Introduction
Pulmonary alveolar microlithiasis (PAM) is a
hereditary lung disease in which calcium phosphate microliths,
termed calcospherites, accumulate in the alveolar spaces (1). Mutations in the solute carrier family
34 member 2 (SLC34A2) gene, which encodes the type IIb
sodium-phosphate cotransporter in alveolar type II cells, are
responsible for the pathogenesis of PAM (2,3). SLC34A2
serves a crucial role in the transportation of phosphate ions from
the alveolar spaces into alveolar type II cells (4). Mutations in the SLC34A2 gene that cause
the dysfunction of alveolar type II cells result in the
accumulation of phosphate and the formation of microliths in the
alveolar spaces (1,4). The majority of patients with PAM are
asymptomatic at the time of diagnosis and the disease is usually
identified incidentally following radiographic examination of the
chest for other purposes (2). The
typical presentation of PAM on a chest X-ray is by a ‘sandstorm’
appearance. In contrast to the radiological findings, which are
often severe, the clinical presentation of PAM is often relatively
mild or absent (5). In symptomatic
patients shortness of breath is the most common symptom, followed
by a dry cough, chest pain, sporadic hemoptysis and asthenia
(1). Given the remarkable
clinico-radiological dissociation, a positive diagnosis should be
considered in a patient presenting with the typical radiological
features of the disease without clinical symptoms, particularly in
the presence of a family history or consanguinity (6). To the best of our knowledge, there is
currently no definitive treatment available to prevent the
progression of PAM, although lung transplantation has been used to
treat patients with end-stage PAM (7) and disodium etidronate has been
administered due to its alleged calcium phosphate
precipitation-reducing effect in PAM; however, its use is
controversial and its effectiveness is disputed (8,9).
In the present study three cases of confirmed PAM
are reported. All patients provided written informed consent. The
epidemiology, cause, clinical features, radiological appearance,
pathology, genetic mutations and treatment strategies for PAM were
all considered.
Case report
Case 1
A 13-year-old female whose parents are first cousins
(sharing 1/8 of their genes) presented to Peking Union Medical
College Hospital, Beijing, China (PUMCH) in April 2016. Diffuse
scattered nodules had been detected on a chest X-ray taken 4 years
previously at her local hospital, however she was not diagnosed
with PAM at that time. At the time of presentation the patient's
physical examination was normal and they had no clinical complaints
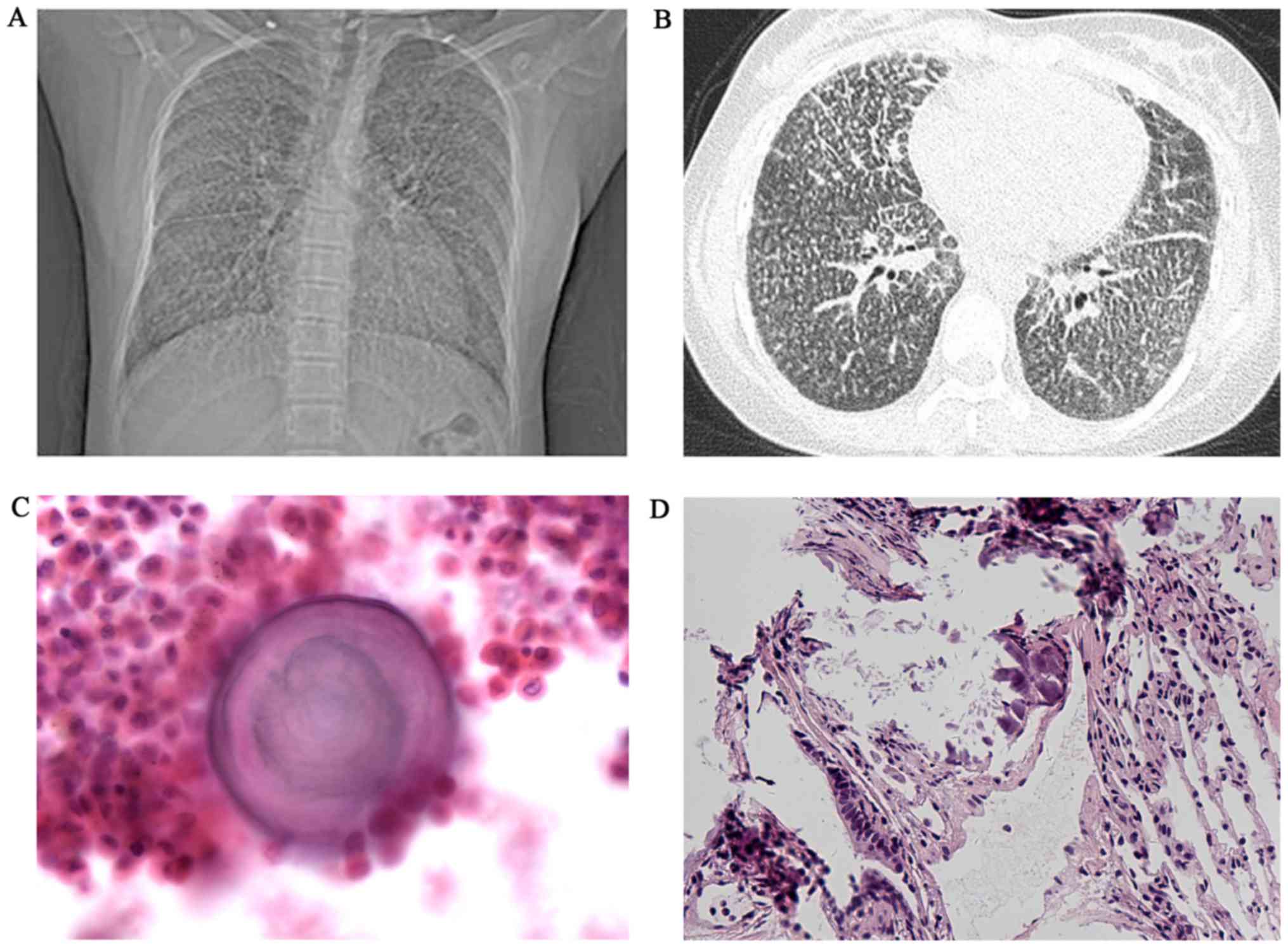
except exertional dyspnea. A chest X-ray revealed a bilaterally
diffuse, fine sand-like micronodular appearance predominantly in
the lower zones; high resolution computed tomography (HRCT)
revealed widespread micro-calcifications throughout the lungs and
pericardium with ground-glass opacities, interlobular septal
thickening and pleural calcification (Fig. 1A and B). Broncho alveolar lavage
fluid (BALF) was performed by squirting three 50 ml samples of
sterile saline into the right middle lobe of the lung and then
immediately collecting it for testing. The sample was subsequently
centrifuged at 250 × g for 5 min at 4°C. The supernatant was
discarded and the sediments were prepared into smears and then
fixed in 95% alcohol (Beijing Jiu Zhou Bai Lin Biological
Technology Co., Ltd., Beijing, China) at room temperature (20–25°C)
for 10 min. The smears were stained with hematoxylin for 1 min and
eosin for 20 sec at room temperature (20–25°C). Images were
captured under an Eclipse 80i microscope (magnification, ×400;
Nikon Corporation, Tokyo, Japan). The quantities and
classifications of inflammatory cells in the BALF sample were
normal, but a concentrically laminated calcified body was
identified (data not shown). Lung biopsy specimens were fixed in
10% neutral-buffered formalin at room temperature (20–25°C; Beijing
Jiu Zhou Bai Lin Biological Technology Co., Ltd.) overnight, cut
into slices and dehydrated through a graded alcohol series, cleaned
with dimethylbenzene and embedded in paraffin (Beijing Jiu Zhou Bai
Lin Biological Technology Co., Ltd.). The samples were then cut
into 4-µm-thick sections. The sections were stained with
hematoxylin and eosin for 10 min at room temperature (20–25°C) and
images were captured under a microscope (magnification, ×100). A
lung biopsy revealed numerous calcified bodies in the alveolar
spaces (Fig. 1C and D). A
99mTechnetium-methylene diphosphonate (Tc-MDP) bone scan
revealed increased in take of radiation in the lungs (data not
shown). A pulmonary function test (PFT), a SLC34A2 gene test, an
oxygen saturation (SO2) test and a tuberculin skin test
were all normal (data not shown). Routine blood biochemistry tests,
including serum calcium and phosphorus concentrations, and hepatic,
renal and parathyroid functions were also normal (data not shown).
Arterial blood gas (ABG) measurement and echocardiography were not
performed due to refused consent by the patient. The patient was
born naturally with no asphyxia at birth or dysgnosia during
growth. However, the patient's height and weight were notably lower
than the national average for Chinese children (10). Based on the typical appearance on
HRCT and the calcified bodies identified in the BALF and alveolar
spaces, the patient was diagnosed with PMA. However, as there were
no serious clinical symptoms at the time of hospital admittance,
the patient did not accept any treatment. At present no follow up
has occurred.
Case 2
A 31-year-old female from a non-consanguineous
family presented to PUMCH in April 2015 due to an abnormal
appearance on a chest X-ray observed during a medical examination 1
year prior. The patient did not present with any respiratory
symptoms such as dyspnea on exertion or a cough. Physical
examination revealed decreased pulmonary sounds in the lower zones
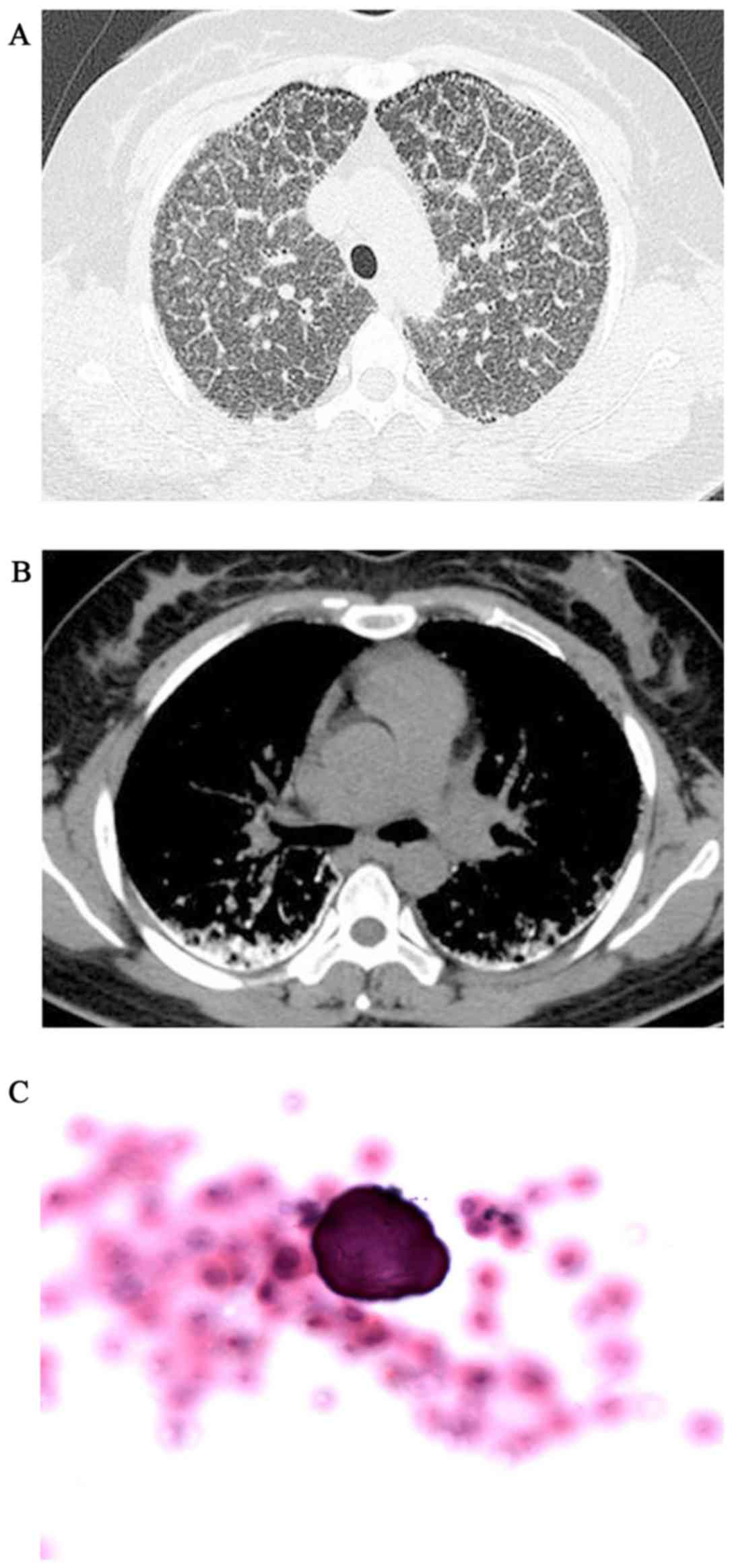
and P2>A2 was heard during heart auscultation. The HRCT revealed
diffuse, scattered, hyperdense micronodules throughout the whole
lungs, accompanied by interlobular septal and bilateral pleural
calcific thickening, subpleural cysts between the ribs and
calcified parenchyma, referred to as the ‘black pleura’ sign
(Fig. 2A and B). The BAFL
examination (performed as above) revealed that the classification
of inflammatory cells was normal and the ratio of cluster of
differentiation (CD)4+/CD8+ T cells was 0.9
(reference range, 0.9–2.0), but a calcified body was detected
(Fig. 2C). ABG measured pH 7.378,
PaCO2 37.0 mmHg, PaO2 88.1 mmHg,
SO2 96.4% and HCO3 22 mmol/l of room air
(data not shown). Lung tissue pathology and SLC34A2 gene testing
were lacking as a positive diagnosis of PMA had been reached based
on the tests performed making invasive surgery unnecessary Gene
testing was not performed on this patient due to economic
constraints. A 99mTc-MDP bone scan revealed a diffusely
increased intake of radiation in the lungs. An echocardiograph
revealed mild enlargement of the right ventricle (anteroposterior
diameter, 32 mm), with mild pulmonary hypertension (pulmonary
systolic pressure, 37 mmHg) (data not shown). PFT and routine blood
biochemistry were normal (data not shown). In the patient's local
hospital they were misdiagnosed with pulmonary tuberculosis (TB)
due to the appearance of miliary nodules on HRCT imaging. Following
a positive diagnosis for PAM no medication was prescribed. At the 3
year follow-up the clinical course of the patient's disease
remained stable.
Case 3
A 35-year-old male whose parents were consanguineous
presented at PUMCH in November 2014 with complaints of a persistent
cough, sputum and occasional chest pain for the last 10 years, and
breathlessness for the last 3 weeks. A physical examination
revealed bibasilar crackles (velcro rales), cyanosis of the lips
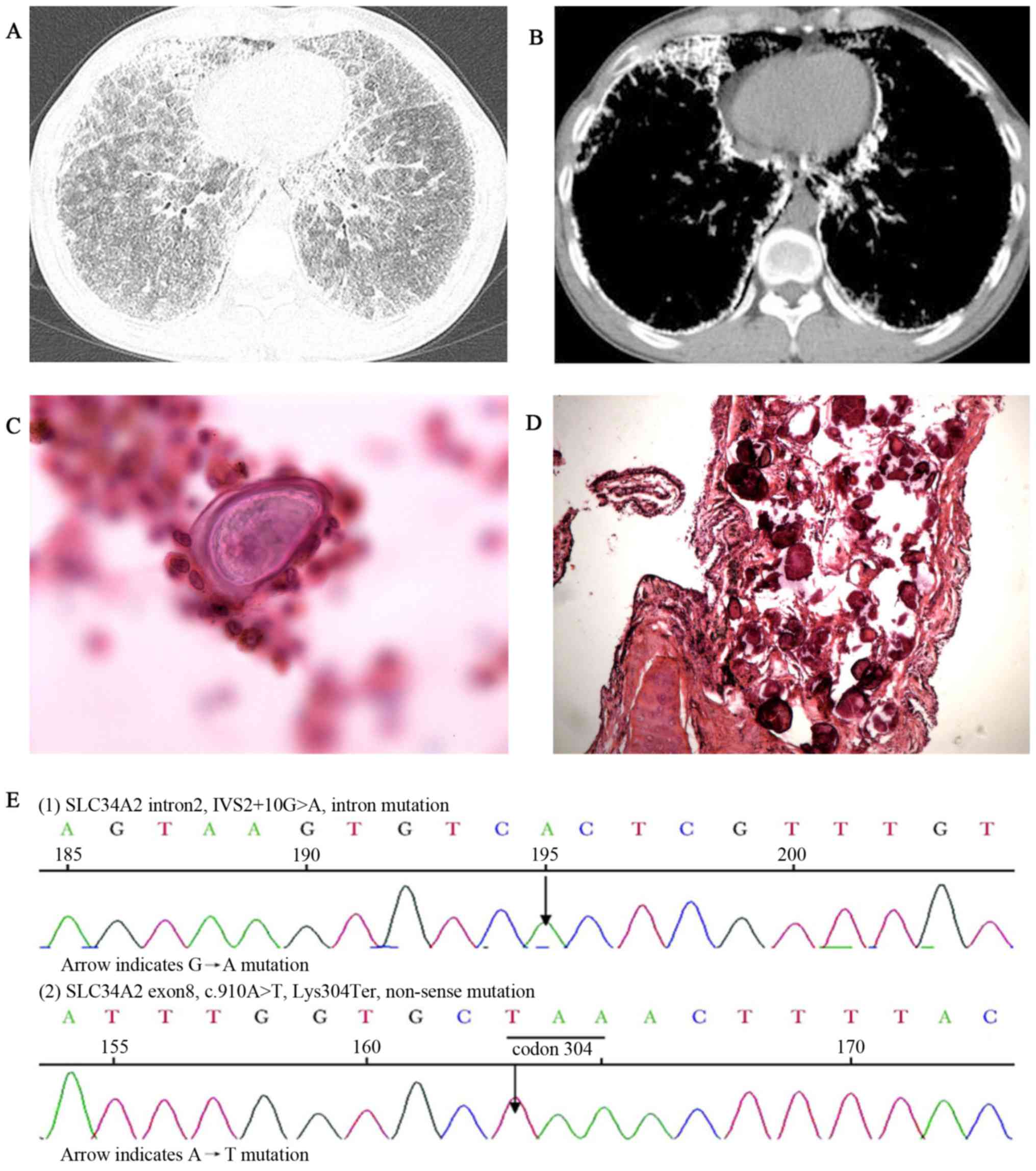
and clubbing of the fingers (data not shown). HRCT imaging revealed
scattered miliary nodules with calcifications, ground-glass
opacities, the ‘black pleura’ sign and pleural calcific thickening
(Fig. 3A and B). BALF examination
(performed as above) detected a calcified body and the ratio of
CD4+/CD8+ T cells was 0.5 (reference range,
0.9–2.0) (data not shown). A biopsy revealed numerous calcified
bodies filling the alveolar spaces of the lungs (Fig. 3C and D). ABG measured pH 7.409,
PaCO2 39.0 mmHg, PaO2 73.1 mmHg,
SO2 95.2% and HCO3 24.5 mmol/l of room air
(data not shown). A PFT revealed defects in the patient's diffusion
capacity and a reduced lung transfer factor for carbon monoxide of
74.5% of the predicted values (normal, ≥80% predicted value), while
ventilatory function was normal (data not shown). Genomic DNA was
extracted from the peripheral blood of the patient using a
GenElute™ Blood Genomic DNA kit according to the manufacturer's
protocol (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Using
Primer3 software version 4.0.0 (primer3.ut.ee/), 12 pairs of
primers were designed (Table I) to
amplify the coding exons of the SLC34A2 gene, as well as the
intronic flanking sequences. Polymerase chain reaction was used to
amplify the DNA according to a previously described method
(11). Direct sequencing was
performed by an ABI 3100 Genetic analyzer (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The obtained
sequence variants were tested for pathogenicity using Polyphen2
(12) and MutationTaster2 (13). This genetic testing identified an
intervening sequence 2+10G>A mutation in intron 2 and
a heterozygous c.910A>T mutation in exon 8 of the SLE34A2 gene
(Fig. 3E). Routine blood
biochemistry, an echocardiograph and a tuberculin test were all
normal (data not shown). The patient intermittently,
self-administered dextromethorphan, licorice root and ambroxol at
undefined doses and frequencies without a marked effect. At the
2-year follow-up, the patient's dyspnea had advanced with their ABG
measuring PaO2 69.3 mmHg and SO2 94.5%.
 | Table I.Primer sequences for polymerase chain
reaction amplification. |
Table I.
Primer sequences for polymerase chain
reaction amplification.
|
| Primer sequence |
|
|---|
|
|
|
|
|---|
| Primer name | Forward | Reverse | Length of amplifie
product (bp) |
|---|
| SLC34A2-1 |
CCGTCGGAGCTTTTCTCTCGG |
GTCGATCGTAAGAGTGTAGCAGC | 477 |
| SLC34A2-2-3 |
GTTGATGCTTTGCAACCAATGG |
TGATGACACCCACAGTGAACG | 500 |
| SLC34A2-4 |
GCTCATTGCCAAACTTCTCAGG |
GCTGGAGAGGGCTTGCTGA | 300 |
| SLC34A2-5 |
GGCCTTGGATGGAGACTTCTG |
TCCCACCCTCAGATAGACAGG | 302 |
| SLC34A2-6 |
GGTAACTTTAGCCTGCCTCCAG |
GCATGTCATCTTTGGCTGGTT | 270 |
| SLC34A2-7 |
GAGGGTGGCAGATGATACAGG |
TGTCAGCTCAGGTAGGGGATG | 357 |
| SLC34A2-8 |
CCCTGGGTTTGTGTCCTAAATC |
CTTCCTTGAAGGCAAGATTTAGTT | 243 |
| SLC34A2-9 |
CATTGCCTCCCATTCCCCACT |
AATAGGTCACCCCCAGACAAC | 241 |
| SLC34A2-10 |
TAACAATCTGTAGCCGTGGTGG |
GAATCTAAAGGACCCCCACAC | 300 |
| SLC34A2-11-12 |
TGTACAACCTCACCCCTAAGCC |
AGAGACCAGTTTGCAAGACCATG | 499 |
| SLC34A2-13-1 |
TGTGATGCCTGCTAGCTTACCT |
CAGCAGCGCATCTGGAAGCAG | 492 |
| SLC34A2-13-2 |
CTGCCGAAGAAACTCCAGAACT |
CCAAAGGGAATCGAGTTAGGTAG | 481 |
Discussion
PAM is a rare genetic lung disease characterized by
widespread sand-like intra-alveolar calcifications (calcospherites)
composed of calcium and phosphorus (5), and clinical-radiological dissociation
is a hallmark of this disease (14).
PAM is present globally and there have been >1,000 cases
reported to date (3). PAM exhibits
no clear sex predilection (2). The
majority of patients with PAM have been from Asia, followed by
Europe (3). The nation with the
highest number of reported cases relative to the population is
Turkey, followed by China, Japan, India, Italy and the USA
(3). Patients with PAM typically
present during the third and fourth decades of life; however, cases
have been reported in neonates and octogenarians (14). Familial incidence of PAM has been
reported in 35–50% of cases in Japan, Turkey and Italy (2).
PAM is considered to be a genetic disease due to the
inactivating mutations within the SLC34A2 gene identified in
patients. This gene is located on chromosome 4p15 and consists of
13 exons (2). SLC34A2 is primarily
expressed in alveolar type II cells (4), but it is also expressed in other
epithelial tissues, including mammary glands, the small intestine,
kidneys, pancreas, ovaries, liver, testes, placenta and prostate
(14). SLC34A2 is the only known
sodium-dependent phosphate transporter expressed in the lungs, and
it serves a key role in clearing phospholipids from alveolar spaces
by transporting phosphate ions into alveolar type II cells
(4). The wild-type transporter
passages phosphate into the alveolar type II cells in the presence
of Na, with a stoichiometry of
3Na+1:1HPO4−2, whereas mutants do
not (1). Impaired function or
deficiency due to mutations in the SLC34A2 gene leads to a
decreased alveolar type II cell uptake of phosphate. This in turn
may lead to the formation of intra-alveolar microliths as a result
of phosphate-chelating calcium in the extracellular fluid (15). Mutations in the SLC34A2 gene have
been detected in almost all tested patients with PAM (2). Mutations have been identified on
multiple exons in patients from Turkey, but appear to cluster on
exon 8 in cases from China and in exons 7 and 8 in patients from
Japan (2). In case 3 of the present
study, a heterozygous c.910A>T mutation in exon 8 was detected,
which has been reported in previous studies (11,16). PAM
has been reported to occur among siblings and cousins in a
horizontal pattern, and less frequently between parents and
children in a vertical pattern (1).
In the cases being reviewed in the present study, the parents of
two of the patients were consanguineous.
Upon macroscopic examination, lungs affected by PAM
are enlarged, heavy and nonbouyant (2). Sectioning of the lungs often reveals a
diffusely calcified, gritty, pleural surface with a studded, fine
and granular appearance (2). In the
cases reported in the present study, lung biopsies demonstrated
characteristic intra-alveolar lamellar microliths, which were
extensively composed of Ca3
(PO4)2; however, small amounts of
CaCo3, Fe, Zn, Al, SiO2 and Mg are also
encountered frequently in PAM microliths (17). Histologically, PAM microliths are
periodic acid-Schiff stain positive (18) and consist of calcareous concentric
lamellae around a central nucleus with an amorphous or granular
aspect (14). In case 2, the patient
did not undergo a lung biopsy; however, the calcified body
identified in the BALF was consistent with the pathological
diagnosis of PAM. The identification of calcified bodies in BALF
may therefore present an alternative method for the histological
diagnosis of PAM.
PAM has a relatively indolent although
non-modifiable disease progression, and there is often a long
period prior to the presentation of any clinical symptoms. As PAM
progresses patients commonly develop dyspnea on exertion, other
symptoms, including cough, chest pain, hemoptysis, asthenia and
pneumothorax, have also been reported. Cyanosis and clubbing of the
fingers may be identified in serious cases. A typical clinical
manifestation of the disease was observed in case 1, where the
patient only complained of exertional dyspnea. The patient in case
3, who had been suffering from the disease for a longer period of
time and exhibited notable radiographic changes, also suffered from
symptoms associated with long-term PAM. The observation of clear
cyanosis, clubbing and crackles in this patient indicated the
severity of the disease. During the normal progression of PAM
microliths may form in early childhood; however, pulmonary function
tests are often normal in the early phase. When large volumes of
the lung are occupied by microliths it can cause restrictive
ventilatory impairment and lead to deterioration of the perfusion
capacity over time. This results in hypoxemia, increased arterial
CO2 levels (1), pulmonary
fibrosis, respiratory failure and ultimately cor pulmonale
(19). In case 2, the patient had
mild enlargement of the right ventricle and pulmonary hypertension,
these symptoms are indicative of a long-term chronic disease and
potential failure of the right side of the heart. However, the
cases of two patients with PAM reported in a previous study suggest
that the disease can be acquired later in life, as their chest
radiographs were clinically normal 2 years prior to diagnosis
(3). In addition, rapid progression
of the disease has also been reported (14).
Routine blood tests, including serum phosphate and
calcium, are usually normal in patients with PAM (20). Serum monocyte chemotactic protein-1
(21), surfactant protein (SP)-A and
SP-D (22) have been revealed to be
elevated in certain patients with PAM. These proteins may
potentially serve as diagnostic biomarkers or be used to indicate
the activity and progression of PAM.
Extrapulmonary calcifications in the male
reproductive system due to PAM can result in testicular atrophy
(2), obstructive azoospermia
(23) and tumors (24) Calcification of the medullary
nephrocalcinosis, nephrolithiasis (25), lumbar sympathetic chain (1), cholelithiasis (26) and cardiac involvements (27) have also been reported in patients
with PAM. Microlith deposits outside of the lungs are possibly
associated with the penetrance of mutations of the SLC34A2 gene
(20). In the present study the
three cases did not reveal recognized microlith deposits in other
locations.
The typical features of PAM on a plain chest
radiographs are a fine, scattered micronodular pattern, producing a
‘sandstorm’ appearance. Diffuse involvement of both lungs is
generally more evident in the middle and lower zones (28). The borders of the heart, diaphragm
and vascular tree may be obscured as a result of the dense
calcifications (2).
Findings on HRCT can be divided into four stages
according to the degree of radiologic severity (29). The first phase, considered as
pre-calcific, is without a typical appearance due to the small
number and lesser size of the calcifications of the microliths
(3). The second phase exhibits the
characteristic features of a ‘sandstorm’ appearance with diffusely
scattered calcific micronodules (<1 mm diameter). The microliths
tend to distribute throughout the lungs, a greater concentration is
usually observed in the medial and inferior regions, although the
border of the heart and diaphragm may still be outlined clearly
(3). In childhood or adolescent
patients these nodules may be larger (~2–4 mm). The third phase
reveals a greater number and volume of opacifications that often
lead to the outline of the heart and diaphragm being obscured, as
well as interstitial thickening (3).
Ground-glass opacity with thickening interlobular septa can
sometimes produce the so-called ‘crazy-paving’ sign (30). The fourth stage is characterized by a
notable advance in the number and size of calcific deposits,
resulting in intense calcification of the interstitium and pleural
serosa, which gives the overall appearance of ‘white lungs’
(3). The first stage is usually
observed in children, the second in adolescence, and the last two
stages in the later years of life (3).
A positive diagnosis of PAM can usually be
established by observation of a typical sandstorm appearance on
chest radiological examinations. A lung biopsy identifying
intra-alveolar microliths with a concentric appearance are required
in doubtable cases (17). In certain
cases microliths can be identified in the BALF or sputum (31), suggesting that this may be a
potential alternative to a lung biopsy as the diagnostic procedure
for PAM. In case 2, the diagnosis of PAM was established by a
combination of radiological observations and BALF, which is
consistent with a previous study (32). Testing for a mutation in the SLC34A2
gene may assist when screening family members of the proband
patient, but it is not necessary for the diagnosis of PAM.
When dense micronodular and ground-glass opacities
are observed in the radiological images, miliary TB, sarcoidosis,
pneumoconiosis, pulmonary alveolar proteinosis, pulmonary
hemosiderosis and amyloidosis should all be considered as a
potential alternative diagnosis to PAM. As in case 2, PAM is often
misdiagnosed as TB, particularly in regions where consanguineous
marriage is common and TB is highly prevalent (2). Metastatic and dystrophic pulmonary
calcifications may be distinguished by a history of chronic renal
failure or pathogenic infections (2). The histological appearance of
calcareous concentric lamellae around a central nucleus with an
amorphous or granular aspect can be also used for distinguishing
PAM from metastatic and dystrophic pulmonary calcifications.
To the best of our knowledge, there are currently no
guidelines for the treatment of PAM. Systemic steroids,
calcium-chelating agents and bronchopulmonary lavage have
demonstrated an ineffectiveness at reducing disease progression in
previous studies (3,11). Disodium etidronate has previously
been administered due to its possible ability to inhibit
Ca3(PO4)2 precipitation and
resolve formed calcifications (8);
certain previous studies have demonstrated its ability to improve
lung function and radiographic appearances (8,33).
However, other reports have revealed that this treatment is not
effective (34,35). Lung transplantation can be performed
in the end stages of PAM and has previously led to the improvement
of right ventricular function (7).
The survival rate and recurrence risk following lung
transplantation requires additional study; however, to the best of
our knowledge, no recurrence of PAM following lung transplantation
has been reported to date (3).
The present study described three cases of PAM, a
rare genetic disease caused by mutations in the SLC34A2 gene. A
brief overview of the disease at different states of progression
was also given. PAM reveals a significant clinical-radiological
dissociation, meaning that there can be a mild clinical
presentation alongside severe radiological changes. Therefore, a
diagnosis can be made on the basis of typical radiological features
without substantial clinical presentation. The prognosis for
individuals diagnosed with PAM is poor due to cor pulmonale and
respiratory failure in the later stages of the disease. Lung
transplantation currently remains the only effective treatment for
this disease.
References
|
1
|
Ferreira Francisco FA, Pereira e Silva JL,
Hochhegger B, Zanetti G and Marchiori E: Pulmonary alveolar
microlithiasis. State-of-the-art review. Respir Med. 107:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saito A and McCormack FX: Pulmonary
Alveolar Microlithiasis. Clin Chest Med. 37:441–448. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Castellana G, Castellana G, Gentile M,
Castellana R and Resta O: Pulmonary alveolar microlithiasis: Review
of the 1022 cases reported worldwide. Eur Respir Rev. 24:607–620.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Izumi H, Kurai J, Kodani M, Watanabe M,
Yamamoto A, Nanba E, Adachi K, Igishi T and Shimizu E: A novel
SLC34A2 mutation in a patient with pulmonary alveolar
microlithiasis. Hum Genome Var. 4:160472017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mahmood K, Ubaid M and Mahmood A:
Pulmonary microlithiasis-A case report. Respir Med Case Rep.
19:112–114. 2016.PubMed/NCBI
|
|
6
|
Qian X, Wu X and Liu X: Pulmonary alveolar
microlithiasis with finger clubbing: A case report and literature
review. Exp Ther Med. 11:1381–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Samano MN, Waisberg DR, Canzian M, Campos
SV, Pêgo-Fernandes PM and Jatene FB: Lung transplantation for
pulmonary alveolar microlithiasis: A case report. Clinics (Sao
Paulo). 65:233–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ozcelik U, Yalcin E, Ariyurek M, Ersoz DD,
Cinel G, Gulhan B and Kiper N: Long-term results of disodium
etidronate treatment in pulmonary alveolar microlithiasis. Pediatr
Pulmonol. 45:514–517. 2010.PubMed/NCBI
|
|
9
|
Cakir E, Gedik AH, Özdemir A,
Buyukpinarbasili N, Bilgin M and Ozgen IT: Response to disodium
etidronate treatment in three siblings with pulmonary alveolar
microlithiasis. Respiration. 89:583–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Ji CY, Zong XN and Zhang YQ: Height
and weight standardized growth charts for Chinese children and
adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi. 47:487–492.
2009.(In Chinese). PubMed/NCBI
|
|
11
|
Yin X, Wang H, Wu D, Zhao G, Shao J and
Dai Y: SLC34A2 Gene mutation of pulmonary alveolar microlithiasis:
Report of four cases and review of literatures. Respir Med.
107:217–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chu A, Shaharyar S, Chokshi B and Bhardwaj
N: Pulmonary alveolar microlithiasis ‘stone lungs’: A case of
clinico-radiological dissociation. Cureus. 8:e7492016.PubMed/NCBI
|
|
15
|
Field JA, Zhang L, Brun KA, Brooks DP and
Edwards RM: Cloning and functional characterization of a
sodium-dependent phosphate transporter expressed in human lung and
small intestine. Biochem Biophys Res Commun. 258:578–582. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Yin X, Wu D and Jiang X: SLC34A2
gene compound heterozygous mutation identification in a patient
with pulmonary alveolar microlithiasis and computational 3D protein
structure prediction. Meta Gene. 2:557–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mariotta S, Ricci A, Papale M, De Clementi
F, Sposato B, Guidi L and Mannino F: Pulmonary alveolar
microlithiasis: Report on 576 cases published in the literature.
Sarcoidosis Vasc Diffuse Lung Dis. 21:173–181. 2004.PubMed/NCBI
|
|
18
|
Lauta VM: Pulmonary alveolar
microlithiasis: An overview of clinical and pathological features
together with possible therapies. Respir Med. 97:1081–1085. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Terada T: Pulmonary alveolar
microlithiasis with cor pulmonale: An autopsy case demonstrating a
marked decrease in pulmonary vascular beds. Respir Med.
103:1768–1771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tachibana T, Hagiwara K and Johkoh T:
Pulmonary alveolar microlithiasis: Review and management. Curr Opin
Pulm Med. 15:486–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saito A, Nikolaidis NM, Amlal H, Uehara Y,
Gardner JC, LaSance K, Pitstick LB, Bridges JP, Wikenheiser-Brokamp
KA, McGraw DW, et al: Modeling pulmonary alveolar microlithiasis by
epithelial deletion of the Npt2b sodium phosphate cotransporter
reveals putative biomarkers and strategies for treatment. Sci
Transl Med. 7:313ra1812015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takahashi H, Chiba H, Shiratori M,
Tachibana T and Abe S: Elevated serum surfactant protein A and D in
pulmonary alveolar microlithiasis. Respirology. 11:330–333. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanat F, Teke T and Imecik O: Pulmonary
alveolar microlithiasis with epididymal and periurethral
calcifications causing obstructive azospermia. Int J Tuberc Lung
Dis. 8:12752004.PubMed/NCBI
|
|
24
|
Kim B, Winter TC III and Ryu JA:
Testicular microlithiasis: Clinical significance and review of the
literature. Eur Radiol. 13:2567–2576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pant K, Shah A, Mathur RK, Chhabra SK and
Jain SK: Pulmonary alveolar microlithiasis with pleural
calcification and nephrolithiasis. Chest. 98:245–246. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piesiak P, Kasibowska-Kuźniar K and
Jankowska R: Pulmonary alveolar microlithiasis in a patient with
urolithiasis and cholelithiasis. Pneumonol Alergol Pol. 69:285–289.
2001.(In Polish). PubMed/NCBI
|
|
27
|
Jönsson ÅL, Hilberg O, Bendstrup EM,
Mogensen S and Simonsen U: SLC34A2 gene mutation may explain
comorbidity of pulmonary alveolar microlithiasis and aortic valve
sclerosis. Am J Respir Crit Care Med. 185:4642012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan ED, Morales DV, Welsh CH, McDermott
MT and Schwarz MI: Calcium deposition with or without bone
formation in the lung. Am J Respir Crit Care Med. 165:1654–1669.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castellana G, Castellana R, Fanelli C,
Lamorgese V and Florio C: Pulmonary alveolar microlithiasis:
Clinical and radiological course of three cases according to
conventional radiology and HRCT. A hypothesis for radiological
classification. Radiol Med. 106:160–168. 2003.(In English,
Italian).
|
|
30
|
Gasparetto EL, Tazoniero P, Escuissato DL,
Marchiori E, Frare E Silva RL and Sakamoto D: Pulmonary alveolar
microlithiasis presenting with crazy-paving pattern on high
resolution CT. Br J Radiol. 77:974–976. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chatterji R, Gaude GS and Patil PV:
Pulmonary alveolar microlithiasis: Diagnosed by sputum examination
and transbronchial biopsy. Indian J Chest Dis Allied Sci.
39:263–267. 1997.PubMed/NCBI
|
|
32
|
Monabati A, Ghayumi MA and Kumar PV:
Familial pulmonary alveolar microlithiasis diagnosed by
bronchoalveolar lavage. A case report. Acta Cytol. 51:80–82. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ozçelik U, Gülsün M, Göçmen A, Ariyürek M,
Kiper N, Anadol D and Cobanoğlu N: Treatment and follow-up of
pulmonary alveolar microlithiasis with disodium editronate:
Radiological demonstration. Pediatr Radiol. 32:380–383. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mariotta S, Guidi L, Mattia P, Torrelli L,
Pallone G, Pedicelli G and Bisetti A: Pulmonary microlithiasis.
Report of two cases. Respiration. 64:165–169. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jankovic S, Pavlov N, Ivkosic A, Erceg I,
Glavina-Durdov M, Tocilj J, Dragisic-Ivulic S and Primorac D:
Pulmonary alveolar microlithiasis in childhood: Clinical and
radiological follow-up. Pediatr Pulmonol. 34:384–387. 2002.
View Article : Google Scholar : PubMed/NCBI
|