Introduction
Alzheimer's disease (AD) is a chronic degenerative
brain disorder and the leading cause of dementia in the elderly
(1,2). AD is characterized by gradual neuronal
death, memory loss and impaired cognitive ability, and contributes
to 60–70% of dementia cases (2).
Current advances are able to temporarily improve symptoms, while no
effective treatment is available that can cure AD or reverse the
progressive course (3).
Understanding the risk factors for AD contributes to the
investigations on reducing the disease occurrence. The vast
majority of patients with AD have late-onset disease, and the
susceptible risk is determined by complex mechanisms (2,4).
Previous studies have revealed the roles of genetic,
environmental and epigenetic factors in AD risk (4–6).
Epigenetics refers to DNA modifications that alter gene expression
and phenotype without causing changes to the nucleotide sequences
(7). As the best-studied epigenetic
modification, DNA methylation is closely associated with several
key cellular processes, including cell differentiation, gene
regulation and genomic imprinting (8–10).
Several lines of evidence has indicated the effect of DNA
methylation on the pathogenesis of AD (11–13).
Furthermore, the genome-wide methylation profiling technique has
been applied for the characterization of methylation patterns
across the genome, and has been considered as a reliable tool in
identifying methylation differences associated with complex
diseases (13,14).
Numerous studies have focused on the identification
of specific brain regions that are particularly vulnerable
throughout the progression of AD (15–17).
Haroutunian et al (18)
performed an extensive study on the transcriptional vulnerability
involved in the progression of late-onset AD across 15 brain
regions, and observed that the superior temporal gyrus (STG) region
presented significant transcriptional abnormalities. Thus, the
present study focused on the STG region of patients with late-onset
AD. Recently, Watson et al (19) performed a genome-wide DNA methylation
profiling in the STG region of patients with late-onset AD, and
focused on the identification of differentially methylated regions.
In the present study, the publicly available DNA methylation data
of the study by Watson et al (19) were used to extract AD-associated
methylated genes and identify the underlying biological processes
impacted by hypomethylated and hypermethylated genes. Furthermore,
epigenetic biomarkers in the STG region associated with AD risk
were identified in the current study, and the findings may
contribute to the development of novel diagnostic and therapeutic
targets for late-onset AD.
Materials and methods
Genome-wide DNA methylation data
The genome-wide DNA methylation data of late-onset
AD were obtained from the Gene Expression Omnibus database (GEO;
https://www.ncbi.nlm.nih.gov/geo/),
under the GSE76105 accession number (19). The current study was performed using
the Illumina Infinium Human Methylation 450 array platform
(Illumina, Inc., San Diego, CA, USA). The data analysed from the
database included tissue samples obtained from the STG region of 34
patients with confirmed late-onset AD and 34 controls without
dementia. Detailed information of these tissue samples is described
in a previous study (19).
Prior to analysis, the raw DNA methylation data were
subjected to a rigorous preprocessing procedure following the
method developed by Huynh et al (20). Probes were removed from the data
according to the following criteria: i) Single nucleotide
polymorphisms within 5 bp upstream of the targeted CpG; ii) minimum
allelic frequency of <0.05; iii) probes on X and Y chromosomes;
and iv) cross-hybridising probes. Subsequent to applying the
exclusion criteria, a total of 424,497 CpGs were selected and used
for subsequent analysis. These remaining CpGs were quantile
normalized using the lumi package in R software (21) and the beta-mixture quantile method
(22).
Differential methylation analysis
In the dataset, the methylation values for
individual CpGs in each sample were expressed as β-values. β-value
is a quantitative measure of methylation for each CpG site, ranging
between 0 (completely unmethylated site) to 1 (fully methylated
site). Initially, the β-values of individual CpG sites were
obtained in AD subjects and controls, respectively. Next, the mean
β-value difference of each CpG between two conditions was
calculated, and CpG sites with an absolute mean β-value difference
of >0.05 were included into the study. For each probe, β-values
in AD subjects were compared against the controls using the
Student's t-test. The CpGs with an absolute mean β-value
differences of >0.05, as well as P<0.05, were considered to
be differentially methylated sites.
Previous studies indicated that methylation
alterations rarely exist in genomic regions at the extreme ends of
methylation values (<0.2 or >0.8), but are preferentially
detected in genomic locations at intermediate methylation levels,
which is a range associated with active distal regulatory regions,
such as enhancers (20,23). Thus, in order to reduce the number of
non-variable CpGs and improve the statistical power of subsequent
analyses, the CpG sites with β-values of >0.8 and <0.2 in all
samples were removed from the current study. In addition, it is
reported that numerous differentially methylated CpGs with a
β-value difference of <0.2 may be difficult to reproduce by
alternative methodologies or in replication studies (24). Therefore, only CpGs that had a mean
β-value difference of ≥0.2 were retained in the current study.
Hierarchical clustering analysis
Hierarchical clustering is a method of cluster
analysis that seeks to build a hierarchy of clusters in data mining
and statistics. Theoretically, samples with similar methylation
profiles can be clustered together. To assess the classification
performance of differentially methylated CpGs, a hierarchical
clustering analysis was applied using the Euclidian distance and
average linkage criteria (25).
Ideally, the samples should be classified into two distinct
clusters, namely the AD subjects and controls. In order to assess
the classification efficiency, the accuracy was measured, which is
a fraction of correctly classified samples over all samples,
according to the following formula: Accuracy=(TP+TN)/(TP+FP+TN+FN).
TP represents the number of positive samples correctly predicted as
positive, TN is the number of negative samples correctly predicted
as negative, FP is the number of negative samples incorrectly
predicted as positive, and FN represents the number of positive
samples incorrectly predicted as negative.
Functional enrichment analysis
To further investigate the biological functions of
the genes associated with differentially methylated CpGs,
functional enrichment analysis was performed based on Gene Ontology
(GO; http://www.geneontology.org) and Kyoto
Encyclopedia of Genes and Genomes database (KEGG; http://www.genome.jp/kegg/pathway.html).
In addition, enrichment analysis was conducted using the online
software Database for Annotation, Visualization and Integrated
Discovery (DAVID 6.8; https://david.ncifcrf.gov). Fisher's test was utilized
to measure the significance of GO terms and biological pathways.
The P-values were adjusted using Benjamini-Hochberg false discovery
rate (FDR). Statistically significantly different terms were
determined under the criterion of P<0.01.
Results
Differential methylation analysis
The present study focused on comparing the DNA
methylation in the STG region of 34 late-onset AD subjects with
that in 34 controls without dementia. Following quality control
processing of the DNA methylation data, probes were removed under
the filtering criteria described earlier, and methylation data for
424,497 autosomal CpGs in each of the 68 individuals were used in
subsequent differential methylation analysis. Under the criteria of
absolute mean β-value differences between AD subjects and controls
of >0.05, as well as P<0.05, a total of 17,895 differentially
methylated CpG sites covering 8,678 genes were initially
identified. Among these differentially methylated CpGs, a total of
11,822 CpGs were hypermethylated and 6,073 CpGs were
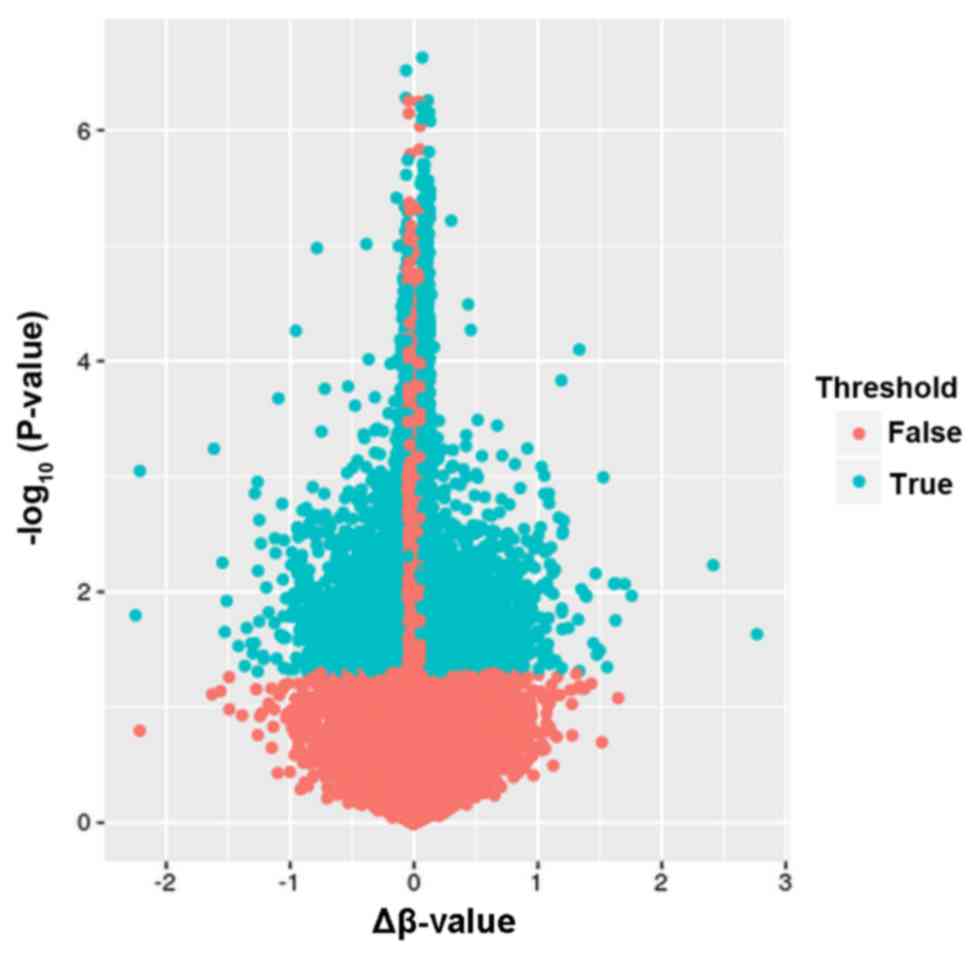
hypomethylated. A volcano plot exhibiting the distribution of
differentially methylated CpGs is shown in Fig. 1.
Since methylation changes were preferentially
detected in genomic regions at intermediate methylation levels,
further selection was performed to reduce non-variable CpGs and
improve the statistical power. By removing CpGs at the extreme ends
of methylation values (β-values of >0.8 and <0.2), 2,225
differentially methylated CpGs covering 2,001 genes were
identified. Furthermore, differentially methylated CpGs with a
β-value difference of <0.2 were considered to be difficult to
reproduce and excluded. Thus, only differentially methylated CpGs
with a mean β-value difference of ≥0.2 were extracted, and 2,211
differentially methylated CpGs (covering 1,991 genes) were finally
identified.
Hierarchical clustering analysis
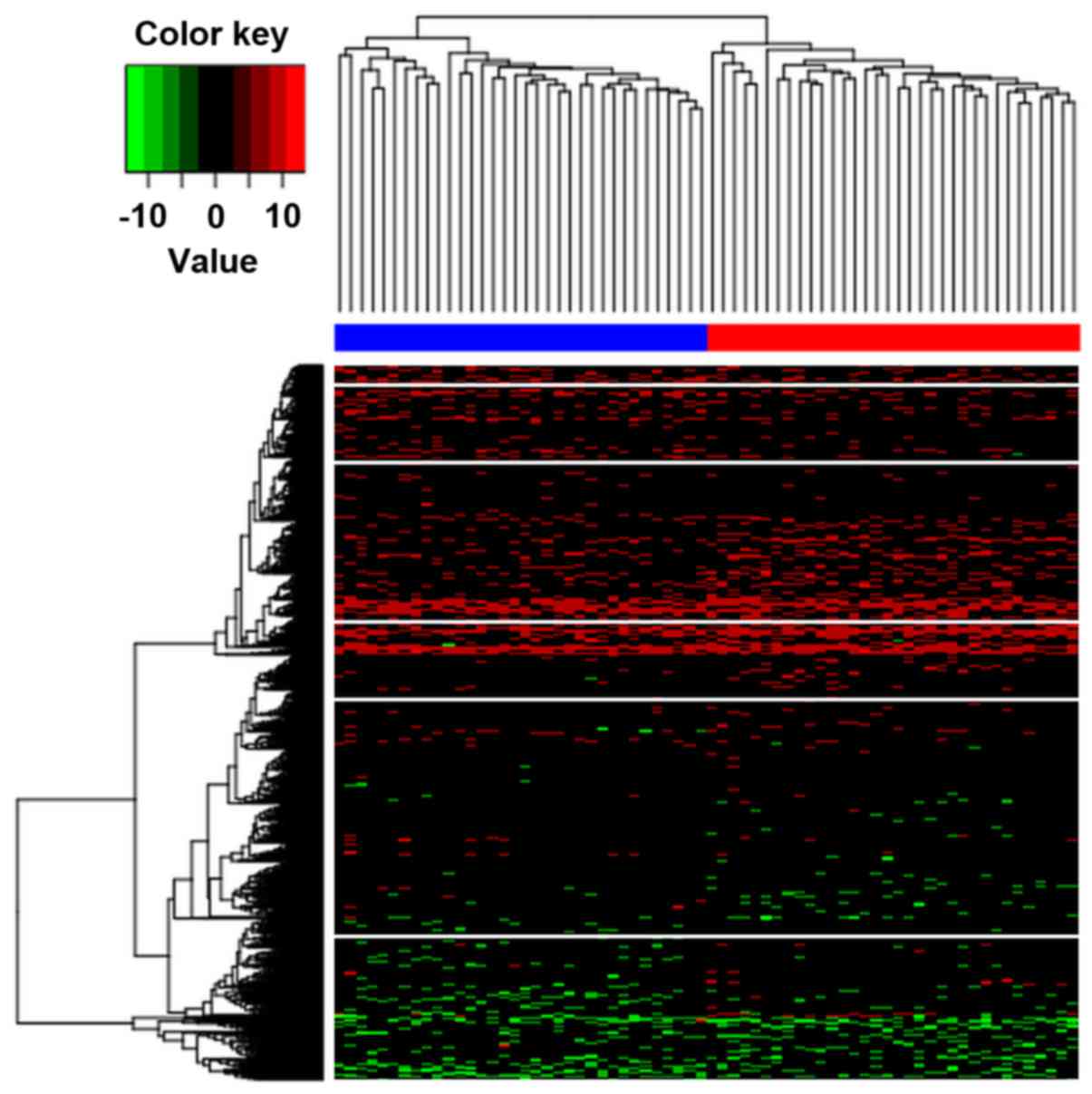
Hierarchical clustering was implemented to determine
whether the identified differentially methylated CpGs can be
applied to distinguish AD subjects from controls. The results
illustrated that these two conditions in the AD and control groups
exhibited distinctive DNA methylation patterns (Fig. 2). In addition, it was observed that
the samples were classified into two distinct clusters by these
differentially methylated CpGs with an accuracy of 1.
Functional enrichment analysis
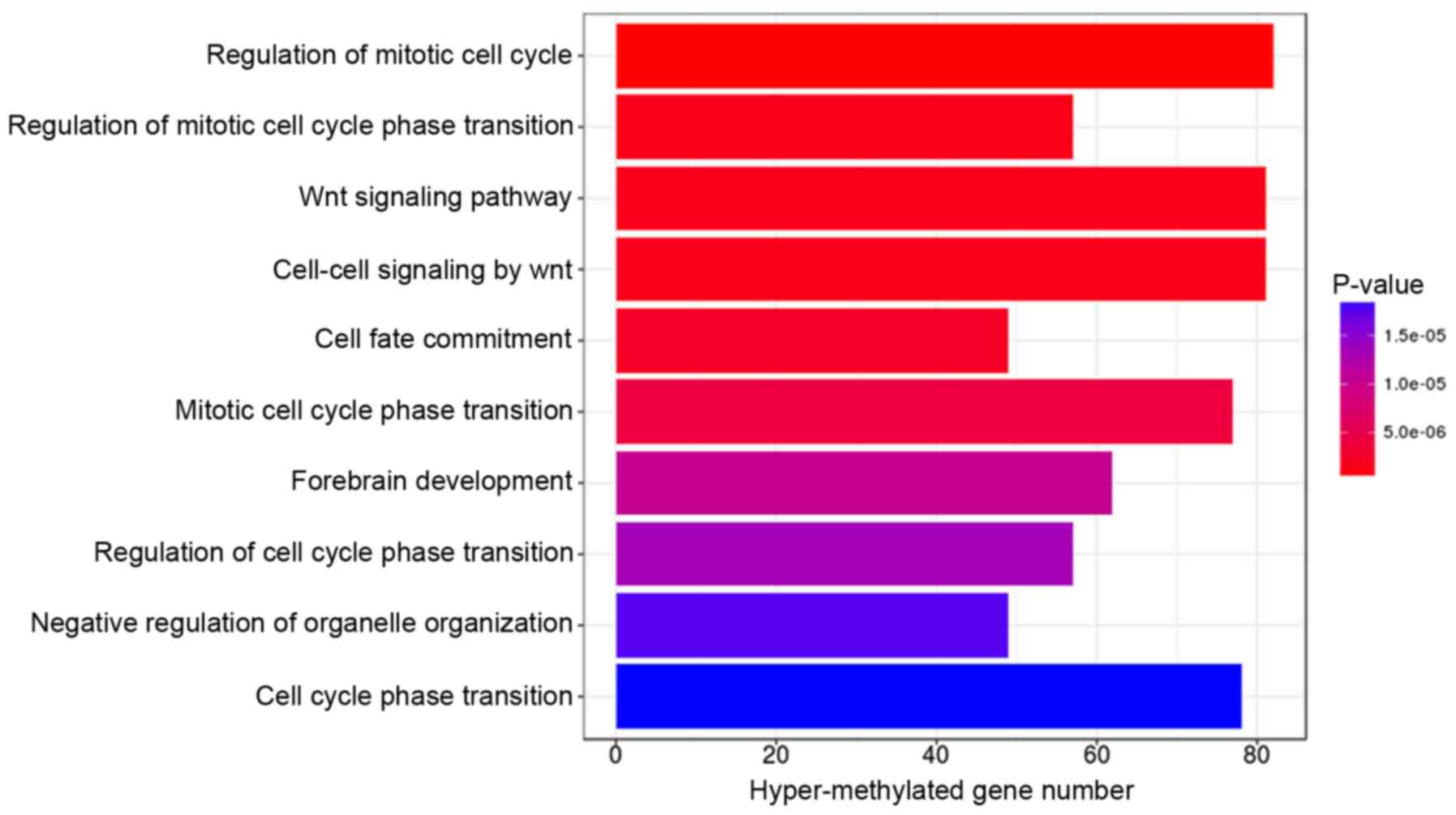
GO terms and KEGG pathway enrichment analysis for
genes associated with the 2,211 differentially methylated CpGs were
performed using the DAVID online tool. Under the criterion of
P<0.01, hypermethylated genes were significantly enriched in 10
GO terms, with the top three (ranked according to their P-value)
involved in the regulation of the mitotic cell cycle, regulation of
the mitotic cell cycle phase transition, and the Wnt signaling
pathway (Fig. 3). The hypomethylated
genes were significantly enriched in 5 GO terms, with the top one
associated with transcription factor binding (Fig. 4). However, KEGG pathway analysis
demonstrated that no statistically significant pathways were
enriched by both the hypermethylated and hypomethylated genes under
an FDR-adjusted P-value of <0.01.
Discussion
Recently, epigenetic modification in diseases has
attracted increasing attention from researchers. Late-onset AD
results from the interactions of multiple genetic and environmental
factors, along with the disruption of epigenetic mechanisms
controlling gene expression (26).
Abnormal DNA methylation patterns have been observed to be
associated with the pathogenesis of AD. In the present study, the
epigenome-wide DNA methylation patterns in the STG of patients with
late-onset AD were assessed, and differentially methylated genes
and biological functions were identified under the AD condition,
which may contribute to the development of novel targets for
late-onset AD diagnosis and therapy.
The present study revealed significant alterations
to the DNA methylation profiles in AD, with 2,211 CpGs displaying
significantly differential methylation levels. The hierarchical
clustering results illustrated that AD subjects exhibited
distinctive DNA methylation patterns compared with the controls,
which was consistent with the observations of a previous study
(19). Initially, the differential
methylation identification demonstrated a strong bias to
hypermethylated alterations in late-onset AD (11,822
hypermethylated and 6,073 hypomethylated CpGs). Given that the
greatest risk factor for late-onset AD is older age (27), it may be hypothesized that CpG
methylation increased with age. Previous studies also indicated
that CpG methylation in AD was significantly associated with age
(13,28).
To further examine the functions of genes associated
with differentially methylated CpGs, GO and KEGG analysis were
performed in the present study. The results revealed that genes
associated with hypermethylated CpGs were enriched in 10 GO terms,
6 of which were cell cycle-associated biological process. The cell
cycle involves a series of events that lead to cell division and
DNA replication to produce two daughter cells. Cell cycle
progression ensures a correct inheritance of epigenetic
modifications. A previous study has indicated that epigenetic
modifications arising within the cells of an individual are
transmitted through multiple cell cycles, which is crucial in
maintaining a given chromatin state (29). In the current study, it was
identified that genes associated with hypermethylated CpGs
participated in two Wnt-associated biological processes, including
the Wnt signaling pathway and cell-cell signaling by Wnt. The Wnt
signaling pathway is a vital signal transduction pathway involved
in various biological activities. Increasing evidence suggested the
protective effects of the Wnt pathway on synaptic modulation and
cognitive processes in neurodegenerative diseases, implying the
potential role of aberrant Wnt signaling in cognitive impairment
(30,31). Riise et al (32) also confirmed an aberrant Wnt
signaling pathway in medial temporal lobe structures of
neurodegenerative AD samples. Recent studies have further affirmed
the potential role of the Wnt signaling pathway as a therapeutic
target for the treatment of AD (33,34).
In the present study, hypomethylation was detected
in genes involved in transcription factor binding and core promoter
binding. Numerous studies have revealed the effects of DNA
methylation on the transcription factor binding (35–37).
Transcription factors serve important roles in the epigenetic
regulation of gene transcription, while DNA methylation prohibits
the recruitment of transcription factors, resulting in
transcription suppression. A protein microarray-based approach
observed that methylation-dependent transcription factor binding
was a widespread phenomenon in biological processes (38). Methylated CpG sites are typically
considered to reduce gene expression by preventing transcription
factors from binding to promoter regions (38). Promoter site-specific CpG methylation
is associated with various biological processes. Cyclical
methylation alterations in promoter CpGs represent a critical event
in achieving transcription (39).
Pieper et al (40) also
identified a tendency for hypomethylation in the tumor necrosis
factor α (TNF-α) core promoter region in neurodegenerative
disorders, resulting in reduced binding of the transcription
factors and suppressed TNF-α promoter activity.
The focus of the present study was the STG region of
patients with late-onset AD. Increasing studies have already
identified unique epigenetic signatures involved in the progression
of AD in various brain regions. For instance, human AD
case-cognitively normal control studies observed global
hypomethylation in the entorhinal cortex (41) and in the temporal neocortex neuronal
nuclei (42). Additionally, Bakulski
et al (13) demonstrated
widespread discordant DNA methylation in AD in the human frontal
cortex. Chouliaras et al (43) identified a consistent decrease in
global DNA methylation and hydroxymethylation in the hippocampus of
AD subjects. Furthermore, the present study demonstrated a strong
bias to hypermethylated alterations in the STG region in AD
patients, implying different methylation changes across various
brain regions.
In conclusion, the present study demonstrated
significant and distinctive DNA methylation patterns in the STG
region of patients with late-onset AD. Hypermethylation was mainly
detected for genes regulating the cell cycle progression, while
hypomethylation was identified in genes involved in transcription
factor binding. These results suggested AD-associated epigenetic
marks, potentially contributing to the understanding of underlying
mechanisms involved in the epigenetic regulation of AD and the
development of novel therapeutic targets for AD.
References
|
1
|
Wilson RS, Segawa E, Boyle PA, Anagnos SE,
Hizel LP and Bennett DA: The natural history of cognitive decline
in Alzheimer's disease. Psychol Aging. 27:1008–1017. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Association As: 2017 Alzheimer's disease
facts and figures. Alzheimers Dement. 13:325–37310. 2017.
View Article : Google Scholar
|
|
3
|
Selkoe DJ: Translating cell biology into
therapeutic advances in Alzheimer's disease. Nature. 399(6738
Suppl): A23–A31. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Migliore L and Coppedè F: Genetics,
environmental factors and the emerging role of epigenetics in
neurodegenerative diseases. Mutat Res. 667:82–97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iatrou A, Kenis G, Rutten BP, Lunnon K and
van den Hove DL: Epigenetic dysregulation of brainstem nuclei in
the pathogenesis of Alzheimer's disease: Looking in the correct
place at the right time? Cell Mol Life Sci. 74:509–523. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gatz M, Reynolds CA, Fratiglioni L,
Johansson B, Mortimer JA, Berg S, Fiske A and Pedersen NL: Role of
genes and environments for explaining Alzheimer disease. Arch Gen
Psychiatry. 63:168–174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bird A: Perceptions of epigenetics.
Nature. 447:396–398. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ziller MJ, Gu H, Müller F, Donaghey J,
Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein
BE, et al: Charting a dynamic DNA methylation landscape of the
human genome. Nature. 500:477–481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith ZD and Meissner A: DNA methylation:
Roles in mammalian development. Nat Rev Genet. 14:204–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Yu JT, Tan MS, Jiang T and Tan L:
Epigenetic mechanisms in Alzheimer's disease: Implications for
pathogenesis and therapy. Ageing Res Rev. 12:1024–1041. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bollati V, Galimberti D, Pergoli L, Valle
E Dalla, Barretta F, Cortini F, Scarpini E, Bertazzi PA and
Baccarelli A: DNA methylation in repetitive elements and Alzheimer
disease. Brain Behav Immun. 25:1078–1083. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bakulski KM, Dolinoy DC, Sartor MA,
Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H and Rozek LS:
Genome-wide DNA methylation differences between late-onset
Alzheimer's disease and cognitively normal controls in human
frontal cortex. J Alzheimers Dis. 29:571–588. 2012.PubMed/NCBI
|
|
14
|
Hannum G, Guinney J, Zhao L, Zhang L,
Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al:
Genome-wide methylation profiles reveal quantitative views of human
aging rates. Mol Cell. 49:359–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bussière T, Gold G, Kövari E,
Giannakopoulos P, Bouras C, Perl DP, Morrison JH and Hof PR:
Stereologic analysis of neurofibrillary tangle formation in
prefrontal cortex area 9 in aging and Alzheimer's disease.
Neuroscience. 117:577–592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
von Gunten A, Kövari E, Rivara CB, Bouras
C, Hof PR and Giannakopoulos P: Stereologic analysis of hippocampal
Alzheimer's disease pathology in the oldest-old: Evidence for
sparing of the entorhinal cortex and CA1 field. Exp Neurol.
193:198–206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ginsberg SD, Hemby SE, Lee VM, Eberwine JH
and Trojanowski JQ: Expression profile of transcripts in
Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol.
48:77–87. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haroutunian V, Katsel P and Schmeidler J:
Transcriptional vulnerability of brain regions in Alzheimer's
disease and dementia. Neurobiol Aging. 30:561–573. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watson CT, Roussos P, Garg P, Ho DJ, Azam
N, Katsel PL, Haroutunian V and Sharp AJ: Genome-wide DNA
methylation profiling in the superior temporal gyrus reveals
epigenetic signatures associated with Alzheimer's disease. Genome
Med. 8:52016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huynh JL, Garg P, Thin TH, Yoo S, Dutta R,
Trapp BD, Haroutunian V, Zhu J, Donovan MJ, Sharp AJ and Casaccia
P: Epigenome-wide differences in pathology-free regions of multiple
sclerosis-affected brains. Nat Neurosci. 17:121–130. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du P, Kibbe WA and Lin SM: lumi: A
pipeline for processing illumina microarray. Bioinformatics.
24:1547–1548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stadler MB, Murr R, Burger L, Ivanek R,
Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ,
Gaidatzis D, et al: DNA-binding factors shape the mouse methylome
at distal regulatory regions. Nature. 480:490–495. 2011.PubMed/NCBI
|
|
24
|
Finer S, Mathews C, Lowe R, Smart M,
Hillman S, Foo L, Sinha A, Williams D, Rakyan VK and Hitman GA:
Maternal gestational diabetes is associated with genome-wide DNA
methylation variation in placenta and cord blood of exposed
offspring. Hum Mol Genet. 24:3021–3029. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sturn A, Quackenbush J and Trajanoski Z:
Genesis: Cluster analysis of microarray data. Bioinformatics.
18:207–218. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Millan MJ: The epigenetic dimension of
Alzheimer's disease: Causal, consequence, or curiosity? Dialogues
Clin Neurosci. 16:373–393. 2014.PubMed/NCBI
|
|
27
|
Hebert LE, Bienias JL, Aggarwal NT, Wilson
RS, Bennett DA, Shah RC and Evans DA: Change in risk of Alzheimer
disease over time. Neurology. 75:786–791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hernandez DG, Nalls MA, Gibbs JR, Arepalli
S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor
BJ and Singleton AB: Distinct DNA methylation changes highly
correlated with chronological age in the human brain. Hum Mol
Genet. 20:1164–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Probst AV, Dunleavy E and Almouzni G:
Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell
Biol. 10:192–206. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ortiz-Matamoros A, Salcedo-Tello P,
Avila-Muñoz E, Zepeda A and Arias C: Role of Wnt signaling in the
control of adult hippocampal functioning in health and disease:
Therapeutic implications. Curr Neuropharmacol. 11:465–476. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arrázola MS, Silva-Alvarez C and Inestrosa
NC: How the Wnt signaling pathway protects from neurodegeneration:
The mitochondrial scenario. Front Cell Neurosci. 9:1662015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Riise J, Plath N, Pakkenberg B and
Parachikova A: Aberrant Wnt signaling pathway in medial temporal
lobe structures of Alzheimer's disease. J Neural Transm (Vienna).
122:1303–1318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sinha A, Tamboli RS, Seth B, Kanhed AM,
Tiwari SK, Agarwal S, Nair S, Giridhar R, Chaturvedi RK and Yadav
MR: Neuroprotective role of novel triazine derivatives by
activating Wnt/β catenin signaling pathway in rodent models of
alzheimer's disease. Mol Neurobiol. 52:638–652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
del Pino J, Ramos E, Aguilera OM,
Marco-Contelles J and Romero A: Wnt signaling pathway, a potential
target for Alzheimer's disease treatment, is activated by a novel
multitarget compound ASS234. CNS Neurosci Ther. 20:568–570. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tenayuca J, Cousins K, Yang S and Zhang L:
Computational modeling approach in probing the effects of cytosine
methylation on the transcription factor binding to DNA. Curr Top
Med Chem. 17:1778–1787. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Banovich NE, Lan X, McVicker G, van de
Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK and Gilad Y:
Methylation QTLs are associated with coordinated changes in
transcription factor binding, histone modifications and gene
expression levels. PLoS Genet. 10:e10046632014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Medvedeva YA, Khamis AM, Kulakovskiy IV,
Ba-Alawi W, Bhuyan MS, Kawaji H, Lassmann T, Harbers M, Forrest AR
and Bajic VB: FANTOM consortium: Effects of cytosine methylation on
transcription factor binding sites. BMC Genomics. 15:1192014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen
HN, Shin J, Cox E, Rho HS, Woodard C, et al: DNA methylation
presents distinct binding sites for human transcription factors.
Elife. 2:e007262013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Métivier R, Gallais R, Tiffoche C, Le
Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F
and Reid G: Cyclical DNA methylation of a transcriptionally active
promoter. Nature. 452:45–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pieper HC, Evert BO, Kaut O, Riederer PF,
Waha A and Wüllner U: Different methylation of the TNF-alpha
promoter in cortex and substantia nigra: Implications for selective
neuronal vulnerability. Neurobiol Dis. 32:521–527. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mastroeni D, Grover A, Delvaux E,
Whiteside C, Coleman PD and Rogers J: Epigenetic changes in
Alzheimer's disease: Decrements in DNA methylation. Neurobiol
Aging. 31:2025–2037. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mastroeni D, McKee A, Grover A, Rogers J
and Coleman PD: Epigenetic differences in cortical neurons from a
pair of monozygotic twins discordant for Alzheimer's disease. PLoS
One. 4:e66172009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chouliaras L, Mastroeni D, Delvaux E,
Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP and
van den Hove DL: Consistent decrease in global DNA methylation and
hydroxymethylation in the hippocampus of Alzheimer's disease
patients. Neurobiol Aging. 34:2091–2099. 2013. View Article : Google Scholar : PubMed/NCBI
|