Introduction
The mitochondrial arginyl-tRNA synthetase 2
(RARS2) encodes the mitochondrial arginyl-transfer RNA
synthetase (1), which is essential
for the translation of all proteins synthesized in the
mitochondria. In 2007, mutations in RARS2 were first
identified in pontocerebellar hypoplasia type 6 (PCH6; MIM no.
611523) with autosomal recessive inheritance (2). To the best of our knowledge, 23 cases
with RARS2 mutations have been reported in the literature
thus far (1–11). PCH6 is a rare mitochondrial disease.
The age of disease onset varies from birth (6,11,14,15)
to 9 months old (8). The common
initial symptoms of mitochondrial disease include hypotonia
(1,2,4,6–8),
epileptic seizures (5,6,8,9,11),
encephalopathy (1,2,4,6,10) and
feeding difficulties (2,3). Furthermore, other uncommon initial
presentations have been suggested, including hypoglycemia (6,11),
tachypnea (2,10) and cyanosis (7). Almost all patients developed
early-onset epileptic seizures (1–3,5,6,9,11) and
the age of onset may vary from 1 day (5) to 9 months old (8). Seizures have been typically described
as intractable to a variety of antiepileptic drugs. In addition,
feeding difficulties have been suggested a prominent problem in the
patients with PCH6 (1–6,8,11) and some cases have been diagnosed with
gastroesophageal reflux (3,6). The majority of patients have presented
with severe developmental delay (1–3,5,6,8,9,11) and microcephaly (1–6,8–11).
Hyperlactacidemia has also been determined a common clinical
feature in PCH6 (1–3,5–7,11).
Additionally, hearing loss and optic atrophy have been observed in
some cases (1,8). The most characteristic finding in
magnetic resonance imaging (MRI) is early onset or progressive
cerebral/pontocerebellar atrophy (1–3,5–8,11). Furthermore, one case has been
reported with subdural effusions in MRI (7).
In the present study, a PCH6 case with distinctive
MRI presentations in an infant was reported. In addition, the
characteristics of previous published cases of PCH6 in the
literature were reviewed. The present case enriched the phenotypic
and genotypic spectrum of the RARS2 mutations.
Case report
Case
The present case involves a boy at the age of 3
months that presented at the Peking University First Hospital
(Beijing, China) at September, 2014 with complaint of frequent
convulsions. The patient was a full-term infant (birth weight,
2,250 g) with non-consanguineous parents, and failed a neonatal
hearing screening. Frequent multifocal myoclonic seizures and focal
seizures were reported, which began at the age of 3 months. The
patient did not respond to antiepileptic drug treatment for the
seizures, including valproic acid, levetiracetam, topiramate,
phenobarbital and clonazepam. Ketogenic diet therapy was attempted;
however, this was stopped due to poor tolerance. The infant
gradually presented with hypotonia and feeding difficulty from 5
months of age, lethargy and progressive microcephaly from 7 months
of age.
Hyperlactacidemia was determined from blood samples
with the range of 3.2–9.5 mmol/l (normal range is 0~2.5 mmol/l),
which was observed since 4 months of age. An electroencephalogram
at the age of 6 months demonstrated diffuse slow waves mixed with
multifocal spikes and fast waves in the two hemispheres, with
persistent focal seizures observed during the monitoring. An
auditory brainstem response test at the age of 4 months revealed no
response to the stimulus with an intensity of 100 db.
Serial brain MRI scans were performed at 4, 5, 7 and
13 months after birth, respectively (Fig. 1). Various distinctive features were
observed in these scans, whereas pontocerebellar dysplasia or
atrophy was absent. A high diffusion-weighted imaging (DWI) signal
was detected in the frontal and parietal cerebral cortex at 4 and 5
months, which resolved after 7 months. The cortex edema was
possibly associated with the frequent multifocal seizures at that
time. From 5 months of age, a high signal was present on
T2-weighted image (T2WI) and DWI scans in the basal ganglia and
thalamus, followed by progressive atrophy. At 7 months, a symmetric
high signal was detected on DWI in bilateral cerebellar
hemispheres, which was suggestive of intracellular edema. In
addition, in the 7-month scan, atrophy of the cerebral cortex
became prominent with concomitant subdural effusions, while high
DWI signals were observed in the genu and splenium of the corpus
callosum, concomitant with progressive white matter depletion.
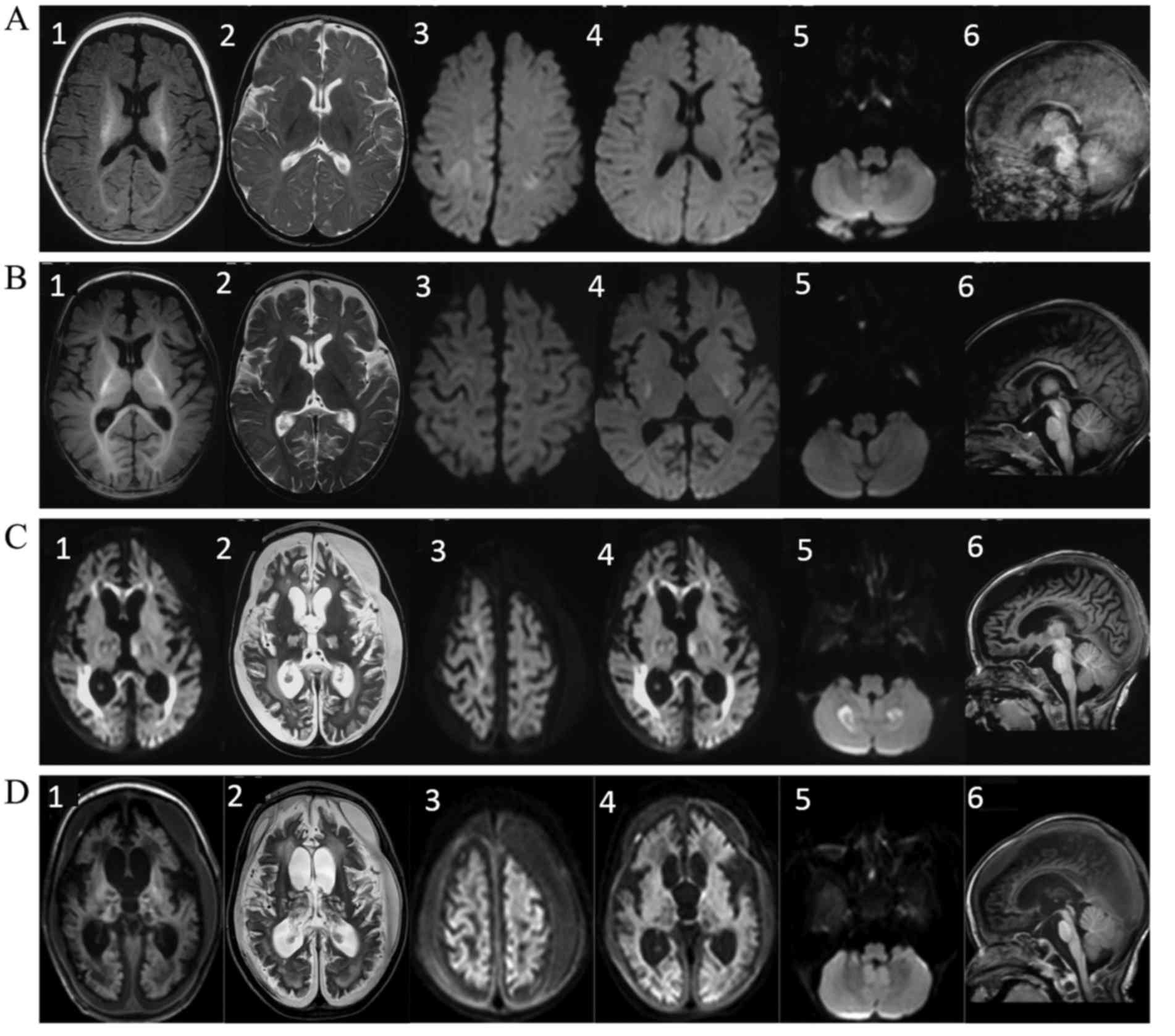 | Figure 1.Serial brain MRI scans performed in
the present case. MRI scanning was performed at (A) 4 months, (B) 5
months, (C) 7 months and (D) 13 months after birth. The images show
the axial T1WI (labeled as 1), axial T2WI (labeled as 2), axial DWI
in the frontal and parietal cerebral cortex (labeled as 3), axial
DWI in the basal ganglia and thalamus (labeled as 4), axial DWI in
the cerebellar hemispheres (labeled as 5) and sagittal T1WI scans
(labeled as 6). The following distinctive features were observed in
the MRI scans: i) Absence of pontocerebellar dysplasia or atrophy
was absent (A6, B6, C6 and D6); ii) symmetric high signal was
observed on the DWI scan in the bilateral cerebellar hemispheres at
7 months of age (C5), which was suggestive of intracellular edema;
iii) a high DWI signal was observed in the frontal and parietal
cerebral cortex at 4 months (A3), which resolved after 7 months of
age (C3); iv) atrophy of the cerebral cortex became prominent at 7
months of age, with concomitant subdural effusions (C1, C2, D1 and
D2) and v) high T2WI and DWI signals were observed in the basal
ganglia and thalamus at 5 months (B4), followed by progressive
atrophy (C4 and D4). vi) A high DWI signal was detected in the genu
and splenium of the corpus callosum at 7 months (C4), concomitant
with progressive white matter depletion (C1-3 and D1-3). MRI,
magnetic resonance imaging; WI, weighted image; DWI,
diffusion-weighted imaging. |
On the basis of the refractory seizures,
encephalopathy, hyperlactacidemia and special MRI features, the
patient was suspected with the diagnosis of early epileptic
encephalopathy and mitochondrial disease since his 9 months. He
received gene analysis at the age of 9 months, which included
analysis of common mitochondrial nuclear genes.
Genomic DNA was isolated from peripheral leukocytes,
fragmented into 150–200 bp length and sonicated. The DNA fragments
were then processed by end-repairing, A-tailing and adaptor
ligation, a 4-cycle pre-capture PCR amplification and enriched by a
custom-designed panel capturing the coding exons of 256 genes
associated with mitochondrial nuclear genes, including
RARS2. Paired-end sequencing (150 bp) was performed on
Illumina HiSeq X-ten platform to provide a mean sequence coverage
of >x100, with >95% of the target bases having at least ×20
coverage. Raw data was processed by the Illumina pipeline (version
1.3.4) for image analysis, error estimation, base calling and
generating the primary sequence data. For the quality control, the
Cutadapt (https://pypi.python.org/pypi/cutadapt) and FastQC
(www.bioinformatics.babraham.ac.uk/projects/fastqc/)
were used to remove 3′-/5′-adapters and low-quality reads,
respectively. The clean reads were mapped to the human reference
genome (UCSC hg19) with the use of the BWA (version 0.7.10,
http://bio-bwa.sourceforge.net)
(12), duplicate sequence reads were
removed by Picard (version 1.85; http://picard.sourceforge.net), and GATK (version 3.1,
https://software.broadinstitute.org/gatk/) (13) was used to detect variants. Variants
were annotated using ANNOVAR software (http://www.openbioinformatics.org/annovar/) as
described previously (14), which
included function implication (gene region, functional effect, mRNA
GenBank accession number, amino acid change and cytoband) and
allele frequency in several databases: dbSNP138, 1000 Genomes
(Phase 3-Variant Frequencies 5b), ExAc (exac.broadinstitute.org/) as described previously
(15). Damaging missense mutations
were predicted by SIFT (sift.bii.a-star.edu.sg/), PolyPhen-2 (genetics.bwh.harvard.edu/pph2/), and
MutationTaster (www.mutationtaster.org/). Interpretation of the
variants was conducted according to the American College of Medical
Genetics and Genomics recommended standards (16), and all the variants were categorized
into ‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’,
‘likely benign’ and ‘benign’. Sanger sequencing was performed to
validate the putative pathogenic variants and segregation analyses
where possible.
Novel compound heterozygous mutations in
RARS2, namely c.1718C>T(p.Thr573Ile) and
c.991A>G(p.Ile331Val), were identified in the patient. SIFT
(sift.bii.a-star.edu.sg/) and Mutation
Taster (www.mutationtaster.org/) predicted that these
mutations would affect the protein function (Table I). At the age of 10 months, the
patient was diagnosed with PCH6 due to RARS2 mutations.
 | Table I.RARS2 mutations in patients with PCH6
in the present and previous studies. |
Table I.
RARS2 mutations in patients with PCH6
in the present and previous studies.
| Patient | Mutation
location | Nucleotide
change | Amino acid
change | (Refs.) |
|---|
| 1 | Exon14 | c.1211T>A | p.Met404Lys | (1) |
|
| Exon7 | c.472_474delAAA | p.Lys158del |
|
| 2 | Intron2 | c.110+5A>G | – | (2) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 3 | Intron2 | c.110+5A>G | – | (2) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 4 | Intron2 | c.110+5A>G | – | (2) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 5 | Exon12 | c.1024 A>G | p.Met342Val | (3) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 6 | Intron2 | c.110+5A>G | – | (4) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 7 | Exon10 | 773G>A | p.Arg258His | (5) |
|
| Intron19 | c.1651–2A>G | – |
|
| 8 | Exon10 | 773G>A | p.Arg258His | (5) |
|
| Intron19 | c.1651-2A>G | – |
|
| 9 | Exon1 | c.25A>G | p.Ile9Val | (6) |
|
| Intron18 | c.1586+3A>T | – |
|
| 10 | Exon1 | c.25A>G | p.Ile9Val | (6) |
|
| Intron18 | c.1586+3A>T | – |
|
| 11 | Exon9 | c.734G>A | p.Arg245Gln | (6) |
|
| Exon16 | c.1406G>A | p.Arg469His |
|
| 12 | Exon9 | c.734G>A | p.Arg245Gln | (6) |
|
| Exon16 | c.1406G>A | p.Arg469His |
|
| 13 | Exon9 | c.721T>A | p.Trp241Arg | (6) |
|
| Exon1 | c.35A>G | p.Gln12Arg |
|
| 14 | Exon1 | c.1A>G; | p.Met1Val | (7) |
|
| Intron8 |
c.613-3927C>T | – |
|
| 15 | Exon1 | c.1A>G | p.Met1Val | (7) |
|
| Intron8 |
c.613-3927C>T | – |
|
| 16 | 5′-UTR | c.-2A>G | – | (8) |
|
| 5′-UTR | c.-2A>G | – |
|
| 17 | 5′-UTR | c.-2A>G | – | (8) |
|
| 5′-UTR | c.-2A>G | – |
|
| 18 | Intron2 | c.110+5A>G | – | (9) |
|
| Intron10 | c.878+5G>T | – |
|
| 19 | Intron2 | c.110+5A>G | – | (9) |
|
| Intron10 | c.878+5G>T | – |
|
| 20 | Exon5 | c.392T > G | p.Phe131Cys | (10) |
|
| Exon5 | c.392T > G | p.Phe131Cys |
|
| 21 | Exon5 | c.392T > G | p.Phe131Cys | (10) |
|
| Exon5 | c.392T > G | p.Phe131Cys |
|
| 22 | Exon7 |
c.472_474delAAA | p.Lys158del | (11) |
|
| Exon10 | c.848T>A | p.Leu283Glu |
|
| 23 | Exon7 |
c.472_474delAAA | p.Lys158del | (11) |
|
| Exon10 | c.848T>A | p.Leu283Glu |
|
| Present case | Exon20 | c.1718C>T | p.Thr573Ile |
|
|
| Exon12 | c.991A>G | p.Ile331Val |
|
The last follow-up was performed when the patient
was 3 years old. The patient was receiving nasogastric feeding,
while severe developmental delay, rigidity and loss of response to
external stimuli were reported. The seizures were partially managed
by combination therapy with topiramate (4 mg/kg/day) and
levetiracetam (42 mg/kg/day).
Written informed consent was obtained from the
patient's family for the publication of the present case study.
Literature review
The databases PubMed (https://www.ncbi.nlm.nih.gov/pubmed), EMbase
(http://www.embase.com), CBM (http://www.sinomed.ac.cn/), CNKI (http://www.cnki.net/) and VIP (http://lib.cqvip.com/) were searched to collect
studies with the key words of ‘RARS2’ and ‘PCH6’, date from
database foundation to December, 2016. The publication language was
limited to English and Chinese. Studies reported of cases with
PCH6, which include detailed clinical data were included in the
present study.
Discussion
Since the identification of RARS2 mutations
in PCH6 in 2007 (2), a total of 23
cases with RARS2 mutations have been reported in 11 studies
(1–11), as identified according to a
literature review performed in the present study. The clinical
features of the reported cases are summarized in Table II.
| Table II.Clinical features of 23 previously
reported cases and the present case with pontocerebellar hypoplasia
type 6. |
Table II.
Clinical features of 23 previously
reported cases and the present case with pontocerebellar hypoplasia
type 6.
|
|
Patient |
|
|---|
|
|
|
|
|---|
|
Characteristics | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | Present case |
|---|
| Study
reference | (1) | (2) | (2) | (2) | (3) | (4) | (5) | (5) | (6) | (6) | (6) | (6) | (6) | (7) | (7) | (8) | (8) | (9) | (9) | (10) | (10) | (11) | (11) | – |
| Basic
characteristics |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sex | F | F | M | F | F | – | F | M | M | M | F | F | M | F | F | M | F | F | M | M | F | M | M | M |
| Small
for gestational age | N | N | – | N | N | – | N | N | Y | Y | N | Y | N | N | N | N | N | N | N | N | N | N | N | Y |
| Symptoms |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Age at
disease onset | 16 h | 1 d | 19 h | 3 w | 19 h | birth | 2-3 w | 1 d | 1 d | 1 d | birth | 11 d | 20 d | birth | birth | 9 m | 6 m | 2 m | 3 m | 1 d | 2 d | 40 h | 8 w | 3 m |
| Initial
symptoms | Hy, | Hy, | Ta, | Hy, | Ta, | Hy, | Se | Se | En | Hy, | Hyp | Se | Hy, Se | Hy, | Hy, | Hy, | Hy | Se | Se | En | Ta | Hyp | Se | Se |
|
| En | Po | Po | En, Po | Po | En |
|
|
| En |
|
|
| Cy | Cy | Se |
|
|
|
|
|
|
|
|
|
Epilepsy | Y | Y | – | Y | Y | – | Y | Y | Y | Y | Y | Y | Y | – | – | Y | N | Y | Y | Y | – | Y | Y | Y |
| Age of
seizures onset | 4 w | 2 m | – | 4 m | 36 h | – | 2-3 w | 1 d | 11 d | 4 m | 20 d | 11 d | 20 d | – | – | 9 m | N | 2 m | 3 m | 3 m | – | 5 w | 8 w | 3 m |
| Feeding
difficulties | Y | Y | Y | Y | Y | Y | N | N | Y | Y | Y | Y | Y | – | – | Y | N | – | – | – | N | Y | Y | Y |
|
Development delay | Y | Y | Y | Y | Y | – | Y | Y | Y | Y | Y | Y | Y | – | – | Y | Y | Y | Y | Y | – | Y | Y | Y |
| Hearing
loss | Y | – | – | – | – | – | N | N | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | Y |
| Optic
atrophy | Y | – | – | – | N | N | N | N | – | – | – | – | – | – | N | Y | Y | – | – | N | – | – | – | N |
| Signs |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hypotonia | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | – | Y | Y | N | N | Y | Y |
|
Microcephaly | Y | Y | – | N | Y | – | Y | Y | Y | Y | Y | Y | Y | – | – | Y | Y | Y | – | Y | N | Y | Y | Y |
| Biochemical
test |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hyperlactacidemia | Y | N | Y | N | Y | – | N | Y | Y | Y | Y | Y | Y | – | Y | – | – | – | – | Y | Y | Y | N | Y |
|
Elevated lactic level in
CSF | Y | – | – | Y | Y | Y | Y | – | Y | Y | Y | – | N | – | Y | – | – | – | – | N | – | N | N | N |
| Brain MRI
findings |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pontocerebellar atrophy | Y | – | Y | Y | Y | – | Y | Y | Y | Y | Y | Y | Y | – | Y | Y | Y | N | N | N | N | Y | Y | N |
|
Subdural effusions | N | N | – | N | N | – | Y | N | N | N | N | N | N | – | N | N | N | N | N | N | N | N | N | Y |
| Basal
ganglia involvement | N | N | – | N | N | – | N | N | N | N | N | N | N | – | N | N | N | N | N | N | N | N | N | Y |
In these previously reported PCH6 cases, the age of
disease onset was 1 day to 9 months. In 52.17% (12/23) of cases
symptoms were detected on the first day of life, while disease
onset was observed between 1 day and 1 month of age in 26.09%
(6/23) of cases, between 1 month and 3 months of age in 13.04%
(3/23) and after 6 months in 8.70% (2/23) of cases. The initial
presentations included symptoms of hypotonia (43.48%; 10/23),
epileptic seizures (34.78%; 8/23), encephalopathy (26.08%; 6/23)
and feeding difficulties (17.39%; 4/23). Other uncommon initial
presentations were hypoglycemia (8.70%; 2/23), tachypnea (13.04%;
3/23) and cyanosis (8.70%; 2/23). Patients often presented with
more than one initial symptom. In the present study, the initial
presentation of the patient was epileptic seizures, which began at
the age of 3 months.
With the exception of 4 cases of neonatal mortality
and 1 patient with an age of 2 months at the last follow-up, almost
all patients (94.44%; 17/18) developed early-onset epileptic
seizures. The age of onset of refractory epilepsy was 1 day to 9
months, and seizures were usually intractable to a variety of
antiepileptic drugs. Furthermore, 14 cases (60.87%; 14/23)
presented feeding difficulties subsequent to birth or in the
process of the follow-up. Of the included cases, 2 patients were
unavailable for evaluation due to mortality within 2 weeks after
birth, while information regarding feeding was unavailable in 3
cases. In total, 6 patients were diagnosed with gastroesophageal
reflux, whereas 4 patients presented no feeding problems until the
last follow-up conducted at 2 months, 1, 1.5 and 3 years of age,
respectively. The present case also presented severe feeding
difficulty and required nasogastric feeding subsequent to the
disease onset.
The majority (82.61%; 19/23) of included cases
presented severe developmental delay, with the exception of 3 cases
of mortality within 14 days (at 14 days of age) and 1 patient with
a last follow-up at the age of 2 months, who were unavailable for
assessment. In addition, with the exception of 4 cases of mortality
within 1 day to 7 weeks (aged 7 weeks old) and unavailable
information of head circumference in 1 case, microcephaly was
observed in 88.89% (16/18) of patients. Only 2 cases had a normal
head circumference until the age of 2 and 4 months, respectively,
which were the last follow-up times.
Mortality was reported in 6 (6 cases succumbed to
fatality at 1, 14 day, 7 weeks, 16 months, 6 days and 4 years) out
of 23 patients in the previous studies included in this literature
review. In 1 case, mortality was reported on the first day
following birth, between 6 days to 2 months in 3 patients, and at
16 months and 4 years in 2 other patients. In total, 73.91% (17/23)
were alive at the last follow-up, with an age of 2 months to 11
years.
Hyperlactacidemia was identified in 76.47% (13/17)
of cases among 17 patients with available data. Other clinical
features included hearing loss that was reported in 4.35% (1/23)
cases and optic atrophy identified in 13.04% (3/23). The clinical
features of the present patient were suggestive of PCH6, with
early-onset epilepsy, feeding difficulty, severe developmental
delay, microcephaly, hearing loss and hyperlactacidemia.
The characteristic finding in MRI was
pontocerebellar dysplasia or progressive cerebral/pontocerebellar
atrophy and in the majority of patients (80.00%, 16/20) were
observed with this feature. Compared with the available MRI data
from 20 reported cases with PCH6, certain distinct MRI features
were noted in the patient of the present study. No pontocerebellar
dysplasia or atrophy was observed prior to 1 year of age in the
present patient. In the previously reported 20 patients with
available MRI data, 4 cases did not present pontocerebellar
dysplasia/atrophy when tested at 10 days (10), 13 months (9), 16 months (9) and 40 months (10), respectively. By contrast,
pontocerebellar dysplasia/atrophy were present in 16 cases, with
25% (4/16) in the neonatal stage, 43.75% (7/16) between 1 and 6
months, 18.75% (3/16) between 1 and 9 months, and 12.5% (2/16)
between 1–24 months (with a long interval between the two MRI
examinations). The last brain MRI scan of the present study patient
was performed at 13 months, therefore it does not exclude that
pontocerebellar atrophy may have occurred at a later time.
MRI examination in the present patient also
demonstrated high signals on T2WI and DWI scans in the cerebral
cortex, cerebellar hemispheres, basal ganglia and thalamus.
Notably, as PCH6 is an encephalopathy resulting from mitochondrial
functional defect, basal ganglia involvement has not been reported
in any previous patients with PCH6. Another MRI feature in the
current study involves atrophy of the cerebral cortex that became
prominent at 7 months after birth, with concomitant subdural
effusions. Subdural effusions have only been reported in 1 previous
case (5). Finally, a high DWI signal
was detected in the genu and splenium of the corpus callosum at 7
months, concomitant with progressive white matter depletion. In
previously reported cases, a high DWI signal was not described,
whereas white matter depletion was present in almost all cases.
A total of 18 mutations in RARS2 have been
identified in the previously reported cases, comprising of 11
missense mutations, one in-frame deletion, one substitution of
nucleotide in the 5′-untranslated region that resulted in decreased
mRNA expression level, and five mutations in introns. In the
present case, two novel missense mutations in RARS2 were
identified, c.1718C>T(p.Thr573Ile) and c.991A>G(p.Ile331Val)
(Table I).
In conclusion, in the current study, two novel
RARS2 mutations were identified in a patient with typical
clinical manifestations of PCH6. Distinctive MRI features were
identified, including pontocerebellar preservation after 1 year of
age, as well as a high DWI signal suggestive of intracellular edema
in the cerebellar hemispheres, basal ganglia, thalamus and corpus
callosum. Furthermore, progressive loss of cerebral white matter
and cortical volume were common features shared by all patients in
similar previously reported cases, which were also observed in the
present patient. Therefore, the findings reported in the present
case enriched the phenotypic and genotypic spectrum of the
RARS2 mutations in PCH6 patients.
References
|
1
|
Glamuzina E, Brown R, Hogarth K, Saunders
D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G and
Grunewald S: Further delineation of pontocerebellar hypoplasia type
due to mutations in the gene encoding mitochondrial arginyl-tRNA
synthetase, RARS. J Inherit Metab Dis. 35:459–467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Edvardson S, Shaag A, Kolesnikova O,
Gomori JM, Tarassov I, Einbinder T, Saada A and Elpeleg O:
Deleterious mutation in the mitochondrial arginyl-transfer RNA
synthetase gene is associated with pontocerebellar hypoplasia. Am J
Hum Genet. 81:857–862. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rankin J, Brown R, Dobyns WB, Harington J,
Patel J, Quinn M and Brown G: Pontocerebellar hypoplasia type: A
British case with PEHO-like features. Am J Med Genet A.
152A:2079–2084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Namavar Y, Barth PG, Kasher PR, van
Ruissen F, Brockmann K, Bernert G, Writzl K, Ventura K, Cheng EY,
Ferriero DM, et al: Clinical, neuroradiological and genetic
findings in pontocerebellar hypoplasia. Brain. 134:143–156. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kastrissianakis K, Anand G, Quaghebeur G,
Price S, Prabhakar P, Marinova J, Brown G and McShane T: Subdural
effusions and lack of early pontocerebellar hypoplasia in siblings
with RARS mutations. Arch Dis Child. 98:1004–1007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cassandrini D, Cilio MR, Bianchi M, Doimo
M, Balestri M, Tessa A, Rizza T, Sartori G, Meschini MC, Nesti C,
et al: Pontocerebellar hypoplasia type 6 caused by mutations in
RARS2: Definition of the clinical spectrum and molecular findings
in five patients. J Inherit Metab Dis. 36:43–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lax NZ, Alston CL, Schon K, Park SM,
Krishnakumar D, He L, Falkous G, Ogilvy-Stuart A, Lees C, King RH,
et al: Neuropathologic characterization of pontocerebellar
hypoplasia type 6 associated with cardiomyopathy and hydrops
fetalis and severe multisystem respiratory chain deficiency due to
novel RARS2 mutations. J Neuropathol Exp Neurol. 74:688–703. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Z, Schonberg R, Guidugli L, Johnson AK,
Arnovitz S, Yang S, Scafidi J, Summar ML, Vezina G, Das S, et al: A
novel mutation in the promoter of RARS2 causes pontocerebellar
hypoplasia in two siblings. J Hum Genet. 60:363–369. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nishri D, Goldberg-Stern H, Noyman I,
Blumkin L, Kivity S, Saitsu H, Nakashima M, Matsumoto N,
Leshinsky-Silver E, Lerman-Sagie T and Lev D: RARS2 mutations cause
early onset epileptic encephalopathy without ponto-cerebellar
ypoplasia. Eur J Paediatr Neurol. 20:412–417. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lühl S, Bode H, Schlötzer W, Bartsakoulia
M, Horvath R, Abicht A, Stenzel M, Kirschner J and Grünert SC:
Novel homozygous RARS mutation in two siblings without
pontocerebellar hypoplasia-further expansion of the phenotypic
spectrum. Orphanet J Rare Dis.
|
|
11
|
Ngoh A, Bras J, Guerreiro R, Meyer E,
McTague A, Dawson E, Mankad K, Gunny R, Clayton R, Mills PB, et al:
RARS2 mutations in a sibship with infantile spasms. Epilepsia.
57:e97–e102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinforma.
26:589–595. 2010. View Article : Google Scholar
|
|
13
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|














