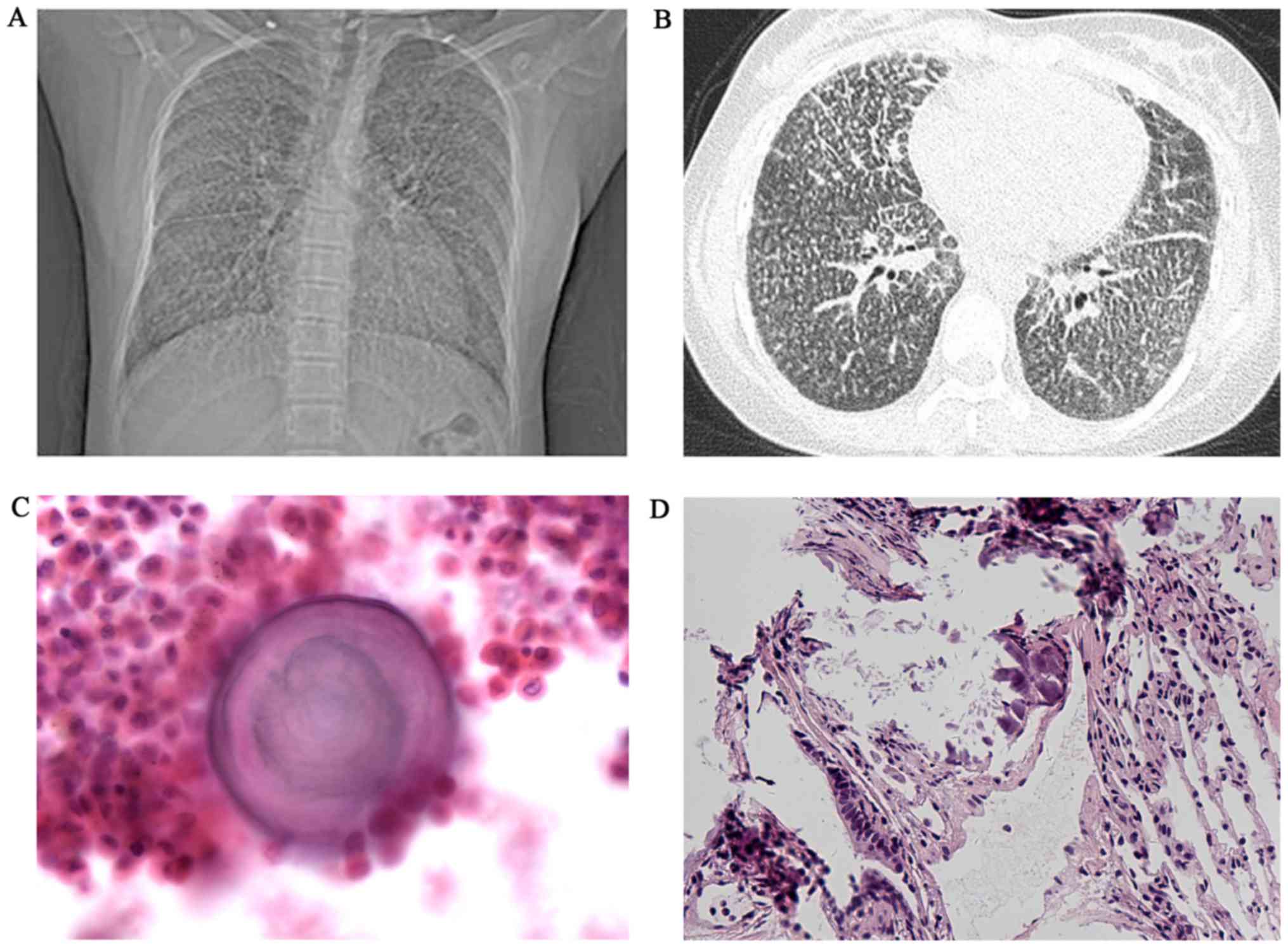
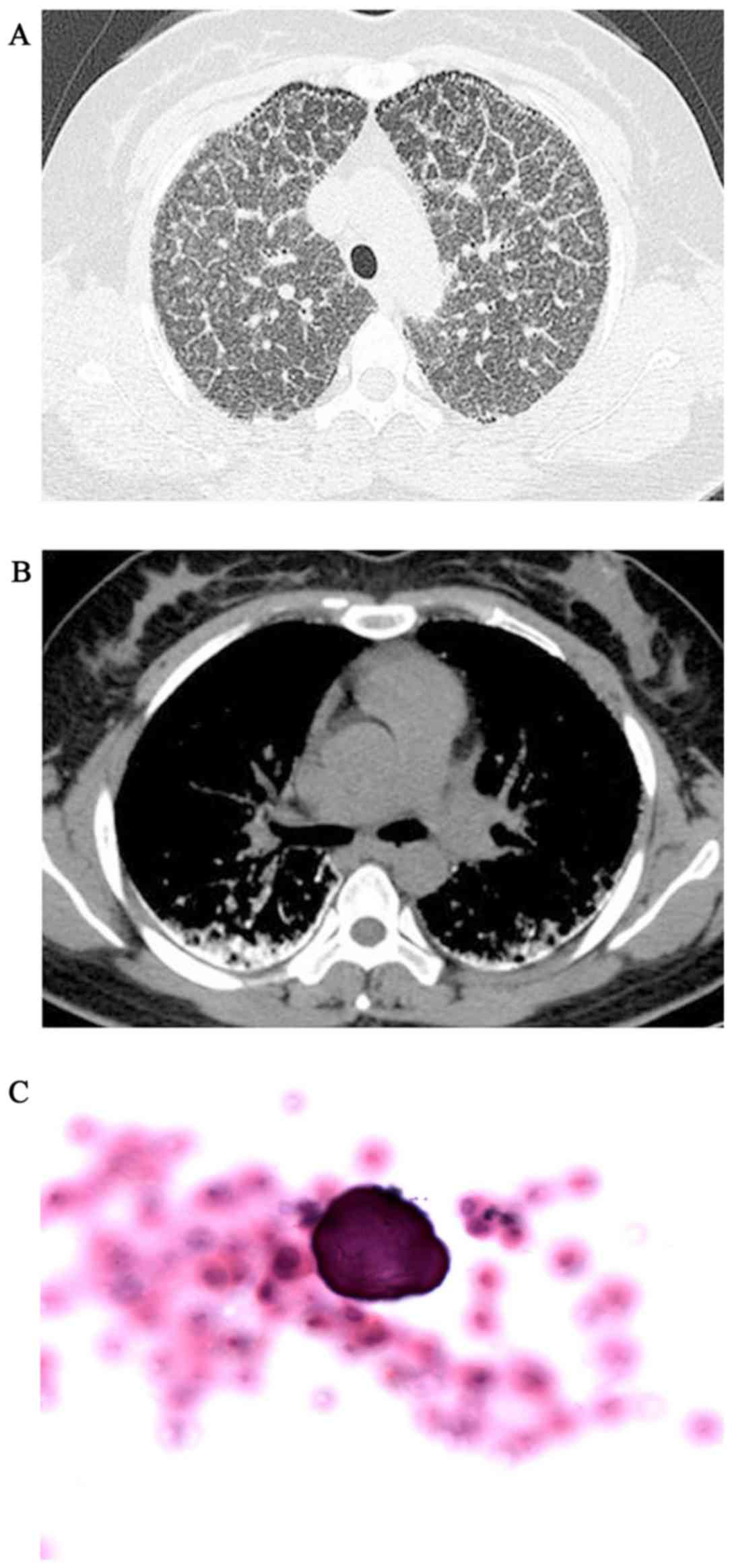
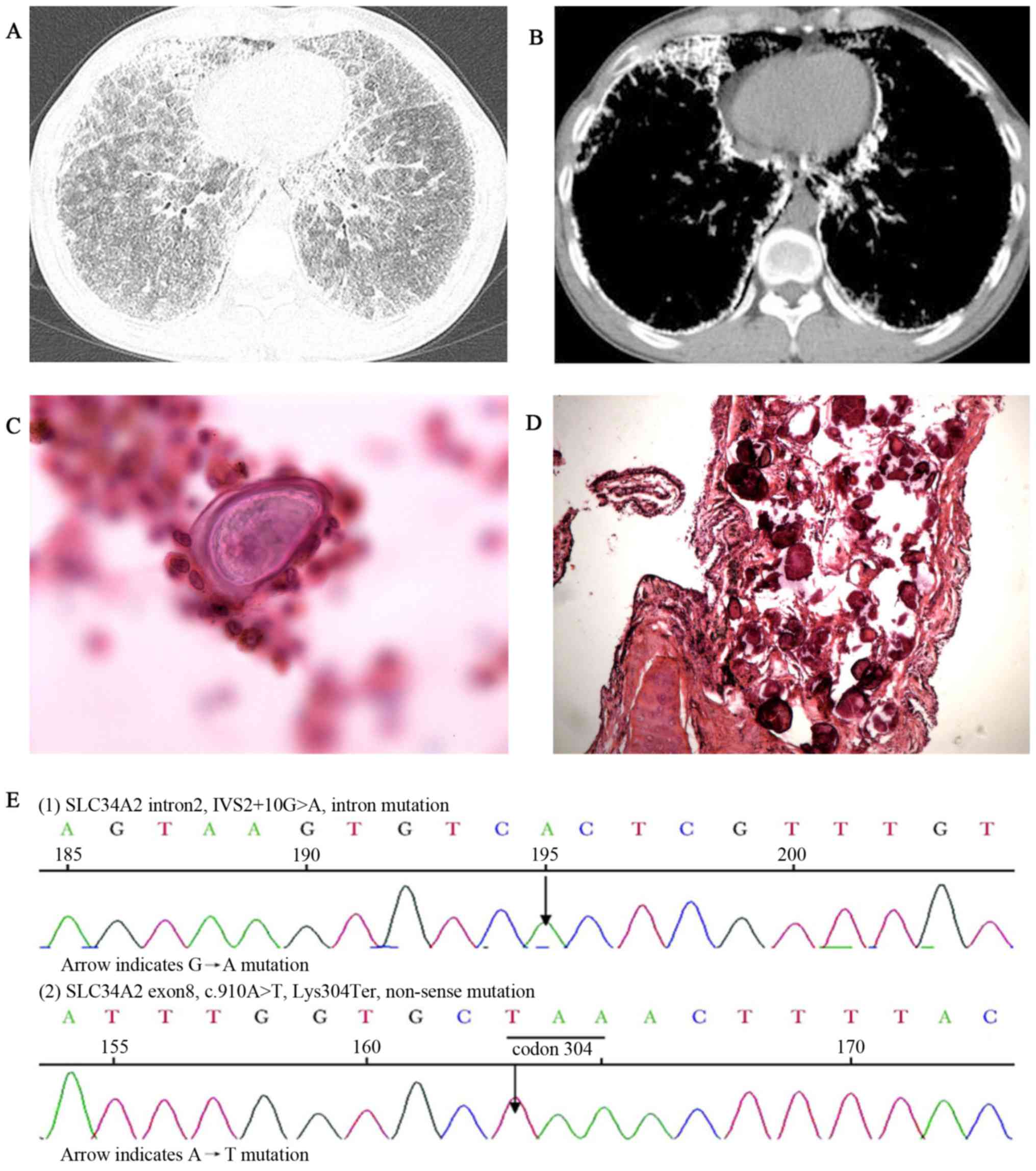
|
1
|
Ferreira Francisco FA, Pereira e Silva JL,
Hochhegger B, Zanetti G and Marchiori E: Pulmonary alveolar
microlithiasis. State-of-the-art review. Respir Med. 107:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saito A and McCormack FX: Pulmonary
Alveolar Microlithiasis. Clin Chest Med. 37:441–448. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Castellana G, Castellana G, Gentile M,
Castellana R and Resta O: Pulmonary alveolar microlithiasis: Review
of the 1022 cases reported worldwide. Eur Respir Rev. 24:607–620.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Izumi H, Kurai J, Kodani M, Watanabe M,
Yamamoto A, Nanba E, Adachi K, Igishi T and Shimizu E: A novel
SLC34A2 mutation in a patient with pulmonary alveolar
microlithiasis. Hum Genome Var. 4:160472017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mahmood K, Ubaid M and Mahmood A:
Pulmonary microlithiasis-A case report. Respir Med Case Rep.
19:112–114. 2016.PubMed/NCBI
|
|
6
|
Qian X, Wu X and Liu X: Pulmonary alveolar
microlithiasis with finger clubbing: A case report and literature
review. Exp Ther Med. 11:1381–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Samano MN, Waisberg DR, Canzian M, Campos
SV, Pêgo-Fernandes PM and Jatene FB: Lung transplantation for
pulmonary alveolar microlithiasis: A case report. Clinics (Sao
Paulo). 65:233–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ozcelik U, Yalcin E, Ariyurek M, Ersoz DD,
Cinel G, Gulhan B and Kiper N: Long-term results of disodium
etidronate treatment in pulmonary alveolar microlithiasis. Pediatr
Pulmonol. 45:514–517. 2010.PubMed/NCBI
|
|
9
|
Cakir E, Gedik AH, Özdemir A,
Buyukpinarbasili N, Bilgin M and Ozgen IT: Response to disodium
etidronate treatment in three siblings with pulmonary alveolar
microlithiasis. Respiration. 89:583–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Ji CY, Zong XN and Zhang YQ: Height
and weight standardized growth charts for Chinese children and
adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi. 47:487–492.
2009.(In Chinese). PubMed/NCBI
|
|
11
|
Yin X, Wang H, Wu D, Zhao G, Shao J and
Dai Y: SLC34A2 Gene mutation of pulmonary alveolar microlithiasis:
Report of four cases and review of literatures. Respir Med.
107:217–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chu A, Shaharyar S, Chokshi B and Bhardwaj
N: Pulmonary alveolar microlithiasis ‘stone lungs’: A case of
clinico-radiological dissociation. Cureus. 8:e7492016.PubMed/NCBI
|
|
15
|
Field JA, Zhang L, Brun KA, Brooks DP and
Edwards RM: Cloning and functional characterization of a
sodium-dependent phosphate transporter expressed in human lung and
small intestine. Biochem Biophys Res Commun. 258:578–582. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Yin X, Wu D and Jiang X: SLC34A2
gene compound heterozygous mutation identification in a patient
with pulmonary alveolar microlithiasis and computational 3D protein
structure prediction. Meta Gene. 2:557–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mariotta S, Ricci A, Papale M, De Clementi
F, Sposato B, Guidi L and Mannino F: Pulmonary alveolar
microlithiasis: Report on 576 cases published in the literature.
Sarcoidosis Vasc Diffuse Lung Dis. 21:173–181. 2004.PubMed/NCBI
|
|
18
|
Lauta VM: Pulmonary alveolar
microlithiasis: An overview of clinical and pathological features
together with possible therapies. Respir Med. 97:1081–1085. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Terada T: Pulmonary alveolar
microlithiasis with cor pulmonale: An autopsy case demonstrating a
marked decrease in pulmonary vascular beds. Respir Med.
103:1768–1771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tachibana T, Hagiwara K and Johkoh T:
Pulmonary alveolar microlithiasis: Review and management. Curr Opin
Pulm Med. 15:486–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saito A, Nikolaidis NM, Amlal H, Uehara Y,
Gardner JC, LaSance K, Pitstick LB, Bridges JP, Wikenheiser-Brokamp
KA, McGraw DW, et al: Modeling pulmonary alveolar microlithiasis by
epithelial deletion of the Npt2b sodium phosphate cotransporter
reveals putative biomarkers and strategies for treatment. Sci
Transl Med. 7:313ra1812015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takahashi H, Chiba H, Shiratori M,
Tachibana T and Abe S: Elevated serum surfactant protein A and D in
pulmonary alveolar microlithiasis. Respirology. 11:330–333. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanat F, Teke T and Imecik O: Pulmonary
alveolar microlithiasis with epididymal and periurethral
calcifications causing obstructive azospermia. Int J Tuberc Lung
Dis. 8:12752004.PubMed/NCBI
|
|
24
|
Kim B, Winter TC III and Ryu JA:
Testicular microlithiasis: Clinical significance and review of the
literature. Eur Radiol. 13:2567–2576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pant K, Shah A, Mathur RK, Chhabra SK and
Jain SK: Pulmonary alveolar microlithiasis with pleural
calcification and nephrolithiasis. Chest. 98:245–246. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piesiak P, Kasibowska-Kuźniar K and
Jankowska R: Pulmonary alveolar microlithiasis in a patient with
urolithiasis and cholelithiasis. Pneumonol Alergol Pol. 69:285–289.
2001.(In Polish). PubMed/NCBI
|
|
27
|
Jönsson ÅL, Hilberg O, Bendstrup EM,
Mogensen S and Simonsen U: SLC34A2 gene mutation may explain
comorbidity of pulmonary alveolar microlithiasis and aortic valve
sclerosis. Am J Respir Crit Care Med. 185:4642012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan ED, Morales DV, Welsh CH, McDermott
MT and Schwarz MI: Calcium deposition with or without bone
formation in the lung. Am J Respir Crit Care Med. 165:1654–1669.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castellana G, Castellana R, Fanelli C,
Lamorgese V and Florio C: Pulmonary alveolar microlithiasis:
Clinical and radiological course of three cases according to
conventional radiology and HRCT. A hypothesis for radiological
classification. Radiol Med. 106:160–168. 2003.(In English,
Italian).
|
|
30
|
Gasparetto EL, Tazoniero P, Escuissato DL,
Marchiori E, Frare E Silva RL and Sakamoto D: Pulmonary alveolar
microlithiasis presenting with crazy-paving pattern on high
resolution CT. Br J Radiol. 77:974–976. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chatterji R, Gaude GS and Patil PV:
Pulmonary alveolar microlithiasis: Diagnosed by sputum examination
and transbronchial biopsy. Indian J Chest Dis Allied Sci.
39:263–267. 1997.PubMed/NCBI
|
|
32
|
Monabati A, Ghayumi MA and Kumar PV:
Familial pulmonary alveolar microlithiasis diagnosed by
bronchoalveolar lavage. A case report. Acta Cytol. 51:80–82. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ozçelik U, Gülsün M, Göçmen A, Ariyürek M,
Kiper N, Anadol D and Cobanoğlu N: Treatment and follow-up of
pulmonary alveolar microlithiasis with disodium editronate:
Radiological demonstration. Pediatr Radiol. 32:380–383. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mariotta S, Guidi L, Mattia P, Torrelli L,
Pallone G, Pedicelli G and Bisetti A: Pulmonary microlithiasis.
Report of two cases. Respiration. 64:165–169. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jankovic S, Pavlov N, Ivkosic A, Erceg I,
Glavina-Durdov M, Tocilj J, Dragisic-Ivulic S and Primorac D:
Pulmonary alveolar microlithiasis in childhood: Clinical and
radiological follow-up. Pediatr Pulmonol. 34:384–387. 2002.
View Article : Google Scholar : PubMed/NCBI
|