Case report
Clinical data
Case 1
The patient, male, 49 years old, was admitted to the
Department of Neurology, Daping Hospital, The Third Military
Medical University on 2016-02-13 due to ‘dizziness with hypomnesis
for one month, and mental and behavior disorder for three days’.
The patient suffered from dizziness and lethargy with myopia and
hypomnesis after catching a cold one month before admission. Three
days previously, his family found that the patient suffered from
balderdash, which was sometimes soliloquized and smirked alone.
Physical examination relieved that the patient had a clear spirit
and smooth communication, but gave irrelevant answers with
hypomnesis; The calculation ability was poor. There was no special
abnormality in the examination of the nervous system. After
admission, cranial MRI suggested that a change happened in the left
parietal lobe and bilateral caudate nucleus heads; infection was
not excluded. Cephalic and cervical CTA displayed no obvious
abnormality. The initial pressure in the lumbar puncture was 130 mm
H2O. The cerebrospinal fluid routine examination,
biochemical examination and virus DNA examination, and three-major
staining examination showed that the protein (0.57 g/l) was in the
normal range. EEG displayed moderate abnormality. The blood routine
examination, hepatic and renal function, blood glucose,
electrolytes, tumor markers, autoantibody spectrum, complete
thyroid function, blood folic acid and VitB12 were normal; TPPA
(−), anti-HIV (−). Blood, cerebrospinal fluid paraneoplastic
markers and complete brain examination were in the normal range. It
was considered that the patients might suffer from a central
nervous system infection - viral encephalitis, so the patient
received antiviral and symptomatic treatment.
After one week, the symptom of dizziness improved,
but the hypomnesis and mental and behavior disorder were not
improved significantly. The patient was discharged from the
hospital upon the request of the families. After one month, the
patient was admitted to the hospital again due to fever and
disturbance of consciousness. According to his families, the
patient suffered from the ‘paroxysmal twitch’ in both upper limbs.
Physical examination revealed that the body temperature was 38.5°C,
and the pulse was 140 times/min. Patient displayed moderately coma,
suspiciously positive neck resistance, abolition of orbital tension
reflex, isometric and circular bilateral pupils, sensitive to light
reflex, and bilateral pathological signs (−). A cranial MRI was
conducted again and showed that the intracranial abnormal DWI
signal were more than before. AEEG indicated periodic synchronous
discharge. The lumbar puncture examination was performed again and
the initial pressure was 160 mm H2O, and the
cerebrospinal fluid routine examination and biochemical examination
suggested that the protein (0.62 g/l) had no significant abnormity.
The cerebrospinal fluid specimens were submitted to the Chongqing
Center for Disease Control (CDC) for inspection, the results
suggested that the cerebrospinal fluid 14-3-3 protein was positive,
and the PRNP gene detection showed that the 129 amino acid
polymorphism was M/M type, and 219 amino acid polymorphism was E/E
type. There was no mutation found related to the genetic
Creutzfeldt-Jakob disease (CJD). According to the National
Surveillance Program on CJD, it was suggested to be diagnosed as
the sporadic CJD. The patient was followed up and deceased
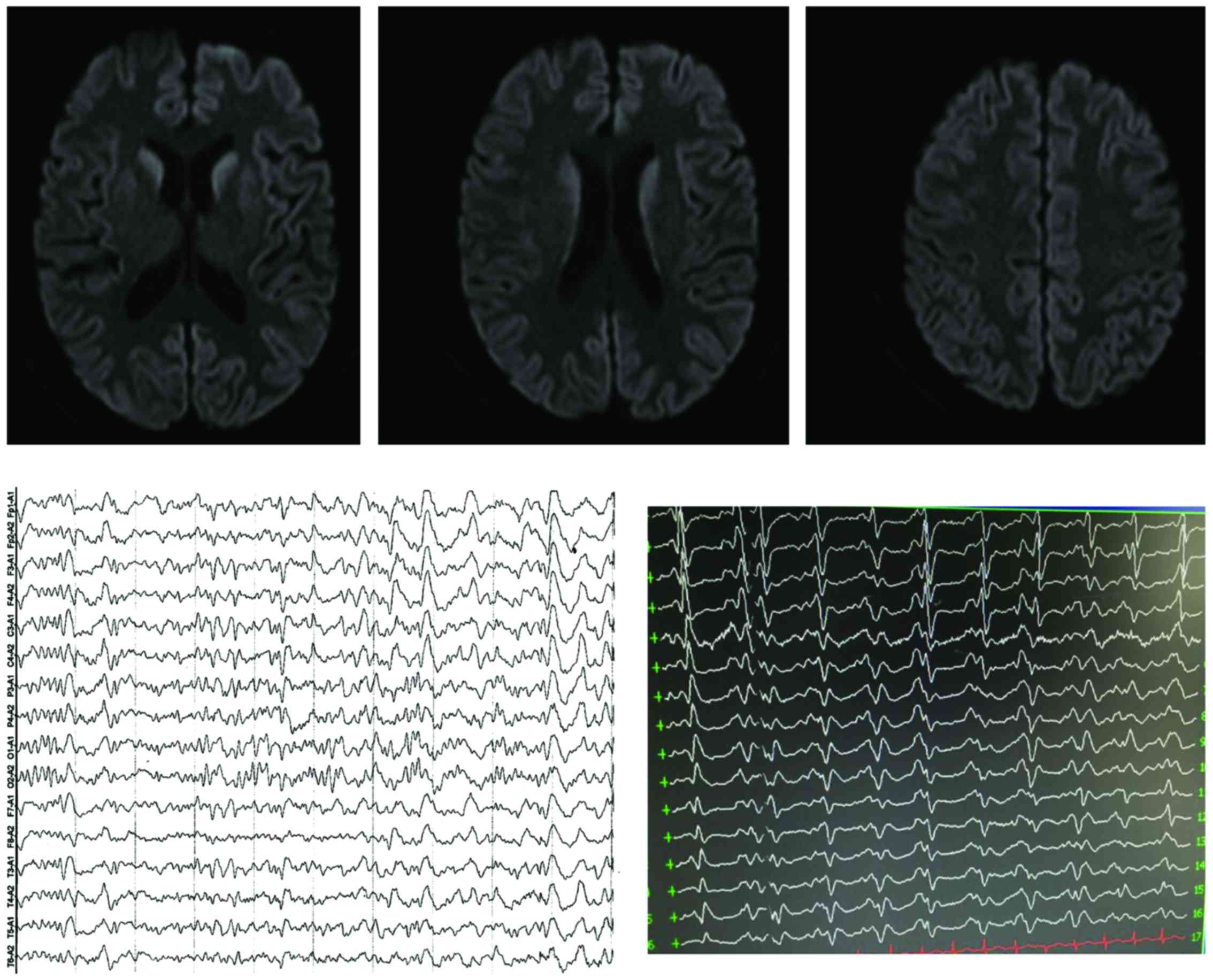
2016-3-16 (Fig. 1).
Case 2
The patient, female, 61 years old, was admitted to
hospital 2016-03-23 due to ‘progressive hypomnesis for half a year,
and exacerbation for more than 20 days’. During the past six
months, the patient suffered from hypomnesis without an obvious
reason, became forgetful and failed to recall what happened that
day or what she said at the moment, remember the name of her
friends, and got lost in a strange environment. She fell by
accident 20 days before admission, and then was slow in reacting
with attention deficit disorder, lack of words, and taciturnity and
communication difficulties with her family. Physical examination
revealed that she had clear consciousness that was slow in speech,
and decreased memory, calculation ability and directional ability,
but without significant abnormal signs in the nervous system. The
MMSE score was 12.
After admission, the cranial MRI suggested that the
change happened in the signal in bilateral cerebral hemisphere
cortices. The EEG demonstrated moderate abnormality. The blood
routine examination, hepatic and renal function, blood glucose,
electrolytes, tumor markers, autoantibody spectrum, complete
thyroid function, blood folic acid and VitB12 were normal, TPPA
(−), anti-HIV (−). The blood and cerebrospinal fluid specimens were
submitted to the CDC and results indicated that the negative
cerebrospinal fluid was 14-3-3 protein. The PRNP gene detection
showed that the 129-amino acid polymorphism was M/M type and
219-amino acid polymorphism was E/E type; the V203I-related
mutation was found in the PRNP gene detection. According to the
National Surveillance Program on CJD, the patient was suggested to
be diagnosed as genetic CJD. The patient was followed up for 40
days, the disease persisted for 7 months, and then the patient died
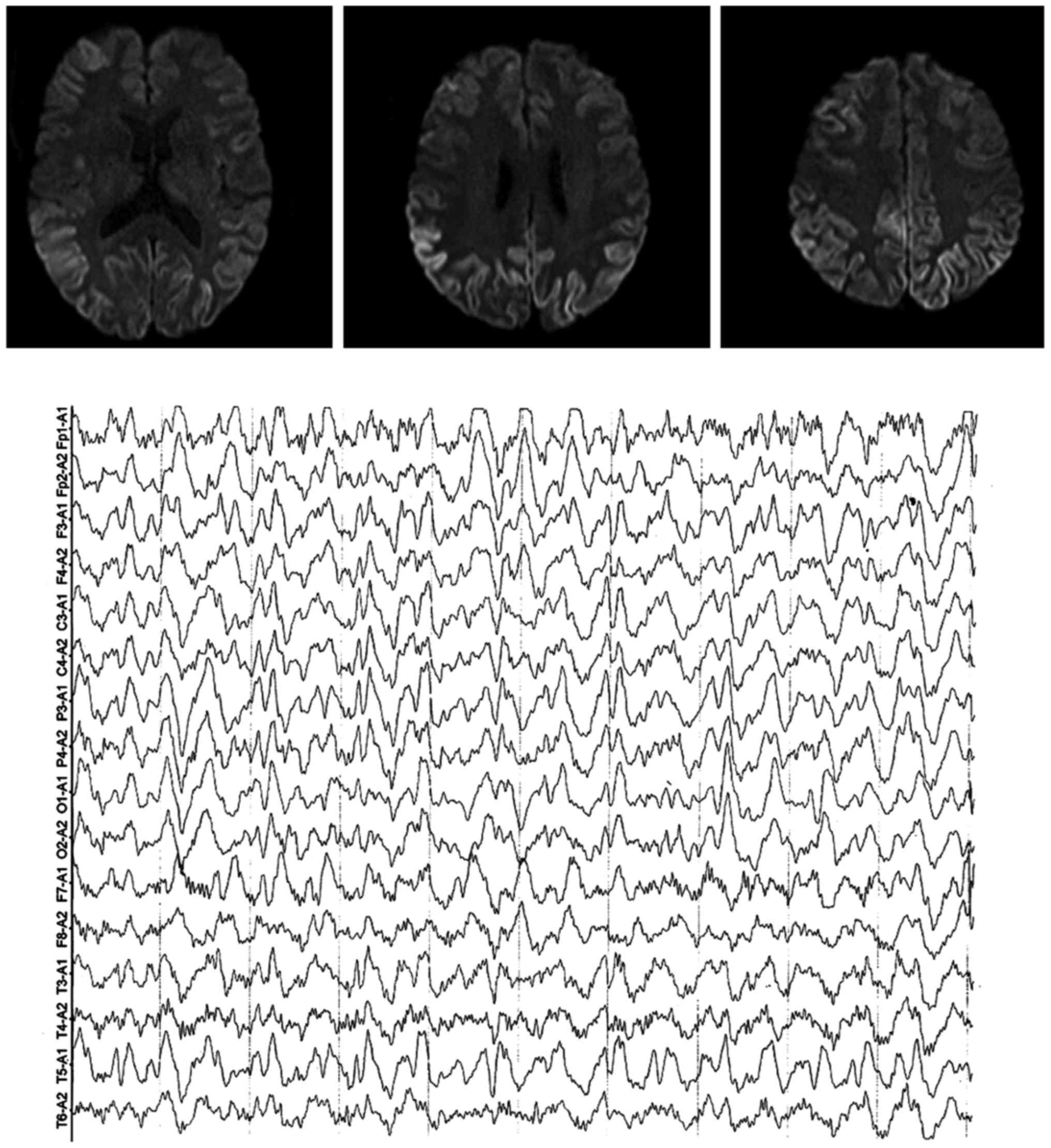
(Fig. 2).
Discussion
Creutzfeldt-Jakob disease (CJD) is reported as early
as in 1929 successively by Creutzfeldt and Jakob, which is one of
the most common types of PrP diseases, and appears mostly in
population aged 50–70 years old in either gender. Its incubation
period after infection is 4–30 years (1). CJD has the clinical features of rapidly
progressive dementia with myoclonus, and change in EEG and MRI,
which is characterized by proliferation, spongy degeneration and
neuronal loss of astrocytes. Abnormal PrP, the pathogenic factor of
CJD, has a very strong transmissibility, and its structure is hard
to be broken by ordinary disinfection measures, thus the early
recognition and diagnosis of CJD has a significant meaning for
preventing its spread.
According to different causes, CJD is divided
clinically into four types: sporadic CJD (sCJD), genetic or
familial CJD (f/gCJD), iatrogenic CJD (iCJD) and variant CJD
(vCJD). The most common type is sCJD, which accounts for 80–90% of
all CJDs. The pathogenesis is often unclear, and it is generally
believed that CJD is caused by PNRP gene mutations or automatic
conversion of normal prion protein due to unknown causes. The
disease develops quickly and the average survival period is
approximately 4 months; 85% of patients die within one year
(1). In this study, the first case
of the patient belongs to sCJD. The early performance of SCJD is
atypical, and is often difficult to identify, therefore, the
patient was misdiagnosed as viral encephalitis after admission, and
the antiviral treatment effect was not obvious. After one month,
the disease was suddenly aggravated with myoclonus, disturbance of
consciousness and typical periodic discharge in EEG, and thus, the
possibility of CJD was considered. Finally, the diagnosis was
confirmed through cerebrospinal fluid biomarker detection. gCJD has
the family history, and the mutant PRNP is obtained through
heredity, including a point mutant gene and insertional mutant
gene. Now, many mutant PRNP sites have been discovered. Unlike
other genetic diseases of the central nervous system, gCJD can
infect others, and gCJD accounts for 5–15% in all CJDs (2).
In this study, the second case of patient belongs to
this type. The patient suffered from progressive dementia, and even
though there was no typical clinical manifestations, such as the
myoclonus, mental disorder and ataxia, in the course of disease,
and there was no specific synchronous discharge in EMG, there were
typical ‘lace signs’ in the cerebral CT, and therefore, the
possibility of CJD was considered; it was confirmed finally through
genetic testing. vCJD was identified in 1996, which was caused by
human consumption of animals with bovine spongiform encephalopathy.
Most cases appeared in the UK, but it is also found in many other
countries (3). The clinical
manifestations and histology between vCJD and sCJD have obvious
differences. Currently, the PrP gene in all patients diagnosed
clearly with variant CJD, methionine homozygote located in the 129
codon was found. vCJD spread through blood transfusion was found,
which suggested that vCJD can be spread through blood products.
iCJD was found as early as in 1974 for corneal transplantation, and
caused by the horizontal transmission between people or between
people and animals; no case of vertical transmission has been found
yet. The routes of iatrogenic transmission mainly include dura
mater transplantation, corneal transplantation, the injection of
growth hormone and gonadotropin from the pituitary in infected
bodies, and the application of infected DEEG probe and
neurosurgical instruments.
The auxiliary examination for CJD include
pathological examination, EEG, imaging and cerebrospinal fluid
examination. Pathological examination is the gold standard in the
diagnosis of CJD. The typical neuronal death, proliferation of
astrocytes and spongiform encephalopathy in brain tissues are
observed microscopically, or the pathogenic-form PrP is found in
immunohistochemical detection. However, the pathological
examination is invasive, and combined with the strong infectivity
of prion, its clinical application is limited. Therefore, other
auxiliary examinations are very important to clinical diagnosis.
Genetic and family history can be used as a reference in diagnosis.
Moreover, the change in EEG is considered to be an important basis
of clinical diagnosis; the specific periodic synchronous discharge
(PSD) occurs in the middle and later periods, but there is only
extensive slow-wave activity in the early or interictal period, and
therefore, the EEG cannot be used as an effective early diagnostic
tool. AEEG examination can improve the positive rate.
In this study, EEG had no specific performance in
the first visit of the first case, however, the paroxysmal PSD was
captured in video EEG in the second visit. Cranial MRI
diffusion-weighted image (DWI) is both highly sensitive and
specific for early diagnosis. For patients with early CJD, the
abnormally high signals in the cortex and (or) basal ganglia region
can appear in DWI; the abnormal banded high signal along the cortex
is known as the ‘ribbon signs’ or ‘lace signs’. In the later period
of the disease, abnormally high signal in DWI will disappear. In
the MRI of vCJD, the ‘pulvinar nodule sign’ or ‘hockey stick sign’
in T2WI or FLAIR sequence has a specific diagnostic significance.
For the second case in this study, the typical change in DWI signal
in the cortex provides strong suggestions for diagnosis. In
addition, the change in multiple biomarkers of cerebrospinal fluid
can be used as auxiliary evidence of CJD diagnosis, such as the
protein Tau, 14-3-3 and S100, among which 14-3-3 is used more
widely. The number of broken cells released into the cerebrospinal
fluid increases in the onset of CJD, which has both high
specificity and sensitivity. There may be a false positive in
patients with subarachnoid hemorrhage, acute cerebral infarction,
infectious diseases of central nervous system, frontotemporal
dementia or Alzheimer's disease, and the false negative in patients
with atypical subtype and variant CJD or gCJD (4). The CJD monitoring network in China is
led by China CDC, and the monitoring network has covered most of
provinces in China (5). Samples of
both cases in this study were detected in CDC.
Geschwind et al (6) analyzed 114 sCJD patients, and found
that the most common initial symptom was cognitive impairment
(39%), followed by cerebellar signs (21%), and symptoms and signs
in behavior (20%), body (20%), feeling (11%), movement (9%) and
visual system (7%). The symptoms in behavior, body and feelings,
such as the headache, discomfort, and dizziness are not included in
the current diagnostic criteria. The diagnosis of CJD is conducted
according to the current standards of WHO (7): i) Patients with progressive dementia;
ii) patients with at least two or more symptoms listed below:
myoclonus, visual impairment, cerebellar ataxia, pyramid sign or
extrapyramidal sign, akinetic mutism; and iii) typical EEG
manifestations, namely the typical three-phase wave 1–2 times per
second. Patients that meet 3 standards are diagnosed as probable
CJD. Patients that meet the first 2 standards with the course of
disease <2 years are diagnosed as possible CJD. The brain
protein (cerebrospinal fluid 14-3-3 protein) detection can be used
to replace the specific change in EEG. In order to make a definite
diagnosis, the brain biopsy or autopsy must be conducted in order
to find specific neuropathologic change. Zerr et al
(8) added the imaging diagnostic
basis in 2009 based on the original diagnostic criteria of the WHO.
The revised new criteria increases the sensitivity of early
diagnosis, which have been adopted by the US CDC. Core symptoms
exist in both cases in this study, namely the rapidly progressive
dementia, and there were abnormal manifestations in different
degrees in head imaging examination.
The clinical manifestation of CJD and some non-PrP
diseases can be rapidly progressive dementia, but the early
diagnosis and treatment of some non-PrP diseases can control the
progression of cognition impairment, and therefore, it is
particularly important to identify this type of disease. In
clinical practice, special attention must be paid to the
identification between CJD and the following diseases: Alzheimer's
disease, frontotemporal dementia, dementia with Lewy body,
autoimmune encephalitis, paraneoplastic syndrome, central nervous
system infection, and Hashimoto's encephalopathy, toxic and
metabolic diseases. The standardized diagnosis process includes an
analysis of medical history, physical examination and auxiliary
examination item by item, which is helpful in diagnosis. The
auxiliary examinations, especially the early change in head MRI,
provide an important basis for identification and diagnosis.
However, very few patients still cannot be diagnosed clearly even
after comprehensive and systematic evaluation.
CJD has a certain infectivity, and therefore,
patients must be isolated and medical personnel should be protected
once it is confirmed. Daily treatment for patients are as follows:
i) The special ward or negative pressure ward is not needed, but a
single room is the best (convenient for treatment and care); ii)
the patient's excretion and urine do not need special disinfection
treatment; and iii) the ward does not need the special disinfection
treatment after the patient's admission or discharge, however, the
place contaminated by patient's blood or other samples can be
soaked in NaClO containing 20,000 ppm free chlorine or 2 mol/l NaOH
for 1–2 h for inactivation. Protection for medical personnel are as
follows: i) Try to avoid direct contact with CJD patient's blood;
ii) avoid direct perforating wound in nursing, examination and
treatment; iii) rinse immediately with plenty of water in the event
of an accident; and iv) wear gloves in daily contact with the
patient's body, but the respiratory protection is not needed. For
treatment with close contacts, no isolation or clinical observation
is needed. Principles of handling instrument and patients'
supplies: i) Try to use disposable instruments and supplies,
instrument contacting surface needs no special disinfection, but
instrument contacting tissues must receive special disinfection;
ii) the reusable non-disposable metal instrument can be inactivated
by high pressure steam at 134–136°C for 1–2 h, and the one-time
sharp instrument can be soaked in 2 mol/l NaOH for 1 h and
incinerated in the appropriate container; iii) the patient should
try to use disposable supplies, and those supplies must be
incinerated after patient's death; clothes and bedding polluted by
blood should be incinerated.
CJD is a fatal disease, and there is no effective
cure yet. The clinical treatment mainly includes symptomatic and
supportive treatment and intensive care. The main pathogenic factor
is the transformation of PrPC to PrPSc, and thus, stopping the
transformation of PrPSc has become the potential therapeutic target
(9). Research hotspots currently
include drug therapy, immunotherapy and RNA interference. In this
study, two cases of patients actively received symptomatic and
supportive treatment, but both of them died soon due to the lack of
efficient drug.
CJD has attracted more and more clinical attention
due to its atypical clinical manifestations and the lack of early
diagnostic method and effective treatment. We should clarify the
diagnosis with the aid of related auxiliary examinations,
especially imaging and cerebrospinal fluid examination. An earlier
diagnosis can provide a time window for treatment and avoid
unnecessary infection and doctor-patient dispute. Currently,
further research on anti-PrP drug, immunotherapy and gene therapy
is the development direction of successful treatment, and further
in-depth study is needed.
References
|
1
|
Sikorska B, Knight R, Ironside JW and
Liberski PP: Creutzfeldt-Jakob disease. Adv Exp Med Biol.
724:76–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosenbloom MH and Atri A: The evaluation
of rapidly progressive dementia. Neurologist. 17:67–74. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ironside JW: Variant Creutzfeldt-Jakob
disease: An update. Folia Neuropathol. 50–56. 2012.PubMed/NCBI
|
|
4
|
Zanusso G, Fiorini M, Ferrari S, Gajofatto
A, Cagnin A, Galassi A, Richelli S and Monaco S: Cerebrospinal
fluid markers in sporadic Creutzfeldt-Jakob disease. Int J Mol Sci.
12:6281–6292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao C, Shi Q, Tian C, Chen C, Han J, Zhou
W, Zhang BY, Jiang HY, Zhang J and Dong XP: The epidemiological,
clinical, and laboratory features of sporadic Creutzfeldt-Jakob
disease patients in China: Surveillance data from 2006 to 2010.
PLoS One. 6:e242312011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geschwind MD, Shu H, Haman A, Sejvar JJ
and Miller BL: Rapidly progressive dementia. Ann Neuro. l64:1–108.
2008.
|
|
7
|
WHO, . Global surveillance, diagnosis and
therapy of human Transmissible Spongiform Encephalopathies: Report
of a WHO consultation. Geneva, Switzerland: 9-11–February. 1998,
World Health Organization; Geneva, Switzerland: 1998.
|
|
8
|
Zerr I, Kallenberg K, Summers DM, Romero
C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B,
Ladogana A, et al: Updated clinical diagnostic criteria for
sporadic Creutzfeldt-Jakob disease. Brain. 132:2659–2668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marandi Y, Farahi N, Sadeghi A and
Sadeghi-Hashjin G: Prion diseases - current theories and potential
therapies: A brief review. Folia Neuropathol. 50:46–49.
2012.PubMed/NCBI
|