Introduction
Ischemic stroke is one of the leading causes of
mortality and morbidity worldwide, accounting for ~87% of all
stroke cases (1). Although several
clinical trials have investigated the effects of different drugs on
ischemic stroke, only intravenous recombinant tissue plasminogen
activator has clinically exhibited protective efficacy (2). Previous studies have demonstrated that
stroke triggers complex cellular molecular events and leads to
neuronal cell necrosis, apoptosis, oxidative stress and
inflammation in the ischemic brain (3–5).
However, the exact mechanisms underlying stroke-induced cell death
and neurological dysfunction remain unclear. It has been
demonstrated that cellular tumor antigen p53 (p53), a tumor
suppressor protein and transcription factor, mediates a number of
intrinsic or extrinsic challenges to cells, serving a pivotal role
in cell cycle arrest and apoptosis (6). Induction of p53 by oxidative stress and
hypoxia results in apoptosis mediated by the mitochondrial pathway
(7). p53 can stimulate the
expression of several Bcl-2 family genes, including Bax and
multiple BH3-only proteins (8).
Glucose deprivation (GD) and combined oxygen-glucose deprivation
(OGD) are common in vitro models of brain ischemia (9–11). Thus,
elucidating the underlying cellular and molecular mechanisms of
OGD/reperfusion (OGD/R)-induced neural death may contribute to the
development of neuroprotective drugs to treat patients with
cerebral ischemia.
MicroRNAs (miRNAs/miRs) are noncoding RNAs that are
19- to 22-nucleotides long and are important post-transcriptional
regulators, which interact with multiple target mRNAs to regulate
their target genes (12). Several
studies have identified that miRNAs serve significant roles in a
wide variety of biological processes, including cell proliferation,
differentiation, apoptosis and signal transduction (13–15). It
has been demonstrated that miRNAs serve an important role in
responding to cerebral ischemia (16). Changes in miRNA expression were
identified in the brains of patients with forebrain ischemia
(17), focal cerebral ischemia
(18) and stroke (19). Furthermore, it has been reported that
the miR-29 family serves an important role in ischemic brain damage
(20,21). However, it remains unclear whether
the miR-29 family comprises pro-survival or pro-apoptotic effects
(22). Studies have demonstrated
miR-29 family members induce anti-apoptotic and pro-apoptotic
effects, potentially as a result of binding to different targets in
different cells or being under different pathological or
physiological conditions (21,23,24). One
study demonstrated that loss of miR-29b at the infarct site is a
pivotal contributor to stroke lesions and indicated that treatment
with miR-29b mimic decreased stroke-induced neural cell death and
the infarct size (20). Although
there are already a number of studies demonstrating the protective
roles of miR-29 family members in cerebral ischemia, the mechanism
of miR-29b in cerebral ischemia remains unknown.
In the present study, the role of miR-29b in the
development of cerebral ischemia was explored. The expression of
miR-29b decreased following OGD/R treatment. Furthermore
p53-mediated apoptosis caused OGD/R-induced injury in N2a cells,
which was reversed by overexpression of miR-29b. The results of the
present study suggest that miR-29b may be developed as a novel
therapeutic strategy to treat cerebral ischemia injury and may be
important in the treatment of neural cell injury and stroke.
Materials and methods
Reagents
The miR-29b mimic, inhibitor and negative control
miRNA sequences were synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). The p53 small interfering (si)RNA sequence and
scrambled siRNA were synthesized by GE Healthcare Dharmacon, Inc.
(Lafayette, CO, USA). The MTT assay kit and Hoechst 33258 were
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The
lactate dehydrogenase (LDH) cytotoxicity assay kit (cat. no. C0016)
and caspase-3 activity kit (cat. no. C1116) were purchased from
Beyotime Institute of Biotechnology (Haimen, China). Rabbit
anti-apoptosis regulator BAX (Bax, cat. no. 14796), rabbit
anti-apoptosis regulator Bcl-2 (Bcl-2) polyclonal antibodies (cat.
no. 3498), p53 (cat. no. 2524), GAPDH (cat. no. 2118) and goat
anti-rabbit horseradish peroxidase (HRP)-conjugated secondary
antibodies (cat. no. 7075) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). All reagents in this study
were of analytical grade.
Mouse N2a neuroblastoma cell
culture
Mouse N2a neuroblastoma cells were supplied by the
American Type Culture Collection (Manassas, VA, USA), maintained in
Dulbecco's modified Eagle's medium (DMEM, Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C in a humidified
atmosphere under normal culture conditions (5% CO2 and
95% O2). The culture medium was replenished every 1–2
days.
Establishment of the OGD/R model and
transfection of miR-29b mimic, miR-29b inhibitors or p53 siRNA
To simulate an in vitro ischemic-like
condition, N2a neuroblastoma cells were exposed to OGD. The culture
medium was replaced with deoxygenated glucose-free DMEM and cells
were incubated in a hypoxic chamber containing 5% CO2,
1% O2 and 94% N2 for 4 h. Subsequently, N2a
neuroblastoma cells were returned to glucose-containing DMEM under
normal culture conditions for 0, 3, 6, 12 or 24 h for reperfusion.
The overall protocol is known as OGD/R treatment. To investigate
the role of miR-29b and p53 in OGD/R-treated N2a cells, the miR-29b
mimics, inhibitors, negative-control miRNA, p53 siRNA or scrambled
siRNA, or two treatments were transfected into cells at working
concentrations [1:1 (v/v)] using Lipofectamine™ 3000
Transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Following 6 h
transfection, N2a cells underwent OGD/R treatment (4 h OGD and 24 h
reperfusion). The transfection efficiencies of the miRNA in N2a
cells were confirmed by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) to measure miR-29b expression.
The transfection efficiencies of p53 siRNA were confirmed by
western blot analysis. The sense and antisense sequences of the
miRNAs used were as follows: hsa-miR-29b mimics, sense,
5-′UAGCACCAUUUGAAAUCAGUGUU-3′ and antisense,
5′-AACACUGAUUUCAAAUGGUGCUA-3; negative-control miRNA, sense,
5′-UUCUCCGAACGUGUCACGUTT-3 and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′; hsa-miR-29b inhibitor, sense,
5′-AGAUGUGGAUUUUGAUAGAGT-3 and antisense,
5′-UCUACACCUAAAACUAUCUCT-3′; p53 siRNA, sense,
5′-GAGAUGUUCCGAGAGCUGA-3′ and antisense, 5′-UCAGCUCUCGGAACAUCUC-3′;
and p53 scrambled siRNA, sense, 5′-GGGGAUAGGUUACAUGCAC-3′; and
antisense, 5′-GUGCAUGUAACCUAUCCCC-3.
MTT assay
N2a neuroblastoma cells were seeded into 96-well
plates at a density of 5×104 cells/ml. Following OGD/R
treatment and transfection, 20 µl MTT (5 mg/ml) was added to each
well and co-incubated for 3–4 h at 37°C. Subsequently, 150 µl
dimethyl sulfoxide was added to dissolve the dark blue crystals.
Absorbance at 490 nm was measured using a microplate reader
(Multiskan MK3; Thermo Fisher Scientific, Inc.). Cell viability was
expressed as 100% in the control group (without any treatment) and
cell viability in other groups were normalized to this value.
LDH assay
The death of N2a neuroblastoma cells was evaluated
using the Lactate Dehydrogenase Cytotoxicity assay kit according to
the manufacturer's protocol. Briefly, N2a cells were cultured in
96-well plates. Following OGD/R treatment and transfection, LDH
levels in the culture supernatant were analyzed by measuring
absorbance at 490 nm using a microplate reader. LDH levels in the
control group were expressed as 100% and the levels in other groups
were normalized to this value.
Hoechst 33258 staining
The morphological characteristics of N2a
neuroblastoma cells during apoptosis were observed using Hoechst
33258 according to the manufacturer's protocol. N2a cells were
seeded onto 24-well plates at a density of 1×105
cells/ml, OGD/R treated and the agents were transfected. N2a cells
were then rinsed twice with ice-cold PBS and fixed with 4%
paraformaldehyde for 15 min at room temperature. Subsequently, N2a
cells were stained with 5 µg/ml Hoechst 33258 at 4°C for 10 min in
the dark. Cells were washed three times with PBS and fluorescence
images of the cells were obtained using a fluorescence microscope
(Olympus IX71; Olympus Corporation, Tokyo, Japan). Cells with
nuclei exhibiting bright fluorescence, shrinkage or pyknosis
morphology and typical phenomena of nuclear condensation, were
considered to be apoptotic. Cells diffusely fluorescent throughout
the cytoplasm were determined to be alive and viable.
Caspase-3 activity assay
Based on the ability of caspase-3 to convert
acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) into the yellow
formazan product pNA, caspase-3 activity in N2a cells was measured
to determine apoptosis. In brief, N2a cells were seeded in 6-well
plates at a density of 1×106 cells/ml. Following OGD/R
treatment and transfection, cells were collected and protein was
extracted using the radioimmunoprecipitation assay lysis buffer
system (Beyotime Institute of Biotechnology) subsequent to
centrifugation at 12,887 × g for 10 min at 4°C. Protein
concentration was then measured using a Bio-Rad Protein assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts of
protein extracts (30 µg) were added to 96-well microtiter plates
and incubated with the caspase-3 substrate (Ac-DEVD-pNA, 100 µM) or
a caspase-3 inhibitor (Ac-DEVD-CHO, 100 µM) for 4 h at 37°C.
Absorbance was measured at 405 nm using a microplate reader. The
assay was also performed with non-induced N2a cells for a
comparative analysis.
RNA isolation and RT-qPCR
Total RNA from cultured N2a neuroblastoma cells was
isolated using TRIzol™ (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's protocol. For
miRNA-expression analysis, first-strand cDNA was synthesized from
total RNA (1 µg) using a TaqMan™ MicroRNA Reverse
Transcription kit (Thermo Fisher Scientific, Inc.) with an
miRNA-specific primer, according to the manufacturer's protocol.
Following the reverse transcription reaction, qPCR analysis to
measure the level of miR-29b was performed using the
All-in-One™ qPCR mix (GeneCopoeia, Inc., Rockville, MD,
USA) on the ABI Prism® 7900HT Sequence Detection system
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The qPCR conditions were as follows: 94°C for 5 min, 40
cycles of denaturation at 94°C for 60 sec, 40 cycles of annealing
at 60°C for 30 sec and extension at 72°C for 60 sec, and a final
extension step of 72°C for 10 min. U6 was used as an internal
control. The primer sequences used for miRNA amplification were as
follows: miR-29b, forward, 5′-GGGTAGCACCATTTGAAATC-3 and reverse,
5′-TTTGGCACTAGCAC-ATT-3; U6, forward, 5′-CTCGCTTCGGCAGCACA-3 and
reverse, 5′-AACGCTTCACGAATTTGCGT-3. All experiments were repeated
three times and results were normalized to that of the internal
control. Data were analyzed using the comparison Cq
(2−ΔΔCq) method to obtain the lg2 (microarray normalized
signal) (25).
Western blot analysis
Following protein extraction using the
radioimmunoprecipitation assay lysis buffer system (Beyotime
Institute of Biotechnology) for 30 min on ice, protein
concentration was measured using a bicinchoninic acid assay. Equal
amounts of protein samples (30–50 µg protein/lane) were separated
using 12% SDS-PAGE (Sigma-Aldrich; Merck KGaA) and transferred to
nitrocellulose membranes (Pall Life Sciences, Port Washington, NY,
USA). Membranes were blocked with 5% with skimmed milk for 2 h at
room temperature, and then incubated overnight at 4°C with the
primary antibodies against Bax, (1:1,000), Bcl-2 (1:500), p53
(1:2,000) and GAPDH (1:2,000). Membranes were washed with
Tris-buffered saline containing 0.1% Tween-20 three times and
incubated for 1 h at room temperature with HRP-conjugated goat
anti-rabbit (1:5,000). Labeled protein bands were detected using
the Pierce™ ECL Western Blotting Substrate (Thermo
Fisher Scientific, Inc). Images were assayed using Quantity One 1-D
analysis software (Bio-Rad Laboratories, Inc.). Western blot
analyses were performed in triplicate.
Statistical analysis
SPSS software (version 16.0; SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis. All values are
expressed as means ± standard deviation of three independent
experiments. Differences among the groups were compared using
one-way analysis of variance analysis followed by a least
significant difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
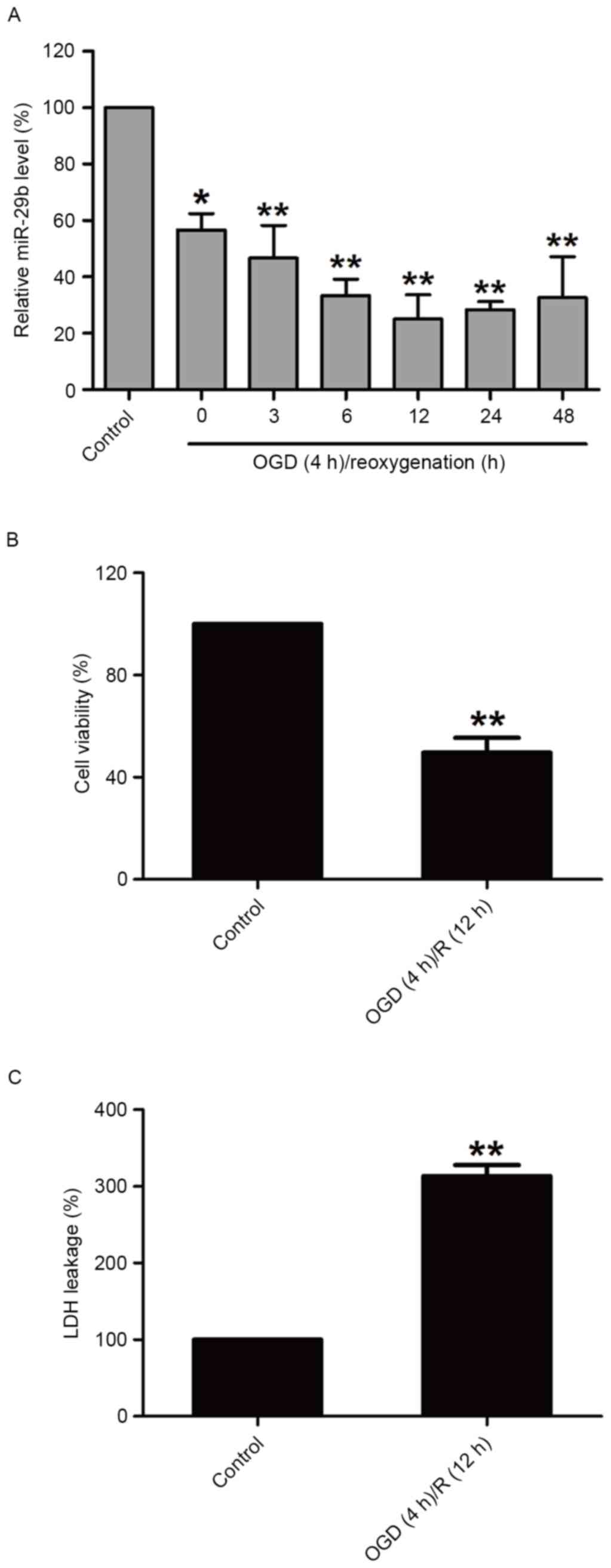
OGD/R treatment decreases miR-29b
expression and induces cytotoxicity in N2a cells
In order to investigate the effect of OGD/R
treatment on miR-29b in N2a cells, miR-29b expression in N2a cells
was measured immediately following 4 h OGD and during different
reperfusion time periods by RT-qPCR. miR-29b levels were
significantly downregulated compared with the normoxic control
group (P<0.05); miR-29b expression was lowest at the 12 h time
point (Fig. 1A). OGD/R treatment
(OGD for 4 h and reperfusion for 12 h) significantly decreased the
viability of N2a cells (P<0.01; Fig.
1B) and increased the release of LDH (P<0.01; Fig. 1C) compared with the normoxic control
group. These results suggest that the downregulation of miR-29b
contributes to cerebral ischemia injury.
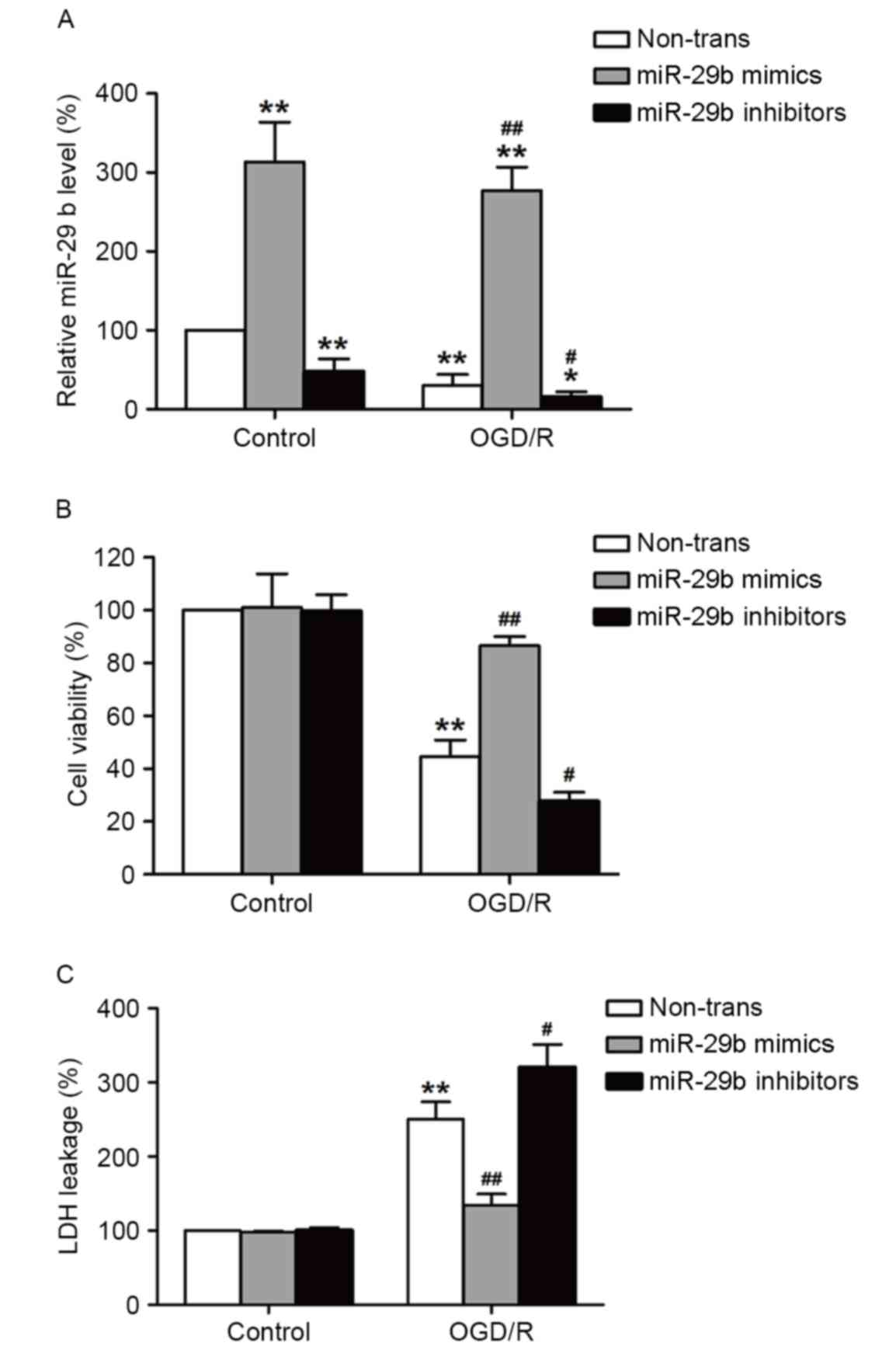
miR-29b mimics suppress and miR-29b
inhibitors enhance cytotoxicity in OGD/R-treated N2a cells
To determine the role of miR-29b in OGD/R-induced
injury in N2a cells, miR-29b mimics or inhibitors were used to
alter the expression of miR-29b. Transfection with miR-29b mimics
significantly increased miR-29b levels (P<0.01), whereas
transfection with miR-29b inhibitors significantly decreased
miR-29b levels (P<0.05) in normoxic and OGD/R-treated cells
compared with their respective controls (Fig. 2A). This demonstrates that the miR-29b
mimics and inhibitors successfully increased and decreased,
respectively, miR-29b levels in cultured N2a cells. The effects of
the miR-29b mimics or inhibitors on OGD/R-induced cell viability
and cytotoxicity were evaluated using MTT and LDH assays,
respectively. ODG (4 h)/reperfusion (12 h) significantly reduced
the viability of N2a cells compared with the normoxic control group
(P<0.01), this effect was significantly reversed by transfection
with miR-29b mimics (P<0.01). By contrast, this effect was
enhanced by transfection with miR-29b inhibitors compared with the
non-transfected ODG/R-treated group (P<0.05; Fig. 2B). miR-29b mimics significantly
suppressed LDH leakage (P<0.01) and miR-29b inhibitors
significantly enhanced the LDH leakage induced by ODG/R treatment
compared with the non-transfected ODG/R-treated group (P<0.05;
Fig. 2C). The miR-29b mimics or
inhibitors alone had no effect on cell viability and LDH leakage.
These results suggest that OGD/R induces N2a cell injury by
suppressing miR-29b expression.
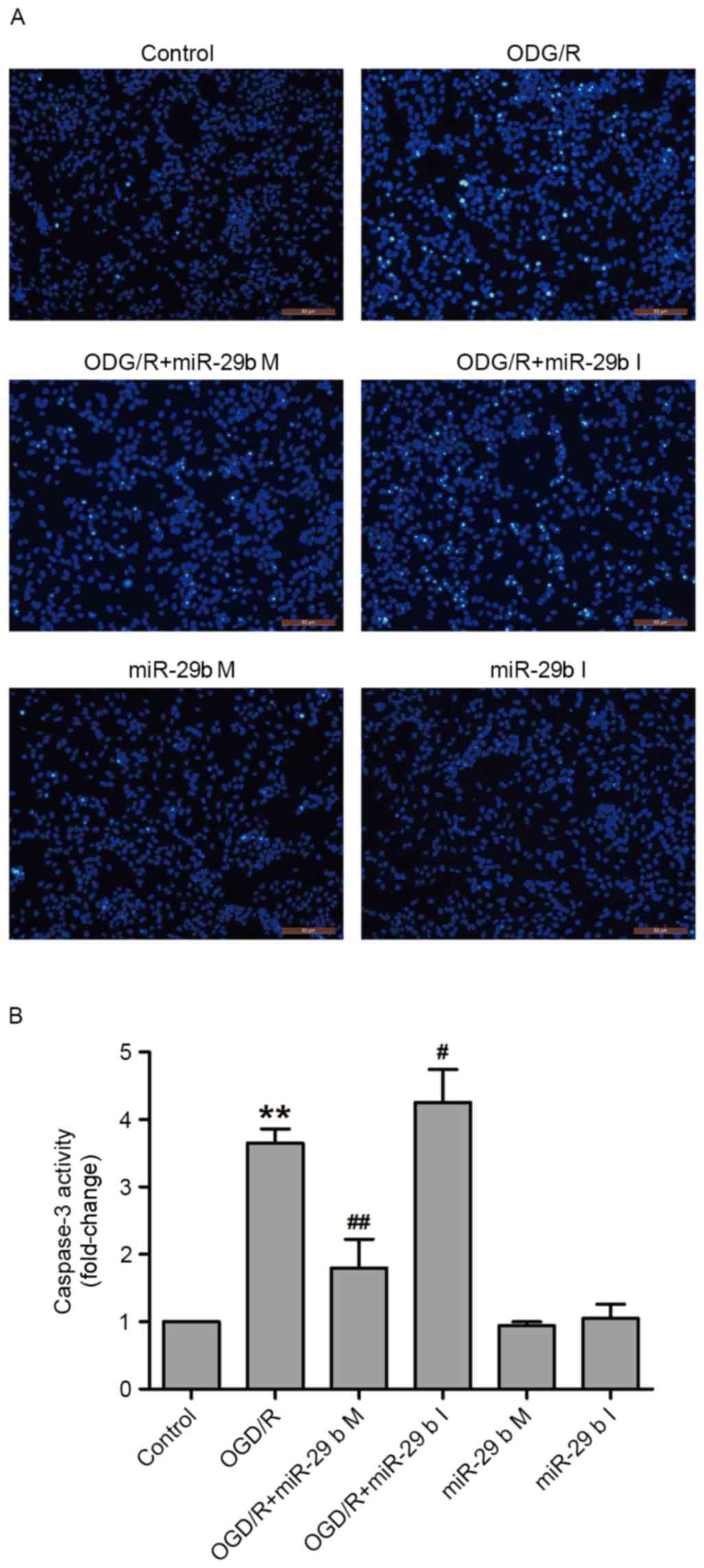
In N2a cells, miR-29b mimics suppress
and miR-29b inhibitors enhance apoptosis induced by OGD/R
To confirm whether miR-29b affected OGD/R-induced
apoptosis in N2a cells, Hoechst 33258 staining and a commercial
caspase-3 kit were used to detect the nuclear morphological
characteristics of apoptotic cells and caspase-3 activity,
respectively. OGD/R treatment increased the phenomenon of nuclear
fragmentation and the number of bright blue nuclei; typical
manifestations of apoptotic cells. These effects were inhibited by
transfection with miR-29b mimics and enhanced by transfection with
miR-29b inhibitors in N2a cells (Fig.
3A). In addition, transfection of miR-29b mimics significantly
attenuated OGD/R treatment-induced caspase-3 activity (P<0.01),
whereas miR-29b inhibitors significantly enhanced OGD/R-induced
caspase-3 activity compared with the non-transfected ODG/R-treated
group (P<0.05; Fig. 3B). Cells
transfected with miR-29b mimics or inhibitors that did not undergo
ODG/R did not undergo apoptosis. These results indicate that the
inhibition of miR-29b contributes to OGD/R-induced apoptosis.
In OGD/R-treated N2a cells, miR-29b
mimics and inhibitors alter the p53-mediated apoptosis signaling
pathway
The effects of miR-29b on the apoptosis-associated
signaling pathway in OGD/R-treated N2a cells were investigated. The
expression of p53 (a tumor suppressor protein), Bax (a
pro-apoptotic protein) and Bcl-2 (an anti-apoptotic protein) were
measured by western blotting (Fig.
4A). Quantification of these results demonstrated that OGD/R
treatment significantly increased the expression of Bax (P<0.05;
Fig. 4B) and reduced the expression
of Bcl-2 (P<0.05; Fig. 4C),
compared with the normoxic control group. These effects were
significantly inhibited (P<0.05) and enhanced (P<0.05) by
transfection of N2a cells with the miR-29b mimics and miR-29b
inhibitors, respectively. p53 is a tumor suppressor protein that
inhibits cellular proliferation by inducing cell cycle arrest and
apoptosis in response to cellular stress, including DNA damage,
growth factor deprivation and hypoxia (26,27).
Transfection with miR-29b mimics and inhibitors significantly
downregulated (P<0.05) and upregulated (P<0.01) p53 protein
expression, respectively, in N2a cells compared with the
non-transfected ODG/R-treated group (Fig. 4D). Transfection with miR-29b mimics
or inhibitors in cells that did not undergo ODG/R produced no
effect on Bax, Bcl-2 or p53 expression. These results indicate that
the protective effect of miR-29b against OGD/R-induced N2a cells
injury may be due to the activation of the p53 signaling pathway
and the subsequent alterations of Bax and Bcl-2 expression.
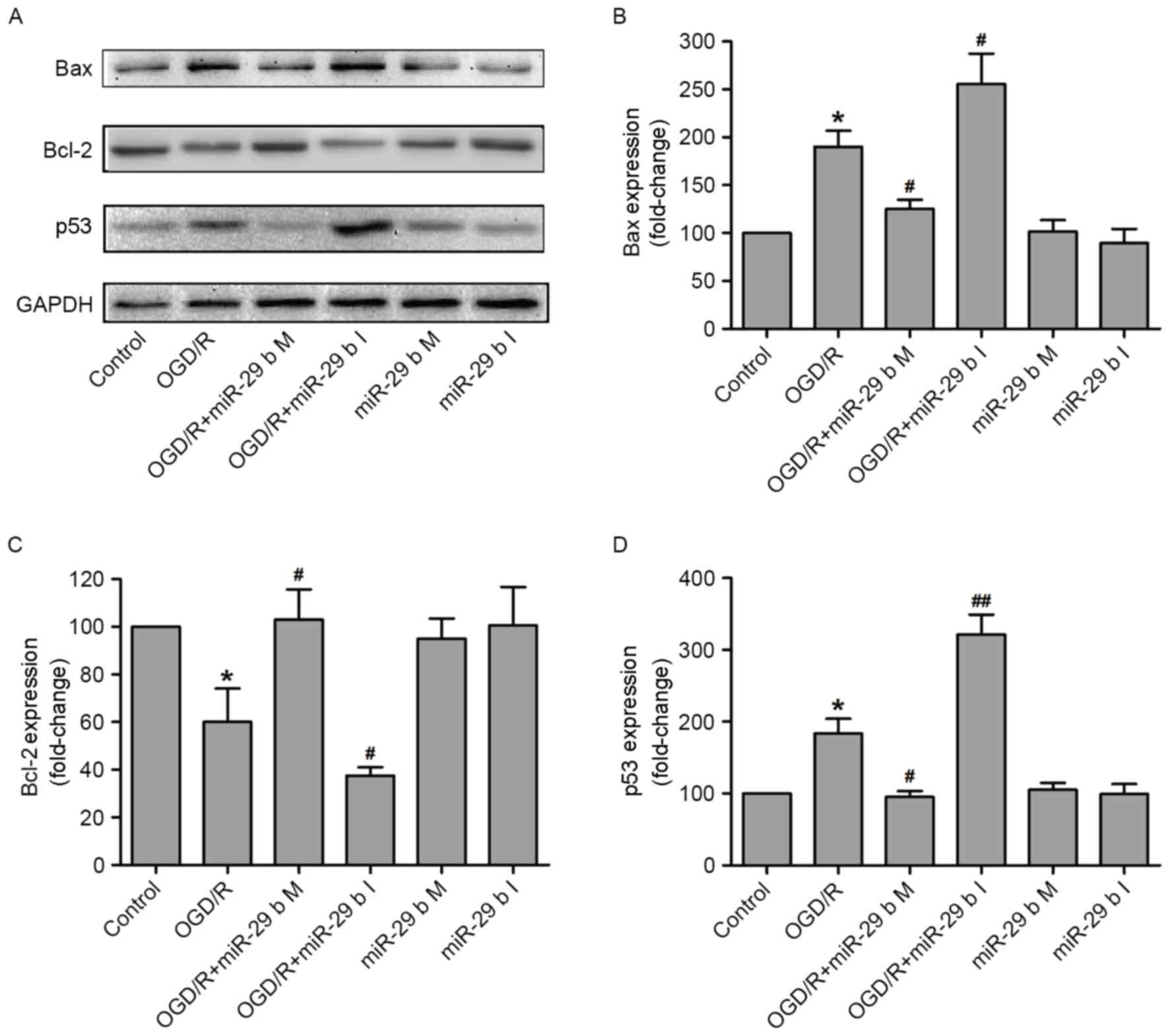 | Figure 4.miR-29b mimics and inhibitors alter
the expression of apoptosis-associated proteins in OGD/R-treated
N2a cells. (A) The apoptosis-associated proteins were measured by
western blotting and quantitative analyses of (B) Bax, (C) Bcl-2
and (D) p53 expression. *P<0.05 vs. the normoxic non-transfected
group, #P<0.05, ##P<0.01 vs. the
OGD/R-treated non-transfected group. OGD/R, oxygen and glucose
deprivation and reperfusion; miR-29b, microRNA-29b; M, mimics; I,
inhibitors; Bax, apoptosis regulator BAX; Bcl-2, apoptosis
regulator Bcl-2; p53, cellular tumor antigen p53. |
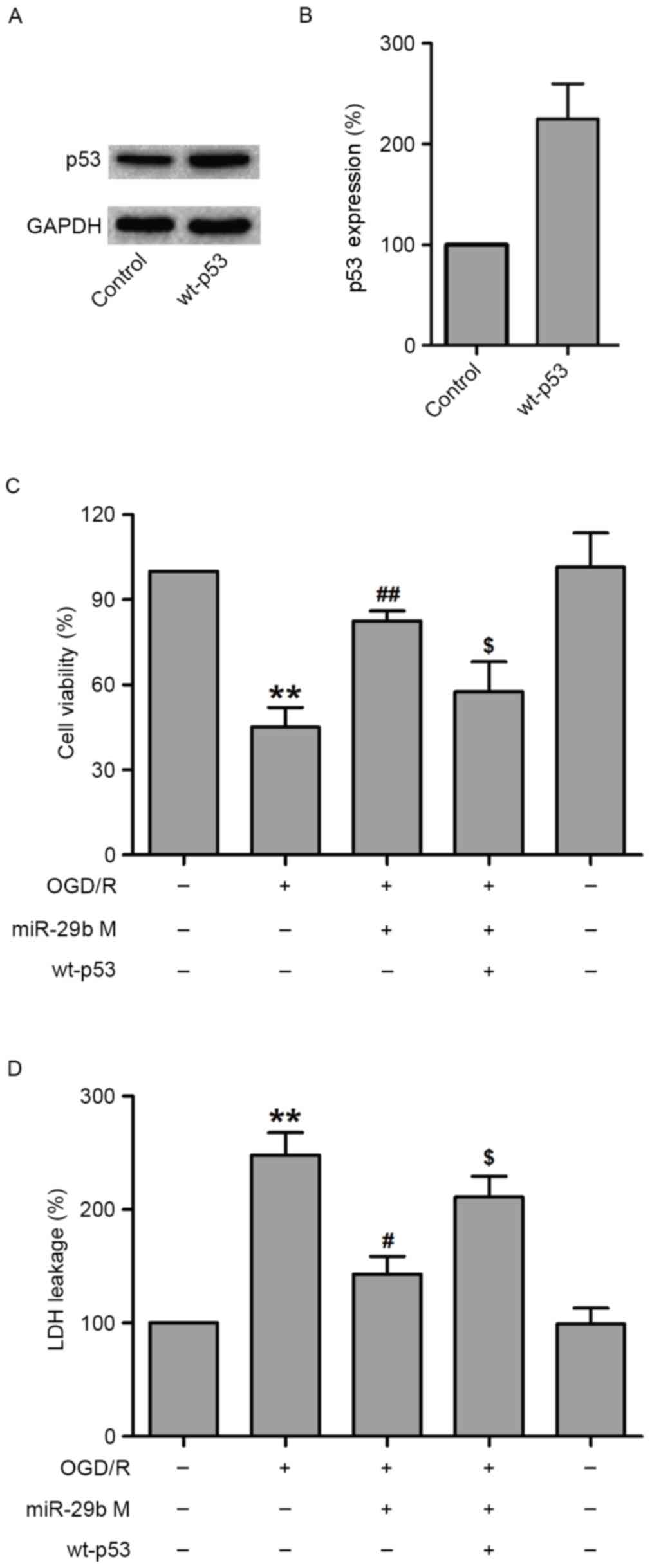
Overexpression of p53 reverses the
protective effect of miR-29b against OGD/R-induced N2a cell
injury
To further assess the role of p53 in the protection
effects of miR-29b against OGD/R-induced N2a cell injury, the
effect of p53 overexpression was examined using a specific plasmid
DNA encoding for wild-type p53 (wt-p53) in OGD/R-treated N2a cells.
To verify this hypothesis, N2a cells were transfected with siRNA
against p53 or scramble siRNA, treated with miR-29b mimics and then
subjected to OGD/R treatment. Western blotting demonstrated that
wt-p53 markedly increased p53 levels in normoxic control N2a cells
(Fig. 5A and B). The results of MTT
indicated that wt-p53 treatment significantly decreased the
viability of N2a cells compared with N2a cells transfected with
miR-29b mimics that underwent OGD/R treatment (P<0.05; Fig. 5C). In addition, transfection with
miR-29b mimics significantly reduced LDH leakage (P<0.05), which
was reversed by wt-p53 transfection in OGD/R-treated N2a cells
(P<0.05; Fig. 5D). These results
suggest that p53 mediates OGD/R-induced N2a cell injury.
Discussion
A complex interplay of various pathways, including
excitotoxicity, mitochondrial dysfunction, oxidative stress and
inflammation, are involved in the molecular mechanism of cerebral
ischemic injury (28). However, the
currently approved therapies, therapeutic targets and biomarkers
for cerebral ischemic injury are not ideal due to not being fully
effective in all cases (28). In the
present study, miR-29b expression was downregulated in
OGD/R-treated N2a cells following reperfusion. Importantly, it was
determined that miR-29b overexpression induced neuroprotection by
negatively regulating p53-associated apoptosis.
Over the past decade, it has been confirmed by a
number of studies that miRs serve an important role in the cellular
response to ischemic injury (11,29). A
recent study investigating the role of miRs in cerebral ischemia
injury established that the miR-29 family is an important mediator
in the assessment of brain injury and determining the prognosis of
patients following stroke (30).
Khanna et al (20)
demonstrated that ischemic stroke induced by middle-cerebral artery
occlusion caused a decrease in the expression of miR-29b in
infarcted tissue. GD and OGD are common in vitro models of
brain ischemia. Consistent with the results of the aforementioned
study, the results of the current study demonstrated that OGD/R
treatment significantly decreased miR-29b levels in N2a cells.
Studies investigating miR-29 have generally focused on its role in
regulating apoptotic signaling pathways and it remains unknown
whether miR-29b is pro-apoptotic or pro-survival (21). A decrease in miR-29b expression also
occurs following cerebral ischemia. It has been demonstrated that
the downregulation of miR-29 contributed to neuronal cell death in
focal ischemia by enhancing the expression of Bcl-2-like protein 2,
an anti-apoptotic member of the BCL-2 protein family (31). By contrast, upregulation of miR-29
protected neurons from apoptosis during neuronal maturation
(32), a well as during forebrain
(24) and focal ischemia (26). Therefore, the expression and function
of miR-29b in cerebral ischemia injury remains unknown.
In the present study, it was demonstrated that the
upregulation of miR-29b by miR-29b mimics significantly increased
cell viability and decreased LDH leakage in OGD/R-treated N2a
cells, indicating that miR-29 upregulation protects neuronal cells
against OGD/R-induced cytotoxicity. The BCL-2 protein family
includes Bcl-2 and Bax, and regulates apoptosis by modulating
mitochondrial membrane integrity, function and apoptotic signaling
(33). Several in vivo and
in vitro studies have reported that the overexpression of
pro-survival BCL-2 protein family members protects against cerebral
ischemia injury (34,35). In the current study, OGD/R treatment
significantly increased caspase-3 activity and Bax expression, and
significantly reduced Bcl-2 expression, while the miR-29b mimics
and inhibitors inhibited and enhanced these effects, respectively.
These results indicate that miR-29b may protect against cerebral
ischemia injury by regulating expression of BCL-2 family
proteins.
p53 is a major orchestrator of the cellular response
to different types of stress by regulating cell cycle arrest,
apoptosis, DNA repair and genetic stability (36). Previous studies have demonstrated
that p53 is involved in the neuronal death that occurs following
stroke and neurodegeneration (37,38). It
has been reported that the expression of p53 is elevated in injured
neurons in acute injury models, including ischemia and epilepsy,
and in brain tissue samples extracted from patients with chronic
neurodegenerative diseases (39).
The results of the current study demonstrated that OGD/R treatment
markedly increased the expression of p53 in N2a cells, which was
consistent with the results of the aforementioned study.
A novel transcription-independent pro-apoptotic
function mediated by p53 was identified in a previous study
(40). Additionally, it has been
demonstrated that p53 is involved in the intrinsic apoptosis
signaling pathway by interacting with the multi-domain members of
the BCL-2 protein family to maintain mitochondrial function
(41). Previous studies indicated
that certain forms of neuronal injury invoke a common signaling
pathway involving signal transduction via p53, Bax, cytochrome
c release, mitochondrial dysfunction and caspase-3
activation (42–44). Furthermore, the results of various
studies support the theory that the activation of p53 signaling,
which precedes the release of pro-apoptotic proteins from
mitochondria, may induce apoptosis in ischemic neurons (27,37).
However, the upstream events that contribute to p53 signaling and
neuronal death remain unclear. The current study demonstrated that
upregulation of p53 expression in N2a cells following OGD/R was
accompanied by an upregulation of Bax expression and downregulation
of Bcl-2 expression. In addition, the transfection of a plasmid DNA
encoding wt-p53 to increase p53 expression decreased the viability
of N2a cells and increased leakage of LDH in co-treated N2a cells
compared with cells that underwent OGD/R and transfection with
miR-29b mimics. Taken together, these results indicate that
p53-mediated apoptosis contributes to OGR/D-induced injury in N2a
cells.
In conclusion, the present study demonstrated that
miR-29b expression was downregulated following OGD/R. The miR-29b
mimics alleviated OGD/R-induced cytotoxicity and apoptosis, whereas
miR-29b inhibitors enhanced OGD/R-induced cytotoxicity, indicating
that miR-29b protects N2a cells against OGD/R injury. In addition,
OGD/R increased the expression of p53 protein and the upregulation
of p53 inhibited the beneficial effects of miR-29b on OGD/R injury,
suggesting that p53 mediates the protective effect of miR-29b
against OGD/R-induced injury in N2a cells. The results of the
present study demonstrate that miR-29b protects against OGD/R
insult and that its mechanism of action is dependent on
p53-mediated apoptosis. The results of the current study may pave
the way for the potential clinical application of miR-29b.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81460276).
References
|
1
|
Jeon JH, Jung HW, Jang HM, Moon JH, Park
KT, Lee HC, Lim HY, Sur JH, Kang BT, Ha J and Jung DI: Canine model
of ischemic stroke with permanent middle cerebral artery occlusion:
Clinical features, magnetic resonance imaging, histopathology, and
immunohistochemistry. J Vet Sci. 16:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shope SR and Schiemann DA: Passive
secretory immunity against Salmonella typhimurium demonstrated with
foster mouse pups. J Med Microbiol. 35:53–59. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anrather J and Iadecola C: Inflammation
and stroke: An overview. Neurotherapeutics. 13:661–670. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang R, Xu M, Wang Y, Xie F, Zhang G and
Qin X: Nrf2-a promising therapeutic target for defensing against
oxidative stress in stroke. Mol Neurobiol. Sep 30–2016.(Epub ahead
of print).
|
|
5
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: Two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller FD, Pozniak CD and Walsh GS:
Neuronal life and death: An essential role for the p53 family. Cell
Death Differ. 7:880–888. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sax JK and El-Deiry WS: p53 downstream
targets and chemosensitivity. Cell Death Differ. 10:413–417. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andrabi SA, Kang HC, Haince JF, Lee YI,
Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM and
Dawson VL: Iduna protects the brain from glutamate excitotoxicity
and stroke by interfering with poly(ADP-ribose) polymer-induced
cell death. Nat Med. 17:692–699. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tu W, Xu X, Peng L, Zhong X, Zhang W,
Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, et al: DAPK1
interaction with NMDA receptor NR2B subunits mediates brain damage
in stroke. Cell. 140:222–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Culmsee C, Zhu C, Landshamer S, Becattini
B, Wagner E, Pellecchia M, Blomgren K and Plesnila N:
Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase
and Bid mediates neuronal cell death after oxygen-glucose
deprivation and focal cerebral ischemia. J Neurosci.
25:10262–10272. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ouyang YB, Stary CM, Yang GY and Giffard
R: microRNAs: Innovative targets for cerebral ischemia and stroke.
Curr Drug Targets. 14:90–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo Y, Luo F, Liu Q and Xu D: Regulatory
non-coding RNAs in acute myocardial infarction. J Cell Mol Med.
21:1013–1023. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujii S, Sugiura T, Dohi Y and Ohte N:
MicroRNA in atherothromobosis: Is it useful as a disease marker?
Thromb J. 14 Suppl 1:S212016. View Article : Google Scholar
|
|
15
|
Zhu K, Liu D, Lai H, Li J and Wang C:
Developing miRNA therapeutics for cardiac repair in ischemic heart
disease. J Thorac Dis. 8:E918–E927. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ouyang YB and Giffard RG: MicroRNAs
regulate the chaperone network in cerebral ischemia. Transl Stroke
Res. 4:693–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin KJ, Deng Z, Huang H, Hamblin M, Xie C,
Zhang J and Chen YE: miR-497 regulates neuronal death in mouse
brain after transient focal cerebral ischemia. Neurobiol Dis.
38:17–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu DZ, Tian Y, Ander BP, Xu H, Stamova
BS, Zhan X, Turner RJ, Jickling G and Sharp FR: Brain and blood
microRNA expression profiling of ischemic stroke, intracerebral
hemorrhage, and kainate seizures. J Cereb Blood Flow Metab.
30:92–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan KS, Armugam A, Sepramaniam S, Lim KY,
Setyowati KD, Wang CW and Jeyaseelan K: Expression profile of
microRNAs in young stroke patients. PLoS One. 4:e76892009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khanna S, Rink C, Ghoorkhanian R, Gnyawali
S, Heigel M, Wijesinghe DS, Chalfant CE, Chan YC, Banerjee J, Huang
Y, et al: Loss of miR-29b following acute ischemic stroke
contributes to neural cell death and infarct size. J Cereb Blood
Flow Metab. 33:1197–1206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ouyang YB, Xu L, Lu Y, Sun X, Yue S, Xiong
XX and Giffard RG: Astrocyte-enriched miR-29a targets PUMA and
reduces neuronal vulnerability to forebrain ischemia. Glia.
61:1784–1794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pekarsky Y and Croce CM: Is miR-29 an
oncogene or tumor suppressor in CLL? Oncotarget. 1:224–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kole AJ, Swahari V, Hammond SM and
Deshmukh M: miR-29b is activated during neuronal maturation and
targets BH3-only genes to restrict apoptosis. Genes Dev.
25:125–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ye Y, Perez-Polo JR, Qian J and Birnbaum
Y: The role of microRNA in modulating myocardial
ischemia-reperfusion injury. Physiol Genomics. 43:534–542. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Zhang X, Li Y, Han S, Howells DW,
Li S and Li J: Conventional protein kinase Cβ-mediated
phosphorylation inhibits collapsin response-mediated protein 2
proteolysis and alleviates ischemic injury in cultured cortical
neurons and ischemic stroke-induced mice. J Neurochem. 137:446–459.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schuler M and Green DR: Mechanisms of
p53-dependent apoptosis. Biochem Soc Trans. 29:684–688. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen Y and White E: p53-dependent
apoptosis pathways. Adv Cancer Res. 82:55–84. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bano S, Chaudhary V and Garga UC: Neonatal
hypoxic-ischemic encephalopathy: A radiological review. J Pediatr
Neurosci. 12:1–6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li L and Stary CM: Targeting glial
mitochondrial function for protection from cerebral ischemia:
Relevance, mechanisms, and the role of MicroRNAs. Oxid Med Cell
Longev. 2016:60323062016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ouyang YB, Xu L, Yue S, Liu S and Giffard
RG: Neuroprotection by astrocytes in brain ischemia: Importance of
microRNAs. Neurosci Lett. 565:53–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y,
Shi J and Jia L: Upregulated miR-29b promotes neuronal cell death
by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res.
216:225–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Annis RP, Swahari V, Nakamura A, Xie AX,
Hammond SM and Deshmukh M: Mature neurons dynamically restrict
apoptosis via redundant premitochondrial brakes. FEBS J.
283:4569–4582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Adams JM and Cory S: Bcl-2-regulated
apoptosis: Mechanism and therapeutic potential. Curr Opin Immunol.
19:488–496. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao H, Yenari MA, Cheng D, Sapolsky RM
and Steinberg GK: Bcl-2 overexpression protects against neuron loss
within the ischemic margin following experimental stroke and
inhibits cytochrome c translocation and caspase-3 activity. J
Neurochem. 85:1026–1036. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu L, Lee JE and Giffard RG:
Overexpression of bcl-2, bcl-XL or hsp70 in murine cortical
astrocytes reduces injury of co-cultured neurons. Neurosci Lett.
277:193–197. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Laptenko O and Prives C: Transcriptional
regulation by p53: One protein, many possibilities. Cell Death
Differ. 13:951–961. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Endo H, Kamada H, Nito C, Nishi T and Chan
PH: Mitochondrial translocation of p53 mediates release of
cytochrome c and hippocampal CA1 neuronal death after transient
global cerebral ischemia in rats. J Neurosci. 26:7974–7983. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saito A, Hayashi T, Okuno S, Nishi T and
Chan PH: Modulation of p53 degradation via MDM2-mediated
ubiquitylation and the ubiquitin-proteasome system during
reperfusion after stroke: Role of oxidative stress. J Cereb Blood
Flow Metab. 25:267–280. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Morrison RS and Kinoshita Y: The role of
p53 in neuronal cell death. Cell Death Differ. 7:868–879. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vaseva AV and Moll UM: The mitochondrial
p53 pathway. Biochim Biophys Acta. 1787:414–420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chong MJ, Murray MR, Gosink EC, Russell
HR, Srinivasan A, Kapsetaki M, Korsmeyer SJ and McKinnon PJ: Atm
and Bax cooperate in ionizing radiation-induced apoptosis in the
central nervous system. Proc Natl Acad Sci USA. 97:pp. 889–894.
2000; View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cregan SP, MacLaurin JG, Craig CG,
Robertson GS, Nicholson DW, Park DS and Slack RS: Bax-dependent
caspase-3 activation is a key determinant in p53-induced apoptosis
in neurons. J Neurosci. 19:7860–7869. 1999.PubMed/NCBI
|
|
44
|
Xiang H, Kinoshita Y, Knudson CM,
Korsmeyer SJ, Schwartzkroin PA and Morrison RS: Bax involvement in
p53-mediated neuronal cell death. J Neurosci. 18:1363–1373.
1998.PubMed/NCBI
|