Introduction
Atherosclerosis is a chronic inflammatory disease,
which is triggered by lipid retention in the arterial wall
(1). It is considered a benign
disease until plaque rupture occurs, leading to severe thrombus
formation (2). The characteristic
component of the atherosclerotic plaque is the differentiation of
monocytes to macrophages that accumulate lipoprotein-derived
cholesterol to form foam cells (3).
The accumulation of foam cells results in atherosclerotic plaque
growth and lipid storage (4). In
addition, the accumulation of excess low-density lipoprotein (LDL)
cholesterol, which is modified in the oxidant-rich environment,
triggers atherosclerosis (5).
Oxidized LDL (ox-LDL) induces the apoptosis of smooth muscle cells
and macrophages, which can be effectively cleared by macrophages
via efferocytosis in early plaques (6). Accompanied by the accumulation of
lipids in macrophages, efferocytosis becomes defective and plaque
vulnerability is promoted (7).
In order to prevent cholesterol accumulation, an
efficient cholesterol efflux mechanism exists in macrophages.
High-density lipoprotein (HDL) and its apolipoproteins participate
in the transfer of cholesterol from the peripheral tissues and
cells to the liver, through a process known as reverse cholesterol
transport (RCT) (8). According to
previous findings on RCT, the proteins ATP binding cassette
transporter G1 (ABCG1), ATP binding cassette transporter A1 (ABCA1)
and scavenger receptor B1 (SR-B1) serve key roles in suppressing
cholesterol accumulation in macrophages (9).
Toll-like receptors (TLRs) are a family of type I
transmembrane glycoproteins that containan extracellular domain
with leucine-rich repeat motifs and a Toll/interleukin-1 receptor
signaling domain (10). To date, 12
and 10 TLRs have been identified in mice and humans, respectively
(11). A previous study demonstrated
increased expression levels of TLR1, TLR2 and TLR4 in human
atherosclerosis and inflammation with downstream signaling of
inflammatory genes (12). TLR2, one
of 10 human TLRs, is able to recognize the lipoproteins that are
anchored to the bacterial membrane by covalent lipid chains, which
are attached to conserved N-terminal cysteines (13). Furthermore, TLR2 is considered as a
novel target for therapeutic intervention in atherosclerosis, since
it mediates responses to lipoproteins derived from multiple
pathogens (14).
Nuclear factor-κB (NF-κB) serves important roles in
stress response and inflammation (15,16). It
contains five family members in mammals, including c-Rel, RelA/p65,
RelB, p52 (NF-κB2) and p50 (NF-κB1) (17). Under non-stimulated conditions,
p50/p65 NF-κB is sequestered in the cytoplasm (18). However, when the cells are
stimulated, it undergoes phosphorylation in the proteasome, leading
to gene transcription (19).
In present study, it was hypothesized that TLR2 may
be involved in the cholesterol efflux in macrophages. Therefore,
the study initially examined the dose-dependent and time-dependent
effect of TLR2 on cholesterol efflux in THP-1 and RAW264.7
macrophage-derived foam cells. Subsequently, the dose-dependent
effect of TLR2 on the expression levels of genes linked to
cholesterol efflux, including ABCA1, ABCG1 and SR-B1, was explored.
Finally, the regulatory mechanisms of TLR2 on NF-κB in cholesterol
efflux were investigated. The present study provided novel insights
for evaluating the potential roles and mechanisms of the TLR2/NF-κB
pathway in the development of atherosclerosis.
Materials and methods
Cell culture
The human monocytic THP-1 (cat no. TIB-202) and
murine macrophage RAW264.7 cell lines (cat no. SC-6003) were
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cell lines were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100
µg/ml) and 0.1% nonessential amino acids in a 5% CO2
chamber at 37°C. Next, the cells were treated with 160 nmol/l
phorbol 12-myristate 13-acetate (Sigma-Aldrich; Merck, Darmstadt,
Germany) for 12 h. Subsequently, the medium was replaced by a
serum-free medium containing 50 µg/ml ox-LDL for 48 h in order to
obtain macrophage-derived foam cells prior to the following
experiments.
In order to examine the dose-dependent and
time-dependent effects of TLR2 on cholesterol efflux, the THP-1 and
RAW264.7 macrophage-derived foam cells were incubated with 0, 50,
100 and 200 ng/ml TLR2 for 24 h or with 100 ng/ml TLR2 for 0, 6,
12, 24 and 48 h.
Drug treatment
THP-1 and RAW264.7 macrophage-derived foam cells
were divided into 3 groups: Control group (cells were incubated in
RPMI-1640 medium for 24 h), LPS group [cells were cultured in 10
ng/ml lipopolysaccharides (LPS; Biosea Biotechnology Co., Ltd.,
Beijing, China) for 24 h] and LPS+PDTC group [cells were incubated
with 50 µM pyrrolidine dithiocarbamate (PDTC; Sigma-Aldrich; Merck
KGaA), and 10 ng/ml LPS for 24 h]. The cells were then
harvested.
Oil red O (ORO) staining
The lipid accumulation of macrophages following
treatment with ox-LDL was examined by ORO staining as described
previously (20). Briefly, cells
were washed twice with phosphate-buffered saline (PBS) and then
fixed with 10% formalin in PBS for 1 h. Next, cells were washed
with water for three times, dried and stained with ORO
(Sigma-Aldrich; Merck) for 15 min. Subsequently, 70% ethanol was
used to remove excess stain and the stained cells were washed with
water. Images of the cells were captured using a light microscope
(Olympus CX23; Olympus Corporation, Tokyo, Japan).
Small interfering RNA (siRNA)
transfection
THP-1 and RAW264.7 macrophage-derived foam cells
(2×106 cells/well) were seeded in 96-well plates. For
knockdown of TLR2, the cells were transfected with siRNA-TLR2
(GenePharma Co., Ltd., Shanghai, China) using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), and siRNA-scramble
(GenePharma Co., Ltd.) was used as the control. After transfection
for 48 h, the cells were harvested and used in further
experiments.
Cellular cholesterol efflux
experiments
For analysis of the cholesterol efflux, cells were
initially labeled with 0.2 µCi/ml [3H] cholesterol
(PerkinElmer, Inc., Waltham, MA, USA). Following cultivation for 72
h, cells were washed with PBS and incubated in RPMI-1640 medium
supplemented with 0.1% (w/v) bovine serum albumin in order to allow
for equilibration of the [3H] cholesterol in all the
cellular pools. Equilibrated [3H] cholesterol-labeled
cells were then washed with PBS and incubated in 2 ml serum-free
RPMI-1640 containing 0.1% bovine serum albumin (fraction V, fatty
acid free; EMD Millipore, Billerica, MA, USA). Next, 150 µl efflux
medium was obtained after a 6 h incubation and passed through a
0.45-µm filter to remove any floating cells. The monolayers were
subsequently washed twice in PBS, and cellular lipids were
extracted with isopropanol. A liquid scintillation counting method
was performed to measure the medium and cell-associated
[3H] cholesterol (21).
The percentage of efflux was calculated as follows: Cellular
cholesterol efflux=[total media counts/(total cellular counts +
total media counts)] ×100% (22).
High-performance liquid chromatography
(HPLC) assay
The lipid analysis was conducted by HPLC as
described previously (23). Briefly,
the protein concentrations in the cell solution were measured using
a BCA kit (Pierce; Thermo Fisher Scientific, Inc.), and 0.1 ml cell
lysate was used to measure the free cholesterol (FC) and total
cholesterol (TC) levels. Next, the samples were dissolved in 100 µl
isopropanol-acetonitrile (v/v, 20:80; Sinopharm Chemical Reagent
Co., Ltd., Shanghai, China), followed by an ultrasound water bath
for 5 min. Subsequently, the samples were used for HPLC analysis
(Agilent 1100; Agilent Technologies, Inc., Santa Clara, CA, USA).
The cholesterol was eluted with isopropanol-acetonitrile solution
(v/v, 20:80) at a speed of 1 ml/min and then detected in terms of
the absorbance at 210 nm. The levels of cholesteryl ester (CE) were
calculated according to the following formula: CE = TC – FC.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells by TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The quality
of total RNA was assessed by spectrophotometry (A260/280 ratio:
1.8–2.0). cDNA was reverse transcribed from 100 ng RNA using a
First-Strand RT-PCR kit (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. qPCR was then
performed using a SYBR Green qRT-PCR kit (Thermo Fisher Scientific,
Inc.) on an Applied Biosystems 7900HT Real-Time PCR system (Thermo
Fisher Scientific, Inc.). The primers used for qPCR were as
follows: ABCA1 forward, 5′-GATTGGCTTCAGGATGTCCATGTTGGAA-3′ and
reverse, 5′-GTATTTTTGCAAGGCTACCAGTTACATTTGACAA-3′; ABCG1 forward,
5′-CAGTGACAGCCATCCCGGTGCT-3′ and reverse,
5′-CGATGAAGTCCAGGTACAGCTTGGC-3′; SR-B1 forward,
5′-GCTGTCTGCTGGGAGAGTC-3′ and reverse, 5′-TTCTGCCCGTGCCTGGAGTC-3′;
GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse,
5′-TGGTGAAGACGCCAGTGGA-3′. The PCR conditions for quantification
were as follows: 10 min at 95°C, 40 cycles of 10 sec at 95°C, 20
sec at 58°C, and 30 sec at 72°C. qPCR was performed using 2 µl
diluted cDNA products, 12.5 µl SYBR Green (Thermo Fisher
Scientific, Inc.), 0.5 µl forward and reverse primers (10 µM) and
9.5 µl nuclease-free water in a final volume of 25 µl. GAPDH was
used as an internal control and the relative expression of mRNA was
calculated using the 2−ΔΔCq method (24).
Western blot analysis
The proteins were isolated from the cells using
radioimmunoprecipitation assay lysis and extraction buffer
(containing 150 mM NaCl, 25 mM Tris-HCl, pH 7.6, 1% sodium
deoxycholate, 1% NP-40, protease inhibitor and 0.1% SDS). The BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.) was used to
calculate the total protein concentration. The total proteins (50
µg/lane) were separated by 10% SDS-polyacrylamide gel
electrophoresis and then transferred to polyvinylidene difluoride
membranes (EMD Millipore). The membrane was blocked with 5% non-fat
dry milk in PBS with 5% Tween-20. Following 3 washes in PBS with 5%
Tween-20, the membranes were incubated with primary antibodies
against TLR2 (1:1,000; ab108998; Abcam, Cambridge, UK), SR-B1
(1:1,000; MAB8114; R&D Systems, Inc., Minneapolis, MN, USA),
ABCG1 (1:500; NB400-132), ABCA1 (1:1,000; NB400-105) (both from
Novus Biologicals, LLC, Littleton, CO, USA), p-NF-κB (1:500; 8214;
Cell Signaling Technology, Inc., Danvers, MA, USA), NF-κB (1:500;
MAB72261; R&D Systems, Inc.) and GAPDH (1:200; 4670; Cell
Signaling Technology, Inc.) overnight at 4°C, followed by
incubation with horseradish peroxidase-conjugated IgG secondary
antibodies for 1 h at room temperature. The bands were subsequently
visualized by enhanced chemiluminescence detection reagents (GE
Healthcare Life Sciences, Little Chalfont, UK), and the images were
analyzed by the NIH ImageJ software (version 1.47t; National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The data are demonstrated as the mean ± standard
deviation. All experiments were performed at least three times.
Comparisons between two groups were evaluated by Student's t-test.
Statistical analysis was performed using the SPSS version 17.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
as an indicator of statistically significant differences.
Results
Effect of ox-LDL on foam cell
formation in THP-1 and RAW264.7 cells
Macrophages are known to transform into foam cells
when incubated with ox-LDL (25). To
evaluate the formation of foam cells, ORO staining and the
intracellular cholesterol contents were measured. THP-1 and
RAW264.7 cells were incubated with 50 µg/ml ox-LDL for 48 h prior
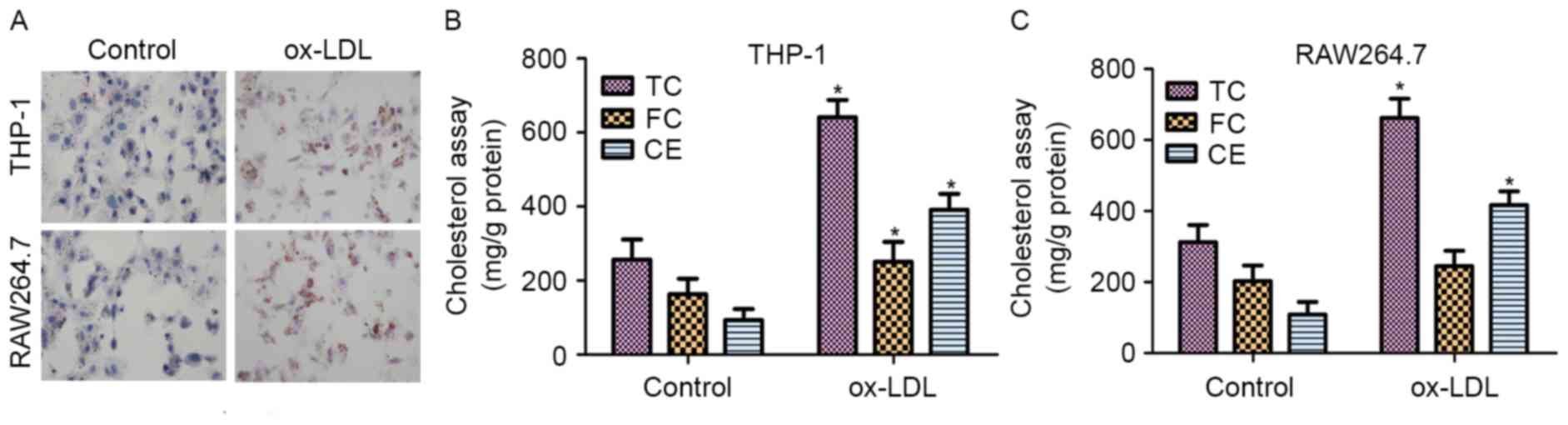
to staining with ORO. As shown in Fig.
1A, THP-1 and RAW264.7 cells treated with ox-LDL exhibited
significant accumulation of lipid droplets.
The contents of TC, FC and CE in normal cells and
foam cells were also detected following incubation with ox-LDL for
48 h. As shown in Fig. 1B, the
results revealed that the TC, FC and CE contents were significantly
increased in THP-1 macrophage-derived foam cells treated with
ox-LDL for 48 h when compared with those in untreated cells. In
addition, the contents of TC and CE were markedly upregulated in
RAW264.7 macrophage-derived foam cells treated with ox-LDL compared
with those in untreated cells (Fig.
1C). These results demonstrated that ox-LDL induced foam cell
formation in the THP-1 and RAW264.7 cells.
TLR2 blocks the efflux of macrophage
cholesterol in THP-1 and RAW264.7 macrophage-derived foam
cells
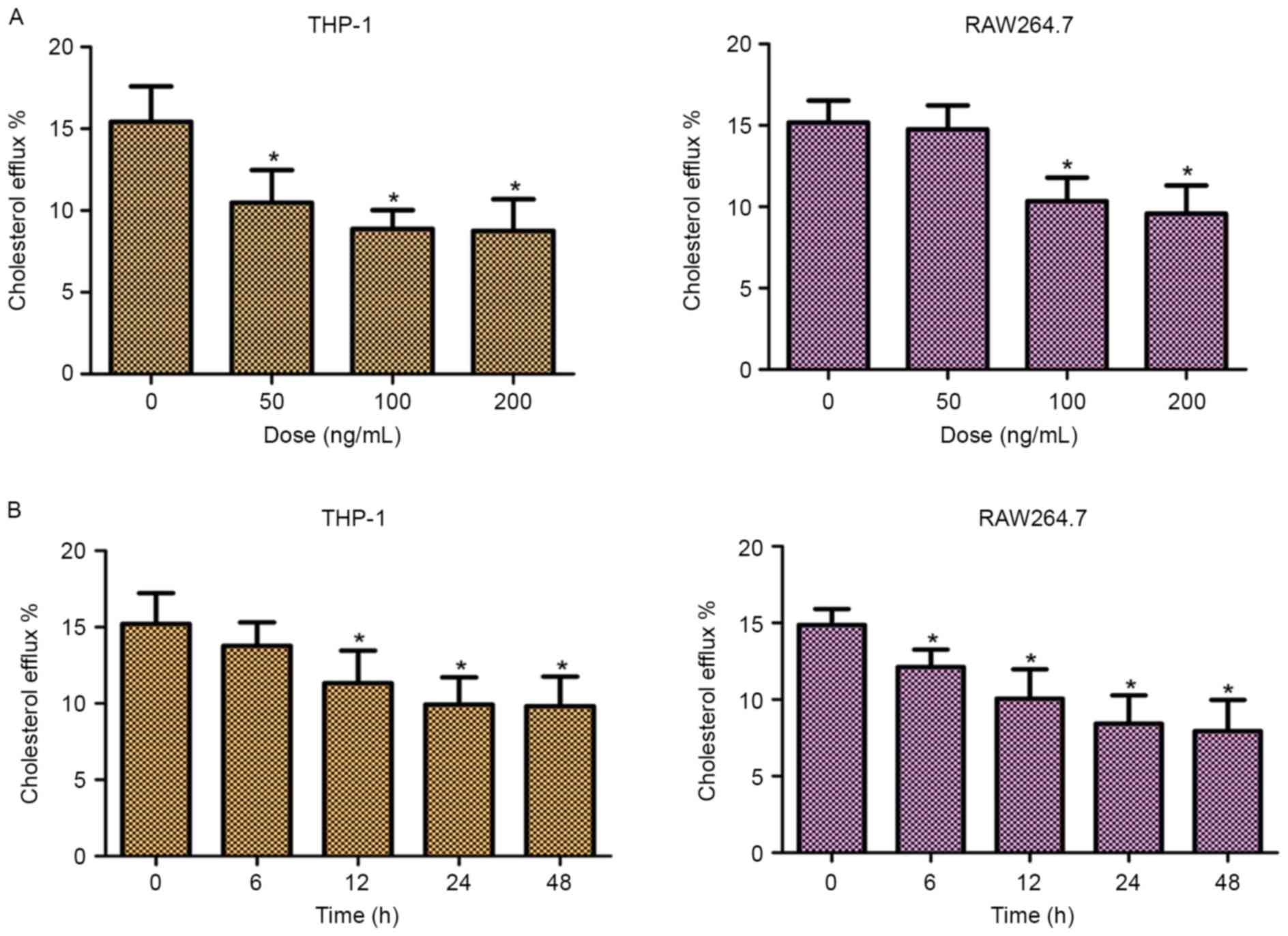
In order to investigate the role of TLR2 in
mediating the efflux of cholesterol, the cellular cholesterol
efflux was measured by liquid scintillation counting. As shown in
Fig. 2, addition of TLR2
significantly decreased the cholesterol efflux of THP-1 and
RAW264.7 cells in dose- and time-dependent manners. These data
suggested that TLR2 was a negative regulator of cholesterol efflux
in THP-1 and RAW264.7 macrophage-derived foam cells.
Knockdown of TLR2 promotes cholesterol
efflux in THP-1 and RAW264.7 macrophage-derived foam cells
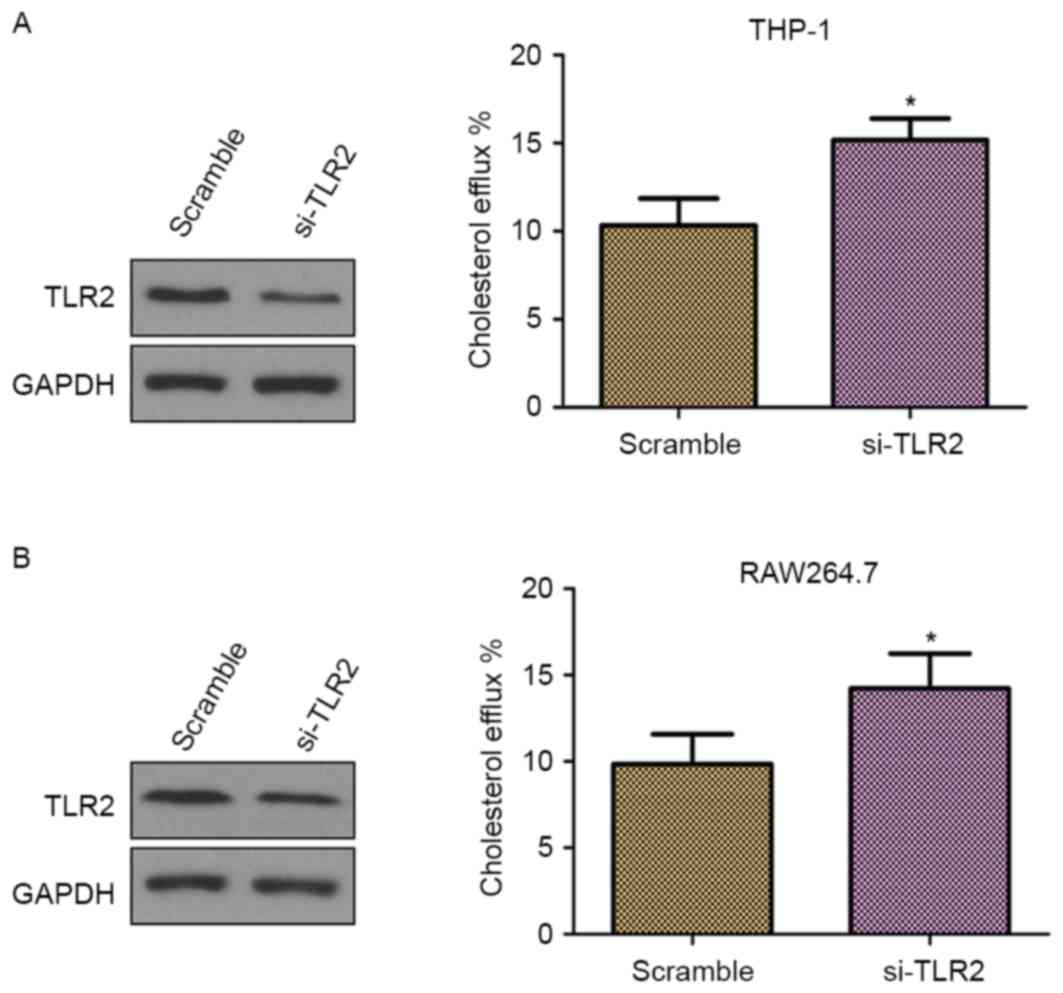
To further confirm whether TLR2 is a negative
regulator of cholesterol efflux, THP-1 and RAW264.7
macrophage-derived foam cells were transfected with TLR2 siRNA. As
demonstrated in Fig. 3A and B (left
panels), the cells transfected with TLR2 siRNA presented inhibited
TLR2 protein expression in comparison with those transfected with
scramble siRNA, which confirmed the knockdown of TLR2. Furthermore,
knockdown of TLR2 by siRNA significantly increased the cholesterol
efflux (Fig. 3; right panels). Thus,
these results supported the involvement of TLR2 in the
downregulation of cholesterol efflux.
TLR2 inhibits ABCA1, ABCG1 and SR-B1
expression in THP-1 and RAW264.7 macrophage-derived foam cells
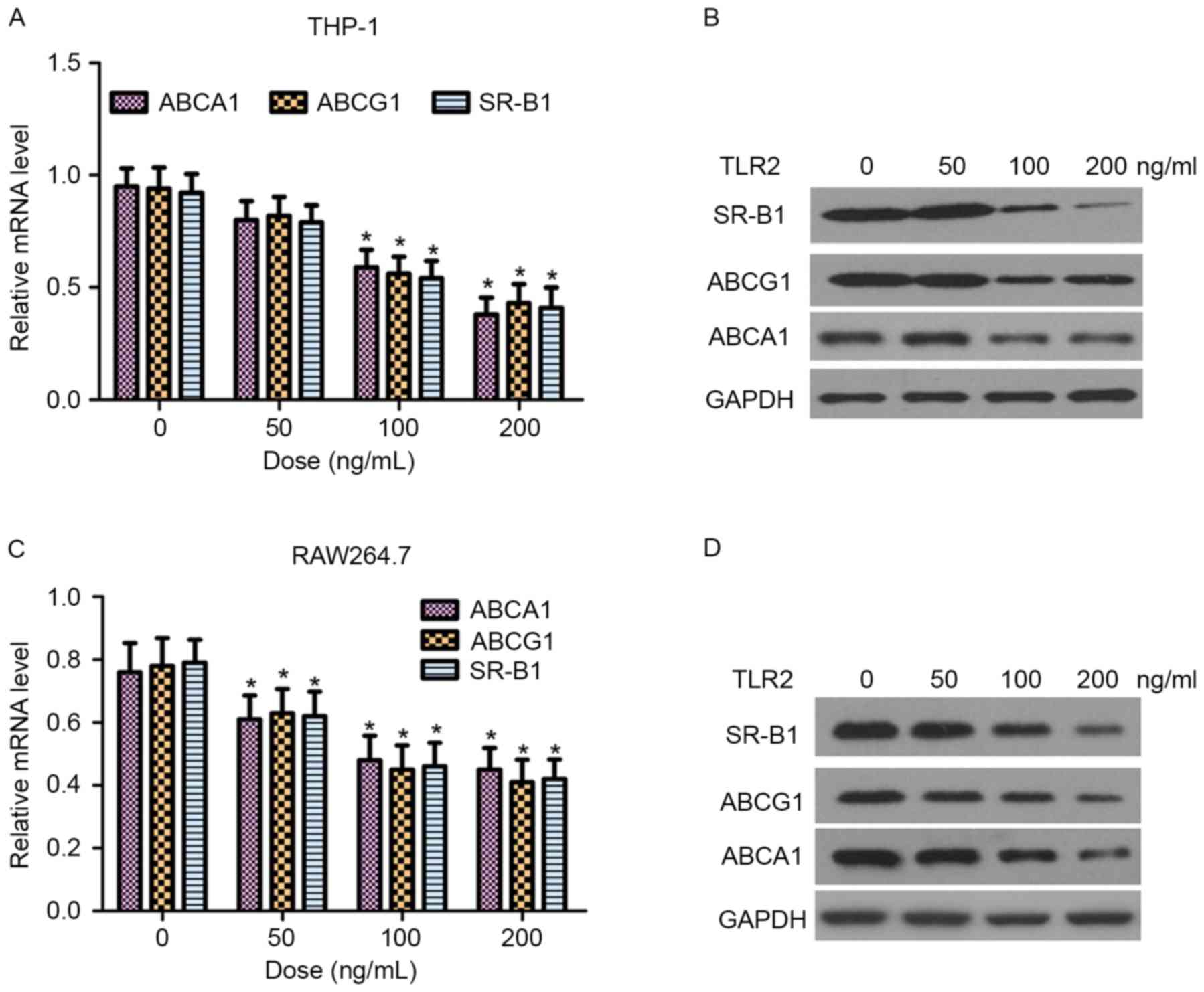
ABCA1, ABCG1 and SR-B1 are critical proteins in the
regulation of cellular cholesterol homeostasis (9). In the present study, the effect of TLR2
on the mRNA and protein expression levels of ABCA1, ABCG1 and SR-B1
in THP-1 and RAW264.7 macrophage-derived foam cells was
examined by RT-qPCR and western blot analysis, respectively. As
shown in Fig. 4, TLR2 reduced ABCA1,
ABCG1 and SR-B1 expression at the transcriptional and translational
levels in a dose-dependent manner.
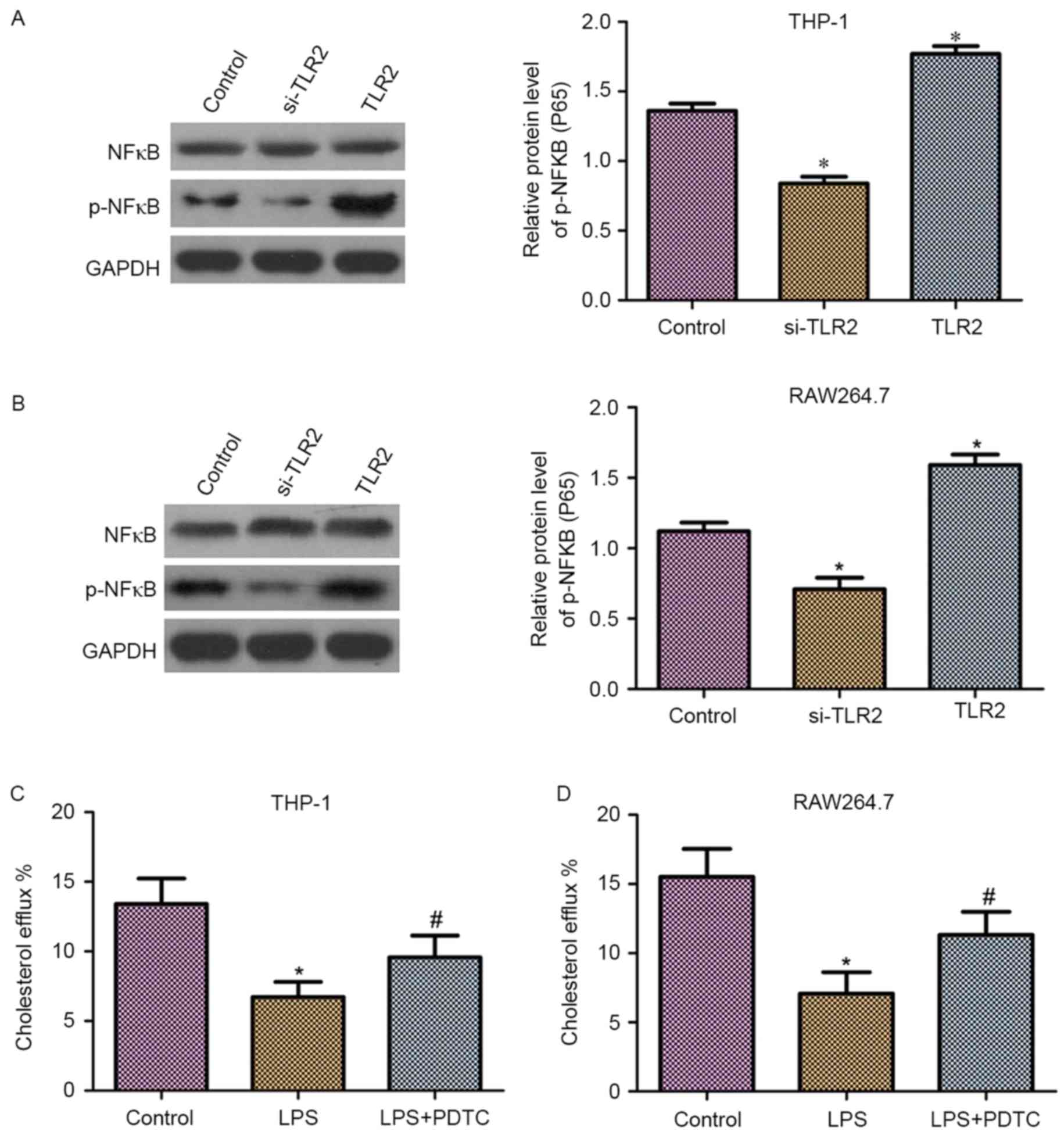
Effect of TLR2 on NF-κB activation and
inhibition of cholesterol efflux by the NF-κB activation in THP-1
and RAW264.7 macrophage-derived foam cells
It has been demonstrated that NF-κB is a downstream
molecule of TLR2 (26). Thus, in the
present study, it was hypothesized that NF-κB may be involved in
the role of TLR2 in the downregulation of cholesterol efflux. As
shown in Fig. 5A and B, western blot
analysis revealed that THP-1 and RAW264.7 macrophage-derived foam
cells transfected with TLR2 siRNA exhibited reduced phosphorylation
levels of NF-κB (p65). By contrast, overexpression of TLR2
increased the phosphorylation levels of NF-κB (p65). Moreover,
upregulation or downregulation of TLR2 had no effect on the protein
expression levels of NF-κB.
To detect the effect of NF-κB on cholesterol efflux,
liquid scintillation counting was performed. As observed in
Fig. 5C and D, the cholesterol
efflux was markedly reduced following treatment with
lipopolysaccharides (LPS) in THP-1 and RAW264.7 macrophage-derived
foam cells. By contrast, the application of PDTC, an NF-κB specific
inhibitor, significantly suppressed the LPS-induced downregulation
of cholesterol efflux.
Discussion
Macrophages take up ox-LDL and other lipids to form
foam cells, resulting in early atherosclerosis (27). The aortic atherosclerotic lesion and
foam cell formation are accelerated by enhanced cholesterol
accumulation (28). In the present
study, the effects of TLR2 on macrophage cholesterol efflux and the
underlying molecular mechanisms were investigated. In addition, the
effect of exogenous TLR2 on cell cholesterol efflux was examined.
Intracellular cholesterol efflux was detected in the THP-1 and
RAW264.7 macrophage-derived foam cells transfected with TLR2 siRNA.
The expression levels of phosphorylated NF-κB (p65) in cells
transfected with TLR2 siRNA and TLR2 overexpression vector were
also determined. The results of the current study provided
convincing evidence for the role of TLR2 in suppressing macrophage
cholesterol efflux through targeting NF-κB.
TLR2 represents an attractive therapeutic target in
atherosclerosis (29). The
proatherogenic effect of TLR2 activation has also been demonstrated
to induce intimal hyperplasia and atherosclerotic lesion
development (30). Mullick et
al (31) revealed that TLR2
participated in the modulation of atherosclerosis in mice, and
complete knockdown of TLR2 led to decreased lesion size, while
exposure to an exogenous TLR2 significantly exacerbated
atherosclerosis. Cao et al (32) identified that Chlamydophila
pneumoniae-induced macrophage foam cell formation was mediated
by TLR2. Similarly, Zhao et al (33) suggested that TLR2 was able to mediate
the effect of C. pneumoniae on cholesterol homeostasis in
human THP-1 macrophages.
Cholesterol efflux transport is mediated by specific
proteins, and has been recently demonstrated to be mediated by
ABCA1, ABCG1 and SR-B1 (34). ABCA1
mediates the transport of phospholipids, cholesterol and other
lipophilic molecules across cellular membranes to lipid-poor HDL
apolipoproteins (23). In addition,
ABCG1 promotes efflux through redistribution of intracellular
cholesterol to the plasma membrane domains accessible for removal
by HDL (35). Furthermore, SR-B1 was
demonstrated to enhance cell cholesterol influx and cholesterol
efflux from HDL, but did not alter cellular cholesterol mass
(36). In the current study, the
decrease in TLR2-mediated cholesterol efflux in dose-dependent
manner was consistent with the downregulated expression of ABCA1,
ABCG1 and SR-B1 at the transcriptional and translational levels in
THP-1 and RAW264.7 macrophage-derived foam cells.
TLR2 has been demonstrated to use the downstream
adaptor MyD88 for signal transmission, and the MyD88-dependent
pathway gives rise to activation of the NF-κB transcription factor,
which controls proinflammatory gene expression (37). Thus, the molecular mechanisms between
TLR2 and NF-κB required to be further investigated. Recent evidence
revealed the potential role of NF-κB in atherosclerosis. For
instance, activated NF-κB has been identified in macrophages and
human atherosclerotic plaques (38),
while the genes regulated by NF-κB have been detected to be
upregulated in plaques (39).
Furthermore, several Toll-like receptors that can signal to NF-κB
have also been identified in lesions (40). NF-κB has been demonstrated to mediate
the inflammatory role of TLR2 in several diseases, such as dry eye
(41,42). However, it remains unclear how the
network of TLR2 and NF-κB signaling controls atherogenesis. In the
current study, the results demonstrated that the cholesterol efflux
was downregulated via the NF-κB activator, LPS. However, cell
treatment with PDTC, an inhibitor of NF-κB, reversed the
LPS-induced downregulation of cholesterol efflux. Additionally,
knockdown of TLR2 attenuated the phosphorylation levels of NF-κB
(p65), while overexpression of TLR2 resulted in the opposite
tendency. Therefore, the role of TLR2 in reducing cholesterol
efflux may partly be through the NF-κB pathway in
macrophage-derived foam cells, and it likely contributes to the
pathogenesis of atherosclerosis.
In conclusion, the present study provided a novel
insight into the role of TLR2 on suppression of cholesterol efflux
via downregulation of ABCA1, ABCG1 and SR-B1 expression levels, and
indicated that the TLR2 effect is mediated by the NF-κB signaling
pathway. Thus, TLR2 may be a potential therapeutic target for the
prevention of atherosclerosis.
References
|
1
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Falk E, Shah PK and Fuster V: Coronary
plaque disruption. Circulation. 92:657–671. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen S, Xiao J, Liu X, Liu MM, Mo ZC, Yin
K, Zhao GJ, Jiang J, Cui LB, Tan CZ, et al: Ibrolipim increases
ABCA1/G1 expression by the LXRα signaling pathway in THP-1
macrophage-derived foam cells. Acta Pharmacol Sin. 31:1343–1349.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu C, Dandapat A, Sun L, Chen J, Marwali
MR, Romeo F, Sawamura T and Mehta JL: LOX1 deletion decreases
collagen accumulation in atherosclerotic plaque in low-density
lipoprotein receptor knockout mice fed a high cholesterol diet.
Cardiovasc Res. 79:287–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karunakaran D, Geoffrion M, Wei L, Gan W,
Richards L, Shangari P, DeKemp EM, Beanlands RA, Perisic L,
Maegdefessel L, et al: Targeting macrophage necroptosis for
therapeutic and diagnostic interventions in atherosclerosis. Sci
Adv. 2:e16002242016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Orekhov AN, Bobryshev YV and Chistiakov
DA: The complexity of cell composition of the intima of large
arteries: Focus on pericyte-like cells. Cardiovasc Res.
103:438–451. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Randolph GJ: Mechanisms that regulate
macrophage burden in atherosclerosis. Circ Res. 114:1757–1771.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Julve J, Llaverias G, Blancovaca F and
Escolàgil JC: Seeking novel targets for improving in vivo
macrophage-specific reverse cholesterol transport: Translating
basic science into new therapies for the prevention and treatment
of atherosclerosis. Curr Vasc Pharmacol. 9:220–237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu YW, Wang Q, Ma X, Li XX, Liu XH, Xiao
J, Liao DF, Xiang J and Tang CK: TGF-beta1 up-regulates expression
of ABCA1, ABCG1 and SR-BI through liver X receptor alpha signaling
pathway in THP-1 macrophage-derived foam cells. J Atheroscler
Thromb. 17:493–502. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gay NJ and Gangloff M: Structure and
function of Toll receptors and their ligands. Annu Rev Biochem.
76:141–165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Nardo D: Toll-like receptors:
Activation, signalling and transcriptional modulation. Cytokine.
74:181–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Medzhitov R: Toll-like receptors and
innate immunity. Nat Rev Immunol. 1:135–145. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hantke K and Braun V: Covalent binding of
lipid to protein. Diglyceride and amide-linked fatty acid at the
N-terminal end of the murein-lipoprotein of the Escherichia coli
outer membrane. Eur J Biochem. 34:284–296. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozinsky A, Smith KD, Hume D and Underhill
DM: Co-operative induction of pro-inflammatory signaling by
Toll-like receptors. J Endotoxin Res. 6:393–396. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mercurio F and Manning AM: NF-kappaB as a
primary regulator of the stress response. Oncogene. 18:6163–6171.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kobayashi H, Hirata M, Saito T, Itoh S,
Chung U and Kawaguchi H: Transcriptional induction of ADAMTS5
protein by nuclear factor-κB (NF-κB) family member RelA/p65 in
chondrocytes during osteoarthritis development. J Biol Chem.
288:28620–28629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun Z and Andersson R: NF-kappaB
activation and inhibition: A review. Shock. 18:99–106. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vallabhapurapu S and Karin M: Regulation
and function of NF-kappaB transcription factors in the immune
system. Annu Rev Immunol. 27:693–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu S, Huang Y, Xie Y, Lan T, Le K, Chen J,
Chen S, Gao S, Xu X, Shen X, et al: Evaluation of foam cell
formation in cultured macrophages: An improved method with Oil Red
O staining and DiI-oxLDL uptake. Cytotechnology. 62:473–481. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang B, Wang X, Yan F, Bian YF, Liu M,
Bai R, Yang HY, Zhang NN, Yang ZM and Xiao CS: Angiotensin-(1–7)
upregulates (ATP-binding cassette transporter A1) ABCA1 expression
through cyclic AMP signaling pathway in RAW 264.7 macrophages. Eur
Rev Med Pharmacol Sci. 18:985–991. 2014.PubMed/NCBI
|
|
22
|
Mo ZC, Xiao J, Liu XH, Hu YW, Li XX, Yi
GH, Wang Z, Tang YL, Liao DF and Tang CK: AOPPs inhibits
cholesterol efflux by down-regulating ABCA1 expression in a
JAK/STAT signaling pathway-dependent manner. J Atheroscler Thromb.
18:796–807. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu XY, Lu Q, Ouyang XP, Tang SL, Zhao GJ,
Lv YC, He PP, Kuang HJ, Tang YY, Fu Y, et al: Apelin-13 increases
expression of ATP-binding cassette transporter A1 via activating
protein kinase C α signaling in THP-1 macrophage-derived foam
cells. Atherosclerosis. 226:398–407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao H, Li L, Li L, Gong B, Dong P,
Fordjour PA, Zhu Y and Fan G: Danshensu promotes cholesterol efflux
in RAW264.7 macrophages. Lipids. 51:1083–1092. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ha T, Liu L, Kelley J, Kao R, Williams D
and Li C: Toll-like receptors: New players in myocardial
ischemia/reperfusion injury. Antioxid Redox Signal. 15:1875–1893.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yao S, Zong C, Zhang Y, Sang H, Yang M,
Jiao P, Fang Y, Yang N, Song G and Qin S: Activating transcription
factor 6 mediates oxidized LDL-induced cholesterol accumulation and
apoptosis in macrophages by up-regulating CHOP expression. J
Atheroscler Thromb. 20:94–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pennings M, Meurs I, Ye D, Out R, Hoekstra
M, Van Berkel TJ and Van Eck M: Regulation of cholesterol
homeostasis in macrophages and consequences for atherosclerotic
lesion development. FEBS Lett. 580:5588–5596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Ukai T, Yurnoto H, Davey M, Goswami
S, Gibson FC III and Genco CA: Toll-like receptor 2 plays a
critical role in the progression of atherosclerosis that is
independent of dietary lipids. Atherosclerosis. 196:146–154. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schoneveld AH, Nijhuis MM Oude, van
Middelaar B, Laman JD, de Kleijn DP and Pasterkamp G: Toll-like
receptor 2 stimulation induces intimal hyperplasia and
atherosclerotic lesion development. Cardiovasc Res. 66:162–169.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mullick AE, Tobias PS and Curtiss LK:
Modulation of atherosclerosis in mice by Toll-like receptor 2. J
Clin Invest. 115:3149–3156. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao F, Castrillo A, Tontonoz P, Re F and
Byrne GI: Chlamydia pneumoniae-induced macrophage foam cell
formation is mediated by Toll-like receptor 2. Infect Immun.
75:753–759. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao GJ, Mo ZC, Tang SL, Ouyang XP, He PP,
Lv YC, Yao F, Tan YL, Xie W, Shi JF, et al: Chlamydia pneumoniae
negatively regulates ABCA1 expression via TLR2-Nuclear factor-kappa
B and miR-33 pathways in THP-1 macrophage-derived foam cells.
Atherosclerosis. 235:519–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adorni MP, Zimetti F, Billheimer JT, Wang
N, Rader DJ, Phillips MC and Rothblat GH: The roles of different
pathways in the release of cholesterol from macrophages. J Lipid
Res. 48:2453–2462. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vaughan AM and Oram JF: ABCG1
redistributes cell cholesterol to domains removable by high density
lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem.
280:30150–30157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji A, Meyer JM, Cai L, Akinmusire A, de
Beer MC, Webb NR and van der Westhuyzen DR: Scavenger receptor
SR-BI in macrophage lipid metabolism. Atherosclerosis. 217:106–112.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen S, Sorrentino R, Shimada K, Bulut Y,
Doherty TM, Crother TR and Arditi M: Chlamydia pneumoniae-induced
foam cell formation requires MyD88-dependent and -independent
signaling and is reciprocally modulated by liver X receptor
activation. J Immunol. 181:7186–7193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brand K, Page S, Rogler G, Bartsch A,
Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA and
Neumeier D: Activated transcription factor nuclear factor-kappa B
is present in the atherosclerotic lesion. J Clin Invest.
97:1715–1722. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Janus P, Stokowy T, Jaksik R, Szoltysek K,
Handschuh L, Podkowinski J, Widlak W, Kimmel M and Widlak P: Cross
talk between cytokine and hyperthermia-induced pathways:
Identification of different subsets of NF-κB-dependent genes
regulated by TNFα and heat shock. Mol Genet Genomics.
290:1979–1990. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Edfeldt K, Swedenborg J, Hansson GK and
Yan ZQ: Expression of toll-like receptors in human atherosclerotic
lesions: A possible pathway for plaque activation. Circulation.
105:1158–1161. 2002.PubMed/NCBI
|
|
41
|
He C, Lai P, Weng J, Lin S, Wu K, Du X and
Liu X: Toll-like receptor 2-mediated NF-κB inflammatory responses
in dry eye associated with cGVHD. Mol Vis. 17:2605–2611.
2011.PubMed/NCBI
|
|
42
|
Xavier RJ and Podolsky DK: Unravelling the
pathogenesis of inflammatory bowel disease. Nature. 448:427–434.
2007. View Article : Google Scholar : PubMed/NCBI
|