates
(15). It has been reported that
>80% of cardiac rhabdomyomas regress completely during infancy
and early childhood (16). Depending
on their size, location and number, cardiac rhabdomyomas cause
serious cardiovascular complications, including intracavitary
obstruction, diminished myocardial function and arrhythmia
(17). Medical and/or surgical
interventions are required for symptomatic patients with
hemodynamically significant cardiac rhabdomyomas or a
life-threatening arrhythmia (18).
In the present review, 2 out of 7 neonates with arrhythmia
succumbed following cardiac arrest but the other 5 neonates
survived following effective antiarrhythmic treatment.
Neurological manifestations including seizures and
mental retardation are the major factors for morbidity in patients
with TSC (19). The current review
indicated that ~20% of neonates with TSC develop seizures in the
first month of life. Early onset of epilepsy in TSC is strongly
associated with mental retardation (20,21). It
has been demonstrated that antiepileptic treatment reduces the
severity of epilepsy and risk of mental retardation in infants with
TSC (22,23). Brain lesions in TSC include cortical
tubers, subependymal nodules (SEN), subependymal giant cell
astrocytomas and white matter lesions (24). In the current review, cortical tubers
and SEN were the two most common brain MRI manifestations in
neonates with TSC.
Cutaneous manifestations of TSC are easily
identified and are present in >90% of patients with TSC
(25). Hypomelanotic macules may be
present at birth, however they also may not appear until later in
life (26). The ocular symptoms of
TSC are retinal hamartomas and these occur in 40–50% of patients.
Retinal hamartomas typically do not cause visual dysfunction
(27) and ocular hamartomas rarely
occur in neonates (28). However, in
the current case, hypomelanotic macules and ocular hamartomas were
present.
Renal lesions serve an important role in the course
of TSC by impairing renal function (29). Renal cysts, angiomyolipomas and renal
cell carcinomas are the most common renal lesions in patients with
TSC (30,31). Isaacs (32) reported that 13.2% of neonates and
fetuses with TSC exhibit renal cysts. Polycystin 1, transient
receptor potential channel interacting (PKD1) is the major gene
responsible for autosomal dominant polycystic kidney disease. The
TSC2 gene lies adjacent to PKD1, suggesting that PKD1 serves a role
in the etiology of renal cystic disease in TSC (33). Deletion of the TSC2 and PKD1 genes is
associated with a severe polycystic phenotype and this occurs in 2%
of patients with TSC (34).
TSC is caused by mutations in either of the two
tumor suppressor genes, TSC1, which encodes hamartin (35) and TSC2, which encodes tuberin
(36). TSC1 and TSC2 inhibit the
mechanistic target of the mTOR-mediated signaling pathway, thus
preventing cell growth and cell cycle progression (37). Dysfunction of TSC1/TSC2, which may be
caused by a mutation, results in the loss of control of mTOR
signaling and subsequently causes cancer. A novel TSC2 mutation was
present in the current case report. The earlier diagnosis of
patients with TSC2 mutations may be beneficial for reducing the
severity of symptoms with earlier intervention (38). Rapamycin inhibits the activation of
the mTOR signaling pathway and has been used to treat patients with
TSC (39). Several studies have
demonstrated successful regression of lesions in the skin, brain,
and kidney (40–42). Canpolat et al (13) demonstrated that rapamycin effectively
controls epilepsy without causing any marked side effects in
children with TSC. However, to the best of our knowledge, there
have been no studies in English investigating the effect of
rapamycin on epilepsy in neonates and the safety of rapamycin in
patients <18 years of age, particularly in infant and neonates,
remains unknown (43). The patient
in the current case study received rapamycin treatment and had a
good prognosis, experiencing regression of cardiac rhabdomyomas and
controlled seizures. Therefore, the current case report indicated
that rapamycin treatment for TSC caused by a TSC2 mutation was
therapeutically beneficial and may be beneficial in treating other
disorders caused by abnormal mTOR signaling, such as cancer.
The current study demonstrates that cardiac
rhabdomyomas, seizures and skin lesions are well established
markers for TSC in neonates. MRI scans of the brain and genetic
screening of TSC1 and TSC2 genes may facilitate an early diagnosis
of TSC.
Acknowledgements
The present study was supported by the Science &
Technology Bureau of Zhejiang Province (Zhejiang, China; grant no.
2015C31101).
References
|
1
|
Curatolo P and Maria BL: Tuberous
sclerosis. Handb Clin Neurol. 111:323–331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crino PB, Nathanson KL and Henske EP: The
tuberous sclerosis complex. N Engl J Med. 355:1345–1356. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wortmann SB, Reimer A, Creemers JW and
Mullaart RA: Prenatal diagnosis of cerebral lesions in Tuberous
sclerosis complex (TSC). Case report and review of the literature.
Eur J Paediatr Neurol. 12:123–126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee KA, Won HS, Shim JY, Lee PR and Kim A:
Molecular genetic, cardiac and neurodevelopmental findings in cases
of prenatally diagnosed rhabdomyoma associated with tuberous
sclerosis complex. Ultrasound Obstet Gynecol. 41:306–311. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shepherd CW, Gomez MR, Lie JT and Crowson
CS: Causes of death in patients with tuberous sclerosis. Mayo Clin
Proc. 66:792–796. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saxena A and Sampson JR: Epilepsy in
Tuberous Sclerosis: Phenotypes, mechanisms and treatments. Semin
Neurol. 35:269–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pascual-Castroviejo I: Neurosurgical
treatment of tuberous sclerosis complex lesions. Childs Nerv Syst.
27:1211–1219. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krueger DA and Northrup H: International
Tuberous Sclerosis Complex Consensus Group: Tuberous sclerosis
complex surveillance and management: Recommendations of the 2012
International Tuberous Sclerosis Complex Consensus Conference.
Pediatr Neurol. 49:255–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Go CY, Mackay MT, Weiss SK, Stephens D,
Adams-Webber T, Ashwal S and Snead OC III: Child Neurology Society;
American Academy of Neurology: Evidence-based guideline update:
Medical treatment of infantile spasms. Report of the Guideline
Development Subcommittee of the American Academy of Neurology and
the Practice Committee of the Child Neurology Society. Neurology.
78:1974–1980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Press Announcements-FDA approves Afinitor
for non-cancerous kidney tumors caused by rare genetic disease.
https://elbiruniblogspotcom.blogspot.ca/2012/04/press-announcements-fda-approves_27.htmlApril
27–2012
|
|
11
|
French JA, Lawson JA, Yapici Z, Ikeda H,
Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz
N, et al: Adjunctive everolimus therapy for treatment-resistant
focal-onset seizures associated with tuberous sclerosis (EXIST-3):
A phase 3, randomised, double-blind, placebo-controlled study. The
Lancet. 388:2153–2163. 2016. View Article : Google Scholar
|
|
12
|
AG Novartis International: Novartis drug
Votubia® receives EU approval to treat refractory
partial-onset seizures in patients with TSC. https://www.novartis.com/news/media-releases/novartis-drug-votubiar-receives-eu-approval-treat-refractory-partial-onsetJanuary
31–2017
|
|
13
|
Canpolat M, Per H, Gumus H, Yikilmaz A,
Unal E, Patiroglu T, Cinar L, Kurtsoy A and Kumandas S: Rapamycin
has a beneficial effect on controlling epilepsy in children with
tuberous sclerosis complex: Results of 7 children from a cohort of
86. Childs Nerv Syst. 30:227–240. 2014. View Article : Google Scholar
|
|
14
|
Gipson TT, Gerner G, Srivastava S, Poretti
A, Vaurio R, Hartman A and Johnston MV: Early Neurodevelopmental
Screening in TSC: A Potential Window of Opportunity. Pediatr
Neurol. 51:398–402. 2014. View Article : Google Scholar
|
|
15
|
Isaacs H Jr: Fetal and neonatal cardiac
tumors. Pediatr Cardiol. 25:252–273. 2004. View Article : Google Scholar
|
|
16
|
Bader RS, Chitayat D, Kelly E, Ryan G,
Smallhorn JF, Toi A and Hornberger LK: Fetal rhabdomyoma: Prenatal
diagnosis, clinical outcome, and incidence of associated tuberous
sclerosis complex. J Pediatr. 143:620–624. 2003. View Article : Google Scholar
|
|
17
|
Verhaaren HA, Vanakker O, De Wolf D, Suys
B, Francois K and Matthys D: Left ventricular outflow obstruction
in rhabdomyoma of infancy: Meta-analysis of the literature. J
Pediatr. 143:258–263. 2003. View Article : Google Scholar
|
|
18
|
Stiller B, Hetzer R, Meyer R, Dittrich S,
Pees C, Alexi-Meskishvili V and Lange PE: Primary cardiac tumours:
When is surgery necessary? Eur J Cardiothorac Surg. 20:1002–1006.
2001. View Article : Google Scholar
|
|
19
|
Napolioni V, Moavero R and Curatolo P:
Recent advances in neurobiology of Tuberous Sclerosis Complex.
Brain Dev. 31:104–113. 2009. View Article : Google Scholar
|
|
20
|
Chu-Shore CJ, Major P, Camposano S,
Muzykewicz D and Thiele EA: The natural history of epilepsy in
tuberous sclerosis complex. Epilepsia. 51:1236–1241. 2010.
View Article : Google Scholar
|
|
21
|
Curatolo P, Aronica E, Jansen A, Jansen F,
Kotulska K, Lagae L, Moavero R and Jozwiak S: Early onset epileptic
encephalopathy or genetically determined encephalopathy with early
onset epilepsy? Lessons learned from TSC. Eur J Paediatr Neurol.
20:203–211. 2016. View Article : Google Scholar
|
|
22
|
Jozwiak S, Kotulska K, Domańska-Pakieła D,
Lojszczyk B, Syczewska M, Chmielewski D, Dunin-Wasowicz D, Kmieć T,
Szymkiewicz-Dangel J, Kornacka M, et al: Antiepileptic treatment
before the onset of seizures reduces epilepsy severity and risk of
mental retardation in infants with tuberous sclerosis complex. Eur
J Paediatr Neurol. 15:424–431. 2011. View Article : Google Scholar
|
|
23
|
Cusmai R, Moavero R, Bombardieri R,
Vigevano F and Curatolo P: Long-term neurological outcome in
children with early-onset epilepsy associated with tuberous
sclerosis. Epilepsy Behav. 22:735–739. 2011. View Article : Google Scholar
|
|
24
|
Mizuguchi M and Takashima S:
Neuropathology of tuberous sclerosis. Brain Dev. 23:508–515. 2001.
View Article : Google Scholar
|
|
25
|
Cardis MA and DeKlotz CM: Cutaneous
manifestations of tuberous sclerosis complex and the
paediatrician's role. Arch Dis Child. 102:858–863. 2017. View Article : Google Scholar
|
|
26
|
Hake S: Cutaneous manifestations of
tuberous sclerosis. Ochsner J. 10:200–204. 2010.
|
|
27
|
Schwartz RA, Fernández G, Kotulska K and
Jóźwiak S: Tuberous sclerosis complex: Advances in diagnosis,
genetics and management. J Am Acad Dermatol. 57:189–202. 2007.
View Article : Google Scholar
|
|
28
|
Knopke S, Olze H, Becker ET, Manthey D,
Lindig-Knopke C, Jöhrens K, Stölzel K and Böttcher A: Head and neck
hamartomas: 10 years of experience at the Charité-University
Medical Center Berlin. HNO. 63:552–556. 2015. View Article : Google Scholar
|
|
29
|
Kingswood JC, Bissler JJ, Budde K, Hulbert
J, Guay-Woodford L, Sampson JR, Sauter M, Cox J, Patel U, Elmslie
F, et al: Review of the Tuberous Sclerosis Renal Guidelines from
the 2012 Consensus Conference: Current Data and Future Study.
Nephron. 134:51–58. 2016. View Article : Google Scholar
|
|
30
|
Korula S, Ekbote A, Kumar N, Danda S,
Agarwal I and Chaturvedi S: Renal manifestations of tuberous
sclerosis among children: An Indian experience and review of the
literature. Clin Kidney J. 7:134–137. 2014. View Article : Google Scholar
|
|
31
|
Rouviere O, Nivet H, Grenier N, Zini L and
Lechevallier E: Kidney damage due to tuberous sclerosis complex:
Management recommendations. Diagn Interv Imaging. 94:225–237. 2013.
View Article : Google Scholar
|
|
32
|
Isaacs H: Perinatal (fetal and neonatal)
tuberous sclerosis: A review. Am J Perinatol. 26:755–760. 2009.
View Article : Google Scholar
|
|
33
|
Martignoni G, Bonetti F, Pea M, Tardanico
R, Brunelli M and Eble JN: Renal disease in adults with TSC2/PKD1
contiguous gene syndrome. Am J Surg Pathol. 26:198–205. 2002.
View Article : Google Scholar
|
|
34
|
Ismail NF, Malik Nik Abdul NM, Mohseni J,
Rani AM, Hayati F, Salmi AR, Narazah MY, Zabidi-Hussin ZA, Silawati
AR, Keng WT, et al: Two novel gross deletions of TSC2 in Malaysian
patients with tuberous sclerosis complex and TSC2/PKD1 contiguous
deletion syndrome. Jpn J Clin Oncol. 44:506–511. 2014. View Article : Google Scholar
|
|
35
|
van Slegtenhorst M, de Hoogt R, Hermans C,
Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A,
Halley D, Young J, et al: Identification of the tuberous sclerosis
gene TSC1 on chromosome 9q34. Science. 277:805–808. 1997.
View Article : Google Scholar
|
|
36
|
European Chromosome 16 Tuberous Sclerosis
Consortium: Identification and characterization of the tuberous
sclerosis gene on chromosome 16. Cell. 75:1305–1315. 1993.
View Article : Google Scholar
|
|
37
|
Kwiatkowski DJ: Rhebbing up mTOR: New
insights on TSC1 and TSC2, and the pathogenesis of tuberous
sclerosis. Cancer Biol Ther. 2:471–476. 2003. View Article : Google Scholar
|
|
38
|
Dabora SL, Jozwiak S, Franz DN, Roberts
PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, et
al: Mutational analysis in a cohort of 224 tuberous sclerosis
patients indicates increased severity of TSC2, compared with TSC1,
disease in multiple organs. Am J Hum Genet. 68:64–80. 2001.
View Article : Google Scholar
|
|
39
|
Kim WS: Mammalian target of rapamycin
inhibitors for treatment in tuberous sclerosis. Korean J Pediatr.
54:241–245. 2011. View Article : Google Scholar
|
|
40
|
Franz DN, Leonard J, Tudor C, Chuck G,
Care M, Sethuraman G, Dinopoulos A, Thomas G and Crone KR:
Rapamycin causes regression of astrocytomas in tuberous sclerosis
complex. Ann Neurol. 59:490–498. 2006. View Article : Google Scholar
|
|
41
|
Hofbauer GF, Marcollo-Pini A, Corsenca A,
Kistler AD, French LE, Wuthrich RP and Serra AL: The mTOR inhibitor
rapamycin significantly improves facial angiofibroma lesions in a
patient with tuberous sclerosis. Br J Dermatol. 159:473–475. 2008.
View Article : Google Scholar
|
|
42
|
Micozkadioglu H, Koc Z, Ozelsancak R and
Yildiz I: Rapamycin therapy for renal, brain, and skin lesions in a
tuberous sclerosis patient. Ren Fail. 32:1233–1236. 2010.
View Article : Google Scholar
|
|
43
|
Rapamune: Prescribing information, .
United States Food and Drug Administration. Wyeth Pharmaceuticals,
Inc.; May. 2015, https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021083s058,021110s075lbl.pdfMay
28–2016
|